ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2007; 3(4):212-224. doi:10.7150/ijbs.3.212 This issue Cite
Research Paper
Characterization of transcriptional regulation of neurogranin by nitric oxide and the role of neurogranin in SNP-induced cell death: implication of neurogranin in an increased neuronal susceptibility to oxidative stress
Jingang Gui#, Yan Song, Nian-Lin Reena Han, Fwu-Shan Sheu ![]()
Department of Biological Sciences, National University of Singapore, 14 Science Drive 4, Singapore 117543, Singapore
# Current address: University of Texas, Health Center at Tyler, Tyler 75708, TX, USA.
Abstract
Neurogranin (Ng), a calmodulin (CaM)-binding protein kinase C (PKC) substrate, regulates the availability of Ca2+/CaM complex and modulates the homeostasis of intracellular calcium in neurons. Previous work showed Ng oxidation by NO donor induces increase in [Ca2+]i. The current study demonstrated that the gene transcription of Ng could be up-regulated by various nitric oxide (NO) donors via a NO-soluble guanylyl cyclase (sGC)-mediated pathway. Furthermore, ectopic expression of neuronal nitric oxide synthase (nNOS) in human embryonic kidney 293 cells (HEK 293) exhibited a nNOS-concentration-dependent biphasic regulatory effect on Ng gene transcription. One of the NO donors, sodium nitroprusside (SNP), however, induced cell death of neuroblastoma Neuro-2a cells. The potency of SNP-induced cell death was shown to be higher in Neuro-2a cells expressing recombinant Ng, as compared with Neuro-2a control cells without Ng expression in cell viability and apoptosis assays. Single-cell fluorescence imaging and site-directed mutagenesis studies suggest that Ng promotes SNP-induced cell death through an amplification of calcium-mediated signaling, which requires the interaction between CaM and IQ motif of Ng. Increased neuronal susceptibility rendered by Ng in response to pathophysiological NO production is suggested to be involved in the selective vulnerability of neurons to oxidative insults in the CNS.
Keywords: neurogranin, Ca2+/CaM complex, nitric oxide, IQ motif, cell death, calcium-mediated signaling.
1. Introduction
Neurogranin/RC3 (Ng) is a postsynaptic protein that binds to calmodulin (CaM) in the absence of or in low levels of Ca2+. As a Ca2+ capacitor, this protein is implicated in learning and memory by mediating long-term potentiation (LTP) and Ca2+/CaM-dependent pathways [1-3]. Like many other CaM-binding proteins, Ng contains an IQ domain interacting with CaM [4]. The CaM-binding affinity of Ng can be modulated by phosphorylation [5] and oxidation [6, 7]. The expression of Ng in the CNS is regulated by thyroid hormone, retinoic acid, and vitamin A [8, 9], insufficient of which are closely associated with an age-related decrease in Ng level [10-12]. A thyroid hormone responsive element located in the first intron of the human Ng gene homolog interferes with T3 trans-activation [13, 14]. Although the presence of several consensus responsive elements for glucocorticoids and retinoic acid were noted in the upstream promoter region of Ng, neither of these hormones influences the transcription of Ng in mammalian cells [15]. The phorbol ester and PKCs were found to stimulate the transcriptional activity directed by the 5'-flanking sequence of the rat Ng gene [16]. Transcription factors Sp1 and Sp3 were recently shown to modulate Ng transcriptional activity [17].
Rat Ng is a redox-sensitive protein containing four cysteine (Cys) residues that are readily oxidized by nitric oxide (NO) [7]. Oxidation of Ng forms an intramolecular disulfide bond and, similar to phosphorylation by PKC, reduces the binding affinity for CaM [18]. As a novel biological messenger synthesized by three main isoforms of NO synthase (NOS), NO can also regulate the expression of various eukaryotic genes [19]. It is of our interests to investigate whether the transcription of Ng gene, like many other genes, could be regulated by NO in addition to its biochemical oxidation effect on Ng.
NO exerts both beneficial and damaging effects in the CNS. Excessive NO production can cause oxidative stress in neurons, ultimately impairing neuronal function, and resulting in the neuronal death, which is implicated in many neuronal diseases [20, 21]. As a potential target of NO, Ng was suggested to promote calcium-mediated signaling [3] and was found to be a pro-apoptotic factor that increases the intracellular calcium concentration [22]. Ng was ranked as the top four abundant transcripts, constituting 0.36% of the total transcripts, more than its binding target calmodulin 1 (0.17%) and 3 (0.12%), in the mice cortex [23], suggesting that pharmacological treatment by NO to neurons expressing Ng is likely to observe an effect of its expression profile and cellular functions. Indeed, exogenous expression of Ng in neuroblastoma cells (Neuro-2a) was shown to amplify the intracellular calcium elevation induced by NO donors [24]. Therefore, it is of great interest to further investigate if Ng exhibits any relevance to NO-induced neuronal death. Our data in the present study not only reveal that NO could modify Ng transcription activity but also support our hypothesis that Ng, by interacting with CaM through its IQ motif, boosts neuronal vulnerability to the pathophysiological NO insults via amplifying calcium-mediated signaling.
2. Materials and Methods
Chemicals
Dimethyl sulfoxide (DMSO) was purchased from Sigma-Aldrich (Singapore). (±)-S-nitroso-N-acetylpenicillamine (SNAP), sodium nitroprusside (SNP), (Z)-1-[N-(3-Ammoniopropyl)-N-[4-(3-aminopropylammonio)butyl]-amino]diazen-1-ium-1,2-diolate (Spermine NONOate), 1-Hydroxy-2-oxo-3-(N-3-methyl-aminopropyl)-3-methyl-1-triazene (NOC-7), (Z)-1-[N-Methyl-N-[6-(N-methylammoniohexyl) amino]]diazen-1-ium-1,2-diolate (NOC-9), 3-morpholinosydnonimine.HCl (SIN-1.HCL), 4-Phenyl-3-furoxancarbonitrile, (+/-)-(E)-4-ethyl-2-[(E)-hydroxyimino]-5-nitro-3-hexenamide (NOR-3), 1H-[1, 2, 4] oxadiazolo [4, 3-a] quinoxalin-1-one (ODQ), A23187, were purchased from Calbiochem/EMD Biosciences (San Diego, CA). Each chemical was freshly dissolved either in phosphate buffered saline (PBS) or DMSO according to the manufacturer's instruction and was used immediately.
Molecular cloning
For constructing the reporter plasmid with a 5'-flanking sequence of the mouse Ng gene, the mouse (Swiss albino) genomic DNA was extracted by NucleoSpin® Tissue Kit (Macherey-Nagel, Düren, Germany). A 258 bp 5'-flanking sequence of mouse Ng gene (+3 to +260) was amplified by PCR using the following primers (forward, 5' GCTTGGCTGTTTGAGGTCC 3'; reverse, 5' GTGTTGAGGGTCCTTGGCT 3'). The PCR product was cloned into the pGEM-T vector (Promega, Madison, WI) and named pGEM-T-pNg (F). Plasmid pGEM-T-pNg (F) was used as a template for PCR and primed with following forward and reverse primers with Sac I and Xho I enzyme linkers, respectively (forward, 5' TAGGAGCTCGGTCCTCGCTCCAGTTCT 3'; reverse, 5' TGTTCTCGAGTGCCGGTGTTGAGGGTC 3'). The PCR product was purified and then digested by Sac I and Xho I and cloned into the pGL3-basic vector (Promega) digested by the same two enzymes. The final reporter construct was named pGL3-pNg (+3) and used in every promoter activity analysis experiment. For constructing a mammalian expression vector encoding wild type (WT) Ng protein, plasmid pET3b-Ng was used for PCR as a template. PCR was carried out using a forward primer corresponding to the start codon region of Ng with an upstream Nhe I site linker. The 3' primer was complementary to the stop codon region of Ng and contained a downstream Sac II site linker (forward, 5' TAAGCTAGCATGGACTGCTGCACGGAGAGCGCCTGCTCCAAGCCA 3'; reverse, 5' TAACCGCGGCTAGTCTCCGCTGG 3'). The PCR product was double digested by Nhe I and Sac II and linked with double cut (Nhe I and Sac II) pIRES2-EGFP vector (Clontech, Palo Alto, CA) by T4 ligase. The final expression plasmid was named pIRES2-WTNg-EGFP. For generating a plasmid encoding Ng with four Cys residues mutation (Cys3, Cys4, Cys9 and Cys51 were mutated to glycine), two consecutive steps were performed. The first step followed the same procedure that was used for generating the WT Ng expression plasmid except the forward primer contained single base substitutions for the codons of Cys3, Cys4, and Cys9 (5' TAAGCTAGCATGGACGGCGGCACGGAGAGCGCCGGCTCCAAGCCA3'). The Cys51 mutation was generated in the second step with the same strategy described previously [6]. The final plasmid containing coding sequence for Ng with mutations of all four Cys residues was named pIRES2-tetra-Ng-EGFP. The plasmid for expressing an I33Q mutation of Ng was made according to a previous method [4] using a GeneTailor™ Site-Directed Mutagenesis System (Invitrogen, Carlsbad, CA).
Cell cultures
HEK293, neuroblastoma Neuro-2a and hypothalamic GT1-7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS), 25 mM HEPES, 3.7 g/L NaH2CO3, 100 u/ml penicillin and 100 μg/ml streptomycin, with a final pH of 7.0-7.1. Cells were cultured in a humidified incubator with 5% CO2 at 37°C. The Neuro-2a clone1 cells were cultured with an addition of 0.5 mg/ml G418 antibiotic in the medium to maintain a selective pressure. For culturing primary cortical neurons, postnatal day 1 mice (Swiss albino) were decapitated and the cortical regions of the brain were carefully taken out. The cortical tissues were minced and immediately put into PBS solution containing 1% penicillin/streptomycin and 10 mM glucose with a steady oxygen perfusion for recovering the cells. After that, the cortical tissue was pelleted and trypsinized in a solution containing 0.1% (w/v) trypsin with 600 μl 10 U/ml DNase I dissolved in DMEM. After 20 min of incubation at 37°C, the tissue was mechanically dissociated and rinsed four times in PBS solution containing antibiotics and glucose. The cells were seeded on poly-L-lysine (50 mg/L) coated 35 mm culture dishes (BD Biosciences, Franklin lakes, NJ) and cultured in DMEM containing F12, N2 supplement and 10% heat-inactivated FBS at 37°C with humidity of 5% CO2. After cells attached to the bottom of the dishes, cytosine arabinonucleoside (Sigma) was added to the medium at a final concentration of 25 μM to inhibit the growth of neuroglia. One half of the medium was replaced with fresh medium every other day.
Transient transfection and luciferase activity measurement
All transfections were preformed at 80% cell confluence using TransIT-LT1 transfection reagent (Mirus, Madison, WI) according to the manufacturer's instructions. For experiments involving luciferase assays, 2 μg reporter plasmid pGL3-pNg (+3) was transfected into mammalian cells cultured on 6-well culture plates (Nunc, Roskilde, Denmark). One hundred and fifty nanograms of the renilla luciferase expression plasmid pRLSV40 (Promega) was co-transfected as a reference plasmid for normalizing transfection efficiency. NO donors were applied to the culture at 24 h post-transfection for 4 h, 12 h or 24 h according to experimental requirements. For pharmacological inhibition experiment, 10 μM ODQ was added in the medium 4 h before NOR-3 application and continue to co-culture with NOR-3. For some transfections, a plasmid expressing neuronal NOS (nNOS), pcDNA3.1-nNOS, was co-transfected with reporter plasmid and pRLSV40. Unless otherwise mentioned, cells were harvested 48 h post-transfection and lysed for luciferase assay and/or Western blotting. The luciferase activity was measured by a Turner TD 20/20 luminometer (Promega) using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions. For transfections of Ng-expressing plasmids for single-cell calcium imaging, 3 μg of each plasmid was used to transfect Neuro-2a cells grown on poly-L-ornitheine (Sigma) coated 13 mm plastic coverslips (Nunc) placed in 35 mm culture dishes. For cell viability assays, 1 μg of each Ng expression plasmid was used to transfect Neuro-2a cells cultured in 48-well culture plates (Nunc).
Western blotting
Total protein was extracted from cultured mammalian cells by modified radioimmunoprecipitation (RIPA) buffer (50 mM Tris-HCl pH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM Phenylmethylsulphonyl fluoride, 1 μg/ml Aprotinin, 1 μg/ml Pepstatin A, 1 mM Na3VO4, 1 mM NaF). One tablet of cocktail protease inhibitor (KPL, Gaithersburg , MD) was added to the buffer immediately before use. In brief, for a 10 mm culture dish, 200 μl RIPA buffer (volume was adjusted accordingly in 6-well and 48-well culture plates) was added and cells were detached by a cell scraper. Mixture was transferred into a 1.5 ml eppendorf tube and incubated on an orbital shaker at room temperature (RT) for 15 min. The mixture was thawed after being frozen overnight at -20°C and centrifuged at 10000 g for 15 min at 4°C. The cell debris was discarded and the concentration of protein in the supernatant was measured by Bradford protein assay reagent (Bio-Rad). An aliquot of 50 μg protein sample was frozen at -80°C or immediately analyzed by Western blotting. For experiment of analyzing nNOS, total protein was boiled at 95°C for 5 min in protein sample buffer (5 x sample buffer: 2 ml 20% SDS, 2.5 ml 0.5 M Tris-HCl pH 6.8, 1 ml β-mercaptoethanol, 2.5 ml glycerol). Denatured protein was chilled on ice for 5 min and separated on 12% SDS-polyacrylamide gel by a SE 250 vertical electrophoresis apparatus (Hoefer, San Francisco, CA). For analyzing Ng expression, protein was separated in non-reducing 10%-20% gradient SDS-polyacrylamide. Bromophenol blue and β-mercaptoethanol were omitted in sample buffer and boiling of sample was avoided. Protein was transferred to Hybond-C nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ). The membrane was then peeled off from the gel and rinsed with 1 x TBST (TBS plus 0.05% Tween 20) once and blocked in 1 x TBST with 5% nonfat dry milk overnight at 4°C on a rocking platform. The blot was then incubated with rabbit polyclonal anti-nNOS (at a 1:5000 dilution; Zymed laboratories, San Diego, CA) or rabbit polyclonal anti-Ng (at a 1:1500 dilution; made from the synthetic peptide of the C terminal 66–78 amino acid sequence of Ng, Biogen, Germany). After washing three times with 1 x TBST, the blot was incubated with horseradish peroxidase-conjugated anti-rabbit IgG (Chemicon, Temecula, CA) at a 1:10,000 dilution for 2 h at RT. Detection of immunoactive bands was performed by incubating the blot with SuperSignal West Pico Chemiluminescent Substrate solution (Pierce, Rockford, IL) for 2 min, followed by exposure to CL-X Posure™ Film (Pierce). For reprobing experiments to correct the protein loading difference, blots were stripped in stripping buffer (1.56 ml 1 M Tris pH 6.7, 175 μl β-mercaptoethanol, 5 ml 10% SDS, bring up to 25 ml with double distilled H2O) and incubated with anti-α-tubulin (clone DM1A, Sigma).
Cell viability and apoptosis assays
MTT reduction assay. Cells were counted by a hemocytometer and equally distributed in 48-well plates at a density of 5 x 104 cells/well and treated with various concentrations of SNP for 12 h, or A23187 for 24 h. To determine cell viability, the DMEM medium was removed and cells were incubated with 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT, Sigma) at a final concentration of 0.5 mg/ml in phenol-red-free RPMI 1640 medium (Hyclone, Logan, UT) containing 10% heat-inactivated FBS for 2 h in the dark at 37°C. Then 200 μl dissolving buffer (11 g SDS in 50 ml of 0.02 M HCl, mixed with 50 ml isopropanol) was added to the wells with gentle trituration. Cultures were incubated with the dissolving buffer at RT in the dark for 1 hour. After the formazan dye was totally dissolved, the solution was transferred to a 96-well Elisa plate (Nunc) and read at 570 nm by a Spectra Max 340 microplate reader (Molecular Devices, Sunnyvale, CA). In MTT assays for the cells transiently transfected with expression plasmid of WT Ng and Ng derivatives bearing I33Q or 4 Cys residues mutation, total protein in the parallel culture plates (with plasmid transfection but without SNP treatment) was extracted and used in Western blotting with polyclonal anti-GFP (Upstate, Milton Keynes, UK). Signals on immunoblot were quantified by densitometric measurement and used to normalize the transfection difference in each MTT assay.
DNA fragmentation assay. Apoptotic cells were collected by centrifugation at 1500 g for 5 min. The cell pellet was washed once with PBS and lysed by a solution containing 10 mM EDTA, 5 mM Tris-HCl (pH 8.0) and 0.5% Triton X-100 for 30 min on ice. Sample was centrifuged and the supernatant (low molecular weight DNA) was separated from the pellet (high molecular weight DNA). Both fractions were digested with RNase A (500 U/ml) for 1 h at 37°C and then digested with proteinase K (0.5 mg/ml) for 3 h at 50°C. The sample was extracted by phenol and phenol/chloroform/isoamyl alcohol (25:24:1, V/V/V), precipitated with 2.5 volume of 95% ethanol and 1/10 volume of sodium acetate (3M, pH 5.2), and then dissolved in Tris-EDTA buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA pH 8.0). DNA was electrophoresed in 1.5% agarose gel in 1 x TBE buffer. The gel was stained with ethidium bromide (2 mg/L) for 15 min and distained 20 min with Milli-Q water. DNA fragments were visualized under UV light and documented.
Quantitative real-time PCR
Primary cortical neurons on the 12th day of culture were incubated either with 200 μM NOR-3 or DMSO in medium for 24 h. After the treatment, the medium containing NOR-3 or DMSO and the medium in the non-treated control were immediately replaced with fresh medium and cultures were kept in a CO2 incubator for another 12 h. The cortical neurons then were washed once with PBS, and total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. DNase I treated RNA was reverse transcribed with Superscript II (Invitrogen). Real-time PCR was performed using syber green real-time PCR mix (Applied Biosystems, Foster City, CA). Primers for Ng are (forward, 5' TCCAAgCCAgACgACgATATT 3'; reverse, 5' CACACTCTCCgCTCTTTATCTTC 3'). Primers for the housekeeping gene β-actin are (forward, 5'CAGCCCTCCTTCCTCGGTAT 3' Reverse, 5'GATGTCCACGTCACACTTCATGAT 3'). In brief, 10 μl of each diluted cDNA was added to 12.5 μl SYBR green PCR mixtures with 1.5 μl H2O, and 1.25 pmol primer mix. PCR reactions were performed with the ABI Prism 7000 sequence detection system (Applied Biosystems). Quantitative analyses of target genes were done by using a standard curve for β-actin, which was compared with a target gene curve using triplicate samples in each cDNA dilution. In all experiments, non-template controls were run together with the samples.
Single-cell fluorescence imaging
Transiently transfected Neuro-2a cells grown on coverslips were washed twice with 1 x PBS and cultured in indicator-free RPMI 1640 medium with 10% heat-inactivated FBS. One micromolar calcium dye x-rhod-1 (AM) (Molecular Probes, Eugene, OR) was dissolved in DMSO and loaded into the cells by incubation at 37°C with 5% CO2 for 1 h. Before fluorescence measurements, cells were washed in indicator-free RPMI 1640 to remove any dye that was nonspecifically attached to the cell surface, and then incubated for a further 30 min to allow complete de-esterification of intracellular AM esters. After that, the medium in the culture dish was removed and replaced with Krebs buffer (20 mM HEPES pH 7.4, 145 mM NaCl, 10 mM glucose, 5 mM KCl, 2 mM CaCl2, 1.3 mM MgCl2, and 1.2 mM NaH2PO4). Fluorescence measurements were performed at RT with an upright E600FN microscope (Nikon, Japan) installed with a HBO 100w/2 mercury bulb (Osram, Munich, Germany) and a Fluor 40× (0.80 W) water-immersion objective lens. Images were acquired by a 12-bit SensiCam cooled CCD camera (PCO, Molecular Devices) and analyzed using Axon Imaging Workbench (AIW) software. Region of Interest (ROI) was drawn using a Chroma 41017 filter set (Chroma, Rockingham, VT). Fluorescence was measured with excitation at 550nm and emission at 580nm using a Chroma 31004 filter set (Chroma). Changes of the fluorescence intensity were recorded as F/F0 by a non-ratiometric protocol defined by AIW software. SNP was added slowly without disturbing the cells on the plastic coverslips.
Statistics
One-way ANOVAs were used to test for differences between experimental groups. Where appropriate, data were compared with Student's t-test. Data are expressed as the mean ± SEM. The p value < 0.05 was considered to be significant.
3. Results
Nitric oxide donors regulate Ng promoter activity in HEK 293 cells and Ng transcription in primary cortical neurons
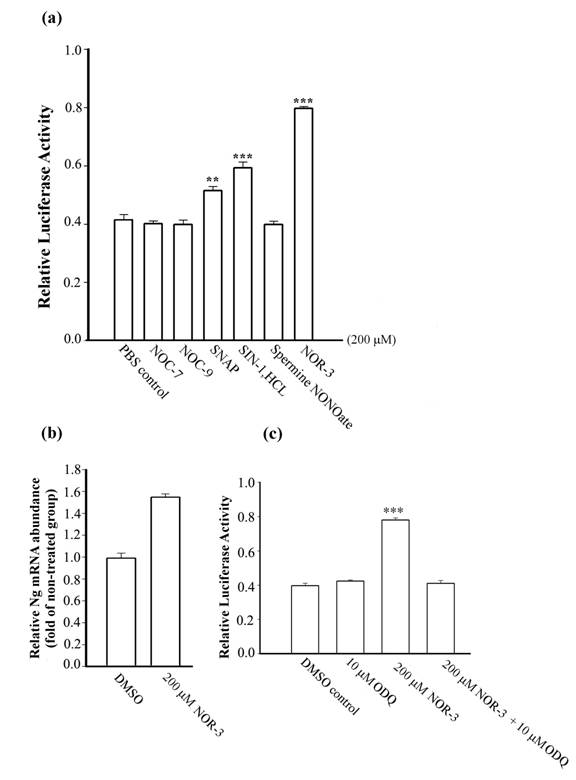
To examine the possibility that NO donors modify Ng transcriptional activity, various NO donors at 200 μM were applied to cell culture for 24 h. Ng promoter activities measured by the firefly luciferase activity were shown to be up-regulated by 1.3 fold with SNAP, 1.4 fold with SIN-1.HCL and 1.9 fold with NOR-3 (Fig. 1a). Other NO donors, such as NOC-7, NOC-9, and spermine NONOate, did not exhibit significant influence on the Ng promoter activity as compared to the control group treated only with PBS. Data were not obtainable due to massive cell death in SNP and 4-Phenyl-3-furoxancarbonitrile treated groups for 24 h at the same cultured conditions. To further confirm our result, we tested whether the NO donor with highest stimulation on Ng promoter, NOR-3 is able to up-regulate Ng transcription in primary cortical neurons that have basal expression of Ng to show its physiological relevance. Real-time PCR showed that the relative abundance of Ng mRNA was increased by 1.6 fold in primary neurons treated with NOR-3 as compared to the non-treated control group (Fig. 1b). Various lines of evidence showed that NO exerts one of its cellular actions by binding to the heme moiety of the enzyme soluble guanylyl cyclase (sGC), which causes an accumulation of cyclic guanosine monophosphate (cGMP) and leads to a wide diversity of biological functions [25, 26]. To study the involvement of sGC on NO-induced Ng transcriptional activity, we examined how NOR-3 would influence on Ng promoter activity in the presence of the sGC inhibitor, ODQ [27], at a concentration of 10 μM. Fig. 1c showed that with 4 h pre-treatment with ODQ and co-incubation with NOR-3, the up-regulation of Ng promoter activity by NOR-3 in HEK 293 cells was markedly inhibited. In our hands, 10 μM ODQ alone was unable to modify Ng promoter activity as compared with the control group which was treated either with DMSO or PBS. That the NO-induced Ng promoter activity could be inhibited by ODQ suggested that the up-regulation of Ng promoter activity by exogenous NO stimulation is through a sGC-dependent pathway.
Analysis of Ng promoter activity upon NO donor stimulation in HEK 293 cells and Ng mRNA abundance in primary cortical neurons. (a) Ng promoter activity was measured in HEK 293 cells stimulated with various kinds of NO donors (indicated below each bar) at 200 μM concentration for 24 h. (b) Relative Ng mRNA abundance in 12-day cultured cortical neurons with NOR-3 treatment for 24 h was assessed by real-time PCR. PCR results were represented as relative folds of mRNA level over non-treated group (arbitrarily assigned as 1 fold). (c) Ng promoter activities were measured in HEK 293 cells with stimulation of ODQ alone or NOR-3 in the presence or absence of ODQ. Data are represented as means ± SEM (n=3) and tested by Student's t-test. ** p < 0.01, *** p < 0.001 (compared with the control group).
nNOS exhibits a concentration-dependent biphasic effect on Ng promoter activity
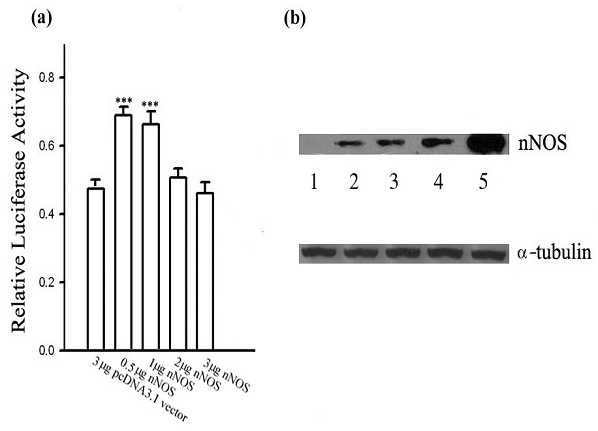
Given the effect of exogenous NO on the transcriptional activity of Ng shown above, it is necessary to know whether endogenous NO produced by nNOS in the same cellular system has similar effects on Ng promoter activity. To answer this question, an expression plasmid encoding for nNOS was cotransfected to HEK 293 cells with the reporter construct containing the Ng promoter, in such a way that the correlation between nNOS expression level and Ng promoter activity could be delineated. As shown in Fig. 2a, changes of Ng promoter activity were largely dependent on the protein level of nNOS ectopically expressed in HEK 293 cells. Ng promoter activity increased significantly when 0.5 or 1 μg of the nNOS cDNA was transfected. However, the up-regulation was attenuated by higher amount of nNOS cDNA (2 μg and 3 μg) transfected. As evidenced by Western blotting in Fig. 2b, the quantity of nNOS expression plasmid transfected was closely in proportion to the expression level of nNOS in the cell. Data illustrated that Ng promoter activity was transactivated by low threshold level of nNOS expression while higher expression of nNOS in HEK 293 cells was unable to further up-regulate Ng promoter activity. Experiments on Neuro-2a cells showed the similar regulatory pattern (data not shown). These results suggest that Ng promoter activity could be regulated with a biphasic pattern depending on a certain threshold level of endogenous NO production by nNOS.
nNOS exhibits a dose-dependent biphasic effect on Ng promoter activity. (a) Ng promoter activities were measured in HEK 293 cells with cotransfection of indicated concentrations of pcDNA3.1-nNOS. The total amount of plasmid transfected was normalized with pcDNA3.1 vector. Data are shown as means ± SEM (n=6) and processed with one-way ANOVA followed by post-hoc analyses of Student's t-test. *** p< 0.001 (compared with the control pcDNA3.1 vector group). (b) Representative nNOS protein expression levels in HEK 293 cells cotransfected with pcDNA3.1 vector in lane 1, 3 μg, or pcDNA3.1-nNOS plasmid in: lane 2, 0.5 μg; lane 3, 1 μg; lane 4, 2 μg and lane 5, 3 μg. Aliquots of extracted protein were used either for luciferase measurement as shown in panel (a), or for Western blotting using nNOS antibody as shown in this panel. Western blotting for α-tubulin was used as a loading control.
High dose of NO donor SNP suppresses Ng promoter activity in the presence of Ng protein
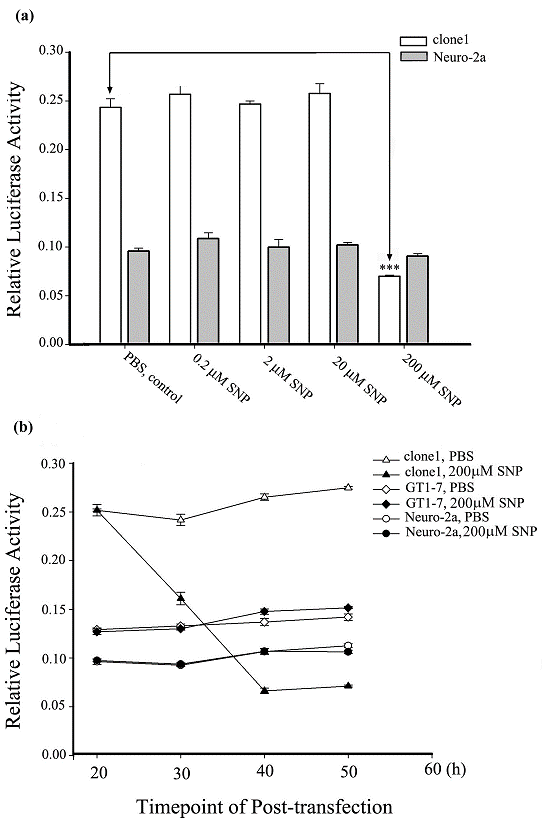
For many years, investigators have been unsuccessful in finding a cell line with endogenous expression of Ng. It was revealed that Ng mRNA was induced by T3 in the hypothalamic cell line GT1-7. Unfortunately, no Ng was detected at a translational level even with a T3 treatment [15]. The clone1 cell used in the present study is a stably Ng-expressing Neuro-2a cell line [24]. Our previous data has shown that upon SNP treatment in short duration of 3 min, Ng-expressing Neuro-2a cells showed amplified calcium elevation versus the control Neuro-2a cells without Ng expression. We suspected that this modulated calcium signaling by Ng expression upon NO treatment might have some influence on Ng transcription. The present results revealed that SNP exhibited a differential effect on the Ng promoter activity (expressed by relative luciferase reporter assay) between the Ng-free Neuro-2a cells and the Ng-expressed clone1 cells. The histogram in Fig. 3a showed that upon treatment with SNP for 4 h at a concentration range from 0.2 to 20 μM, Ng promoter activity was neither significantly changed in Neuro-2a cells nor in clone1 cells. Surprisingly, although 200 µM SNP virtually had no effect on the Ng promoter activity in Neuro-2a cells, it markedly suppressed Ng promoter activity in clone1 cells. Data on high dose of 2 mM SNP stimulation was not obtained because both Neuro-2a and clone1 cells showed typical signs of apoptosis with round and shrunken shape. It is worthy to note that the reporter activity of Ng promoter were more than 2 folds higher in the Ng-expressed clone1 cells than those of the Ng-free Neuro-2a cells for the lower ranges of SNP treatments (0.2 µM to 20 µM) and the control group without SNP treatment group. There was a slight increase in Ng promoter activity for both clone1 and Neuro-2a groups at the lowest 0.2 μM treated SNP although it did not reach a significant level. To observe a dynamics of Ng promoter activity change in response to SNP stimulation, we compared Ng promoter activities in response to 200 μM SNP treatments among Neuro-2a, clone1 and GT1-7 cells with a 30 h time course. As shown in Fig. 3b, Ng promoter activities were determined every 10 h after incubation of SNP in 4 h (i.e. 200 μM SNP was applied during 20 h-24 h post-transfection of the promoter vector and then was washed away with fresh medium). The results revealed that the baseline of Ng promoter activity in different cell lines at the start point was different. The clone1 cells had the highest reporter activity among the three cells tested. However, at 30 h post-transfection, the Ng promoter activity in clone1 cells decreased by 40% of its initial activity that was measured immediately before 200 μM SNP application. Ng promoter activity continued to decrease to approximately 20% of its initial activity at 40 h post-transfection and kept constant from 40 to 50 h. In contrast, Ng promoter activity maintained virtually unchanged throughout the whole time course in either Neuro-2a or GT1-7 cells upon 4 h SNP treatment. The bifurcating response of Ng promoter activity to SNP stimulation between Ng-expressing cells and Ng-free cells implied that Ng transcription was affected either by cell signaling or changed cell conditions caused by Ng expression. Based on our further observation, we found that at the time-point of 40 h post-transfection, most of clone1 cells were shrunk and showed a morphological abnormality. The extremely low promoter activity was actually resulting from the pathophysiological change of clone1 cells.
SNP suppresses Ng promoter activity in the presence of Ng protein. (a) Ng promoter activities were measured in Neuro-2a and clone1 cells with stimulation of indicated concentrations of 4 h SNP treatment. Data were processed by one-way ANOVA and represented as means ± SEM (n=3). *** p < 0.001 (compared with clone1 control). (b) Ng promoter activities were measured at various time points in indicated cell lines with either PBS or 200 μM SNP treatment. PBS or SNP were applied to cultures 20 h post-transfection and incubated for 4 h followed by replacing with fresh medium. Data are shown as means ± SEM (n= 6).
Ng expression promotes SNP-induced cell death
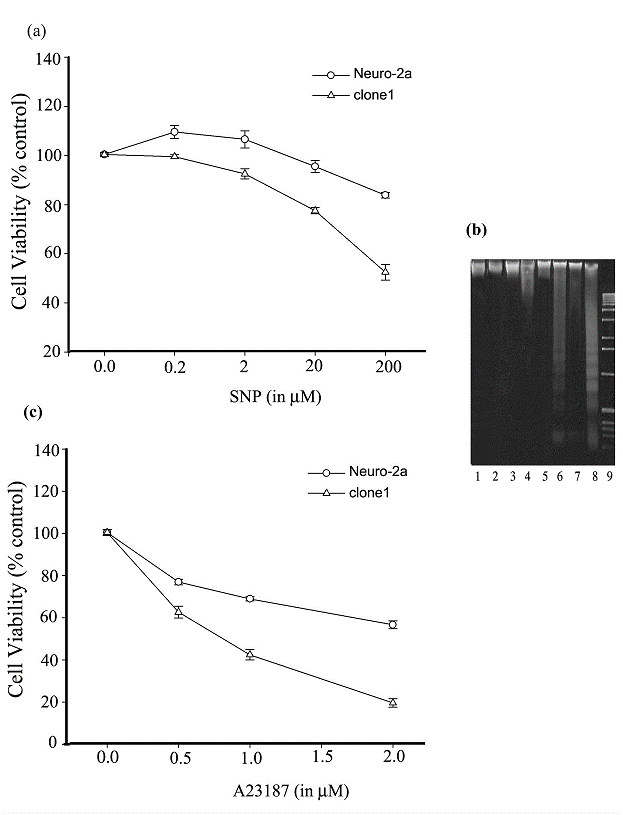
In recent years, NO has received increasing attention for its implication in neuronal death, which is a feature of many neurological disorders. In the present study, we reasoned that Ng protein may enact an additional effect on the dys-regulation of calcium homeostasis resulting from the uncontrolled overdose of NO stimulation, and consequently, promotes NO-induced cell death. To test our conjecture, SNP stimulation of clone1 and Neuro-2a cells was conducted and the percentage of viable cells was determined by an MTT reduction assay. As shown in Fig. 4a, a 12 h prolonged stimulation with 0.2 and 2 μM SNP promoted a slight proliferation of Neuro-2a cells while 20 and 200 μM SNP caused a decrease in the viability of the cells. On the other hand, prolonged SNP stimulation induced cell death of the Ng-expressing clone1 cells in a dose-dependent manner although at 0.2 and 2 μM it only caused a slightly decrease in cell viability. Around 25% of clone1 cells suffered cell death at a 20 μM SNP treatment and virtually 50% of them died at a 200 μM SNP treatment. At each concentration level of SNP, clone1 cells were induced to death with a larger proportion than that of Ng-free Neuro-2a cells. The extreme difference of cell viability between Neuro-2a and clone1 cells occurred at a 200 μM SNP treatment, where cell viability in Neuro-2a cells was up to 85% of its untreated control level in comparison to that in clone1 cells which was only 50% of its control level. Apoptosis analysis revealed that DNA fragmentation was more easily induced in clone1 cells than in Neuro-2a cells upon a prolonged SNP stimulation. As depicted in Fig. 4b, while DNA fragmentation was found neither in clone1 nor Neuro-2a cells in response to 0.2 or 2 μM SNP treatment (lane 1 to 4), it was easily identified in clone1 cells subjected to a 20 or 200 μM SNP treatment (lane 6 and 8). With this same treatment, a lesser degree of DNA fragmentation was found in Neuro-2a cells (lane 5 and 7). These assays revealed that Ng expression synergistically promotes the SNP-induced apoptotic action on Neuro-2a cells. We then asked the question whether a cytotoxicity conferred by elevated intracellular calcium could also be promoted by Ng expression. In so doing, we compared the cell viability of clone1 and Neuro-2a cells upon stimulation of a calcium ionophore, A23187. As shown in Fig. 4c, after 24 h incubation with A23187, a dose-dependent decrease in cell viability was found in both cell lines, but at each concentration level of A23187 (0.5, 1, and 2 μM), clone1 cells exhibited a greater potentiality of death than Neuro-2a cells (60% vs. 80%, 40% vs. 70% and 20% vs. 60%, respectively). These assays indicate that with the expression of Ng the cell becomes more susceptible to a given extracellular calcium influx leading to cell death.
Ng promotes Neuro-2a cell death induced by prolonged SNP or A23187 stimulation. (a) Cell viability of Neuro-2a and clone1 cells was determined by MTT assay after 12 h stimulation with various concentrations of SNP. (b) Detection of DNA fragmentation induced by prolonged SNP stimulation (12 h) in Neuro-2a and clone1 cells. Lanes 1, 3, 5, 7 correspond to Neuro-2a cells treated with 0.2, 2, 20, 200 μM SNP, respectively. Lanes 2, 4, 6, 8 correspond to clone1 cells treated with 0.2, 2, 20, 200 μM SNP, respectively. Lane 9, 1 kb DNA ladder. (c) Cell viability of Neuro-2a and clone1 cells were determined by MTT assay after 24 h stimulation with various concentrations of A23187. For panel a and c, the cell viability was calculated by the following equation: MTT OD value of sample/MTT OD value of control (cell treated with PBS). Data are shown as means ± SEM (n=6).
A functional IQ motif of Ng is required for promoting SNP-induced cell death
Like other IQ motif-containing CaM-binding proteins [28], Ng exerts its functions by interacting with CaM at the IQ motif. Now that our results have shown that Ng expression promotes SNP-induced cell death, we further attempted to elucidate the role of Ng-CaM interaction in promoting cell death. As oxidized Ng has a reduced affinity for CaM, we were also interested in examining which factor, the oxidizable Cys residues or the CaM interacting IQ motif of Ng has any relevance to the Ng-promoted SNP-induced cell death. To answer these questions, we made two site-directed mutants of Ng expression plasmid based on previous reports. One plasmid was to make a specific single amino acid substitution (I33 to Q) in the IQ motif to ablate the binding of Ng to CaM in vivo [4]. The other was to disrupt all four Cys residues in the Ng protein sequence to abolish the oxidation [6]. Hence, we compared SNP-induced cell death among cells expressing wild type (WT), I33Q mutant or Cys mutant Ng. The survivability of Neuro-2a cells transiently transfected with WT or Ng mutants were assessed by MTT reduction assay after 12 h prolonged incubation with 200 μM SNP. The bar chart in Fig. 5a showed that Neuro-2a cells expressing WT Ng suffered a severe NO-induced cell death and the cell viability reached less than 60% of the control level (the control group is Neuro-2a cells transfected with pIRES2-EGFP vector). In contrast, Neuro-2a expressing I33Q mutant Ng obtained cell viability at 90% of the control level. Much to our surprise, Neuro-2a cells expressing Cys mutant Ng also suffered a massive cell death upon prolonged stimulation with 200 μM SNP. The degree of cell death was quite similar to that of cells expressing WT Ng. Cys mutant Ng lacking all four Cys residues was resolved on 10-20% non-reducing gradient SDS-PAGE with disappearance of the fast mobility shift band which is the oxidized form of Ng (Fig 5a, lane 4). Expression of WT Ng or I33Q mutant Ng in Neuro-2a cells resulted in both reduced and oxidized forms (Fig 5a, lane 2 and 3, respectively). This assay in the first place evidenced that Ng with a disrupted IQ motif was unable to promote SNP-induced cell death and implied that a functional IQ motif is indispensable for this promoting effect. Secondly, in this assay, abolishing Ng oxidation did not salvage the SNP-induced cell death. The Cys mutant Ng that still contains a functional IQ motif may be a good indication for this phenomenon.
A functional IQ motif in Ng is required for promoting intracellular calcium increase and NO-induced cell death. (a) Following transfection with different forms of Ng cDNA constructed into pIRES2-EGFP vector, Neuro-2a cells were treated with 200 μM SNP for 12 h. Cell viability was measured by MTT reduction assay. Data were normalized by densitometric scanning of film from Western blotting against EGFP. Data were represented as % of the control (normalized MTT reading from Neuro-2a cells transfected with pIRES2-EGFP vector). Data shown are means ± SEM (n=6). *** p < 0.001 (compared with the vector control by Student's t-test). Transient expression of variations of Ng in Neuro-2a cells were resolved on 10% to 20% gradient non-reduced PAGE and subjected to Western blotting against Ng antibodies. Lane 1, transfected with pIRES2-EGFP vector; 2, transfected with WT Ng cDNA; 3, transfected with I33Q Ng cDNA; 4, transfected with Cys mutant Ng cDNA. (b) Following transfections, Neuro-2a cells were loaded with x-rhod-1, and successfully transfected cells were identified by a green fluorescence filter and chosen as region of interest (ROI). Fluorescence changes were monitored using a filter for x-rhod-1. Representative normalized (F/F0) fluorescence changes versus time are shown in the line plot. Arrow indicates the SNP application time. The bar chart shows averaged peak fluorescence changes from various transfections indicated in response to 200 μM SNP. Data shown are means ± SEM (n=9 for all transfections). ***p < 0.001 obtained from Student's t test (compared with the data from pIRES2-EGFP transfection).
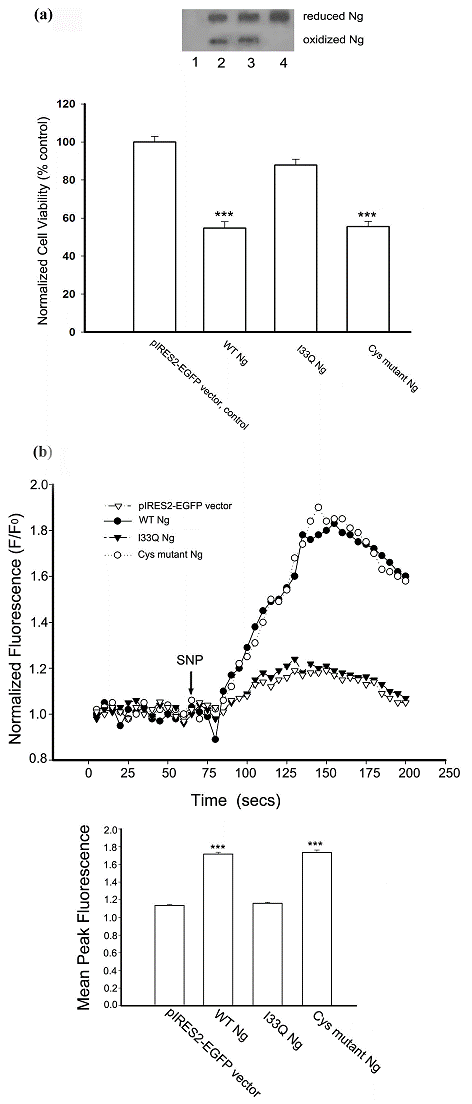
Once it was known that Ng promoted SNP-induced cell death requires the IQ motif/CaM interaction, it was logical to explore if intracellular calcium homeostasis is changed by Ng upon SNP stimulation. We achieved this by using a calcium fluorescence dye rhod-x-1 to monitor the intracellular calcium change upon 200 μM of SNP stimulation in Neuro-2a cells transiently expressing WT Ng or Ng mutants. The fluorescence intensity plot in Fig. 5b showed that 200 μM SNP induced a rapid intracellular calcium increase in Neuro-2a cells expressing WT Ng or Cys mutant Ng, while it only aroused a small and slow calcium increase in Neuro-2a cells transfected with I33Q Ng cDNA or pIRES2-EGFP vector as control. The bar chart in Fig. 5b showed that the average peak of fluorescence derived from a fluorescence intensity plot. Average peak responses from a number of cells (n=9) illustrated that in response to SNP stimulation, there was no significant difference of intracellular calcium changes between Neuro-2a cells transfected with I33Q Ng and those transfected with pIRES2-EGFP control vector. In contrast, the average peak response from cells transfected with WT Ng or Cys mutant Ng showed a dramatically larger increase in intracellular calcium than that of control cells. These results suggest that Ng promotes SNP-induced cell death most likely through amplifying the intracellular calcium increase. Interaction between IQ motif and CaM is implicated in this Ng promoted amplification of calcium signaling.
4. Discussion
NO is thought to be involved in regulating neurogenesis, synaptic plasticity, LTP and learning and memory [29, 30]. Ng is a brain-specific protein implicated in learning and memory and regulates neural plasticity and LTP. The correlated role of NO production and Ng transcriptional regulation possibly accounts for parts of these neuronal functions. The present study showed for the first time that Ng transcription could be regulated either by exogenous NO released by NO donors or endogenous NO production from nNOS. Based on Ng promoter analysis, we demonstrated that NO, released by various NO donors, changes Ng promoter activity in mammalian cells in a differential way. Among the NO donors we tested, SNAP, SIN-1.HCL and NOR-3 were able to up-regulate Ng promoter while NOC-7, NOC-9 and Spermine NONOate were not able to change Ng promoter activity. The other two NO donors, SNP and 4-Phenyl-3-furoxancarbonitrile, at our experiment condition (24 h incubation), showed harmful effect on the cells and caused massive cell death. The most stable NO donor NOR-3 in our experiment stimulated Ng promoter activity with the largest extent. By real-time PCR, Ng mRNA abundance was elevated by NOR-3 stimulation in primary cortical neurons. This is an evidence that Ng transcriptional activity could be enhanced by exogenous NO stimulation. By using a pharmaceutical drug that selectively inhibits sGC, the upregulation of Ng transcription by NO was attributed to the sGC-dependent pathway. Data from the nNOS cotransfection experiment revealed how Ng promoter activity was largely dependent on how much the nNOS expressed in the cell. At lower expression level of nNOS Ng promoter activity was enhanced whereas at higher expression level of nNOS Ng promoter activity was restored to its basal level. In the CNS, NO production is preferentially activated by calcium influx through N-methyl-D-aspartate (NMDA) receptors [31]. PSD-95 assembles a protein complex containing nNOS and NMDA receptors in postsynaptic neurons. The coupled activation of NMDA and nNOS strictly regulates the NO production [32]. It was reported that Ng was oxidized by activation of NMDA receptors in mouse hippocampal slices [33]. It is possible that the activation of NMDA receptors by presynaptic glutamate signaling could up-regulate Ng transcription through nNOS activation while overactivation of NMDA receptors dampens Ng transcription due to overproduction of NO by nNOS. The transcriptional regulation of Ng followed by 24 hr of exogenous and endogenous NO treatment reported here could be an effector to fine-tune the late phase of neuronal plasticity. Recently, Huang et al [34] reported that environmental enrichment boosts Ng expression and in turn enhances hippocampal learning and memory. Even though Ng is an abundant protein in postsynaptic neuron, it is likely the transcription and expression level of Ng in adult could be modulated by certain stimulations. It is also very likely that Ng expression is regulated in certain development stage; especially further studies are needed to clarify whether NO plays the role in the process of Ng up-regulation 1 or 2 weeks after birth when its expression level is rapidly reaching the adult level.
Furthermore, by using a stably Ng-expressing Neuro-2a cell line (clone1), we found that Ng promoter activity could be differentially affected by SNP depending on the presence or absence of Ng. With 200 µM SNP stimulation, the activity of Ng promoter in Ng-expressing cells was dramatically suppressed while it remained unchanged in Ng-free cells. It has been shown that in pathophysiological conditions like ischemia, a dramatic and selective loss of Ng mRNA occurred in the brain [35]. As a matter of fact, after 4 h stimulation with 200 µM SNP some clone1 cells showed a sign of neurite retraction and cell rounding. Thus, we speculated that the SNP-mediated decrease in Ng promoter activity possibly resulted from a pathophysiological change of Ng-expressing cells caused by SNP insult. To test the possibility whether Ng promotes the pathophysiological changes of the cells, we compared the cell viability of Ng-expressing Neuro-2a cells (clone1) and of Ng-free Neuro-2a cells with a 12 h prolonged stimulation of SNP. Our results obtained from cell viability and DNA fragmentation assays revealed that the Ng-expressing Neuro-2a cells are more susceptible to prolonged SNP stimulation. It has been shown that ectopic expression of Ng in IL-2-dependent T cells induces apoptosis [22]. The present study demonstrates another instance that Ng acts as a pro-apoptotic factor in SNP-induced cell death. Interestingly, increased expression of GAP-43, a presynaptic counterpart of Ng, also induces neuronal apoptosis [36]. Transgenic mice with over-expression of GAP-43 were found to have a reduction in the total number of neurons in various brain regions due to apoptosis [37]. Data on transgenic mice with over-expression of Ng are lacking. Yet based on the present data, it is tempting to suggest that it is quite likely that Ng and GAP-43 may perform similar physiological functions to regulate the number of neurons in selected regions during the CNS development.
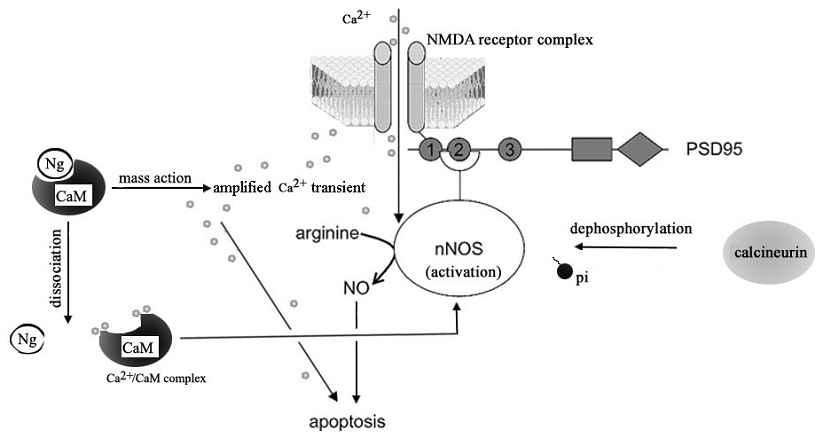
Calcium ion is an important intracellular messenger enabling various physiological processes. In the nervous system, for example, changes in the intracellular free calcium concentration can play a role in postsynaptic physiology, in presynaptic neurosecretion and in the rate of metabolic catabolism [38, 39]. However, uncontrolled increases in cytosolic calcium in neurons can also contribute to excitotoxicity and neuronal death [40, 41]. Study of neurons from Ng -/- mice showed that the absence of Ng alters neuronal Ca2+ dynamics including baseline Ca2+ levels [42]. Heterologous expression of Ng was shown to change the mobilization of intracellular Ca2+ in Xenopus oocytes [43]. It was suggested that the higher the Ng concentration, the more Ng-CaM complexes will be formed, which effectively raises Ca2+ at given Ca2+ influx [3]. By using the calcium inophore A23187 to facilitate the calcium influx of the cell, we demonstrated that the presence of Ng increases the likelihood of cellular apoptosis for a given calcium influx. This finding suggests that an amplified response to a given calcium influx is a cause of Ng-promoted SNP-induced cell death. A number of lines of evidence support that the NO donors exert their various functions through regulating intracellular free Ca2+ homeostasis [44, 45]. We can not exclude the possibility that the SNP-induced cell death may, if not solely, at least partially result from cyanide and iron degraded from SNP other than NO [46,47]. Nevertheless, from our experiment, the evidence of SNP-induced intracellular Ca2+ increase in Ng-expressing cell is substantial. Based on the current findings we suggest that the vulnerability of Ng-expressing cells to prolonged SNP stimulation is a result of Ng-amplified Ca2+ signaling initiated by SNP-induced Ca2+ homeostasis change. Ng has been proposed to modulate [Ca2+]i by the 'mass-action', i.e., the more expression of Ng, the higher Ca2+ transients under given stimulus-induced Ca2+ influx [3]. We proposed that when excitatory glutamate signal was received by NMDA receptors in the postsynaptic neuron, CaM-buffering Ng elevates the Ca2+ transient. This amplified Ca2+ transient itself may directly induce apoptosis or/and promote the dissociation of CaM from Ng to make more Ca2+/CaM complex available for sustaining the activated nNOS, which then make excessive production of NO that causes neuronal death (Fig 6).
A hypothetic role of Ng in NMDA receptor-mediated neuronal death. The activation of NMDA receptor by excitatory presynaptic signal cause Ca2+ transient in neuron which can be amplified by the 'mass action' of Ng. The overacting Ca2+ transients induce neuronal death by itself or cause the dissociation of CaM from Ng and form more Ca2+/CaM complex which sustains the NMDA-mediated activation of nNOS which implicates dephophorylation of nNOS by calcineurin. This sustained over-activation of nNOS then produce overdose NO which induces neuronal apoptosis.
It has been shown that the homeostatic tuning of Ca2+ by Ng implicates the interaction between CaM and the IQ motif of Ng [48, 49]. In the present study, we asked whether Ng promotes SNP-induced cell death through interaction between an IQ motif and CaM. Cell survival experiments indicated that disruption of the Ng IQ motif dramatically inhibited the promotive effect of Ng on SNP-induced cell death. This finding agrees with the result obtained from a study of IL-2 dependent T cells that IQ-disrupted Ng unable to bind CaM eliminates the apoptotic effect of Ng [22]. Single cell calcium imaging further confirmed our hypothesis that an intracellular calcium increase as a result of interaction between CaM and the functional IQ motif of Ng is essential for promoting SNP-induced cell death. Intracellular calcium concentration was dramatically increased by SNP stimulation in Neuro-2a cells expressing WT Ng, while it was basically unchanged in Neuro-2a cells expressing IQ mutant Ng. Since all four Cys residues (oxidization sites) reside outside the IQ motif of Ng, the oxidization of Ng, though reduces Ng affinity for CaM, does not destruct the integrity of IQ motif. Therefore, we interpreted that abolishing the oxidation of Ng by mutating all Cys residues would not attenuate the severity of Ng-promoted SNP-induced death. The cell viability assay for Neuro-2a cells transfected with four Cys mutant Ng confirmed this reasoning. The survival rate of these cells upon prolonged SNP stimulation was comparable to Neuro-2a cells expressing WT Ng. Data from single cell image experiment showed that increase in intracellular calcium concentration in Neuro-2a cells expressing Cys mutant Ng could be normally induced in a way similar to that in Neuro-2a cells expressing WT Ng. These findings suggest that oxidation of Ng does not participate as a critical factor in the process of Ng-promoted SNP-induced cell death. It has been shown that oxidation of Ng in the brain slice occurred only in a transient manner upon NMDA or NO donor stimulation [50] and is responding to physiological signaling. In contrast, a long lasting change of cell status, such as pathophysiological insult due to high oxidative stress, may trigger an apoptotic fate that recruits mechanisms other than the oxidation of Ng.
Since Ng is expressed in specific brain regions critical for learning and memory, this high-demanding function imperatively requires a lower threshold for external stimuli to neurons. Based on the current findings, we proposed that Ng acts as a calcium signal amplifier by interaction with CaM through its IQ motif, rendering the neuron a high sensitivity to calcium change. This high calcium sensitivity, just like a two-edge sword, confers enhanced plasticity of neurons as well as increased susceptibility to pathophysiological stimuli resulted from overacting of NMDA receptors and oxidative stresses.
Acknowledgements
We thank Dr. Huang Kuo-Ping (NIH) for pET3b-Ng plasmid, Dr. Rohini Kuner (University of Heidelberg, Germany) for pcDNA3.1-nNOS plasmid, and Dr. Pamela Mellon (UCSD) for GT1-7 cells. This work was supported in part by a grant from the Ministry of Education Singapore via the University Research Council of the National University of Singapore R-154-000-228-112 to F.-S. S.
Conflict of Interests
The authors have declared that no conflict of interest exists
References
1. Gerendasy D.D, Sutcliffe J.G. RC3/neurogranin, a postsynaptic calpacitin for setting the response threshold to calcium influxes. Mol. Neurobiol. 1997;15:131-163
2. Pak J.H, Huang F.L, Li J, Balschun D, Reymann K.G, Chiang C, Westphal H, Huang K.P. Involvement of neurogranin in the modulation of calcium/calmodulin-dependent protein kinase II, synaptic plasticity, and spatial learning: a study with knockout mice. Proc. Natl. Acad. Sci. USA. 2000;97:11232-11237
3. Huang K.P, Huang F.L, Jager T, Li J, Reymann K.G, Balschun D. Neurogranin/RC3 enhances long-term potentiation and learning by promoting calcium-mediated signaling. J. Neurosci. 2004;24:10660-10669
4. Prichard L, Deloulme J.C, Storm D.R. Interactions between neurogranin and calmodulin in vivo. J. Biol. Chem. 1999;274:7689-7694
5. Huang K.P, Huang F.L, Chen H.C. Characterization of a 7.5-kDa protein kinase C substrate (RC3 protein, neurogranin) from rat brain. Arch Biochem Biophys. 1993;305:570-580
6. Mahoney C.W, Pak J.H, Huang K.P. Nitric oxide modification of rat brain neurogranin. Identification of the cysteine residues involved in intramolecular disulfide bridge formation using site-directed mutagenesis. J Biol Chem. 1996;271:28798-28804
7. Sheu F.S, Mahoney C.W, Seki K, Huang K.P. Nitric oxide modification of rat brain neurogranin affects its phosphorylation by protein kinase C and affinity for calmodulin. J. Biol. Chem. 1996;271:22407-22413
8. Enderlin V, Pallet V, Alfos S, Dargelos E, Jaffard R, Garcin H, Higueret P. Age-related decreases in mRNA for brain nuclear receptors and target genes are reversed by retinoic acid treatment. Neurosci. Lett. 1997;229:125-129
9. Husson M, Enderlin V, Alfos S, Feart C, Higueret P, Pallet V. Triiodothyronine administration reverses vitamin A deficiency-related hypo-expression of retinoic acid and triiodothyronine nuclear receptors and of neurogranin in rat brain. Br. J. Nutr. 2003;90:191-198
10. Etchamendy N, Enderlin V, Marighetto A, Vouimba R.M, Pallet V, Jaffard R, Higueret P. Alleviation of a selective age-related relational memory deficit in mice by pharmacologically induced normalization of brain retinoid signaling. J. Neurosci. 2001;21:6423-6429
11. Mons N, Enderlin V, Jaffard R, Higueret P. Selective age-related changes in the PKC-sensitive, calmodulin-binding protein, neurogranin, in the mouse brain. J. Neurochem. 2001;79:859-867
12. Feart C, Mingaud F, Enderlin V, Husson M, Alfos S, Higueret P, Pallet V. Differential effect of retinoic acid and triiodothyronine on the age-related hypo-expression of neurogranin in rat. Neurobiol. Aging. 2005;26:729-738
13. Martinez de A.C, Morte B, Coloma A, Bernal J. The human RC3 gene homolog, NRGN contains a thyroid hormone-responsive element located in the first intron. Endocrinology. 1999;140:335-343
14. Morte B, Martinez de A.C, Manzano J, Coloma A, Bernal J. Identification of a cis-acting element that interferes with thyroid hormone induction of the neurogranin (NRGN) gene. FEBS Lett. 1999;464:179-183
15. Morte B, Iniguez M.A, Lorenzo P.I, Bernal J. Thyroid hormone-regulated expression of RC3/neurogranin in the immortalized hypothalamic cell line GT1-7. J. Neurochem. 1997;69:902-909
16. Sato T, Xiao D.M, Li H, Huang F.L, Huang K.P. Structure and regulation of the gene encoding the neuron-specific protein kinase C substrate neurogranin (RC3 protein). J. Biol. Chem. 1995;270:10314-10322
17. Gui J, Song Y, Han N.L, Zhou S.F, Sheu F.S. Involvement of the GC-rich sequence and specific proteins (Sp1/Sp3) in the basal transcription activity of neurogranin gene. Biochem. Biophys. Res. Commun. 2006;345:124-32
18. Huang K.P, Huang F.L, Li J, Schuck P, McPhie P. Calcium-sensitive interaction between calmodulin and modified forms of rat brain neurogranin/RC3. Biochemistry. 2000;39:7291-7299
19. Bogdan C. Nitric oxide and the regulation of gene expression. Trends Cell Biol. 2001;11:66-75
20. Dawson V.L, Dawson T.M. Nitric oxide in neurodegeneration. Prog. Brain Res. 1998;118:215-229
21. Guix F.X, Uribesalgo I, Coma M, Munoz F.J. The physiology and pathophysiology of nitric oxide in the brain. Prog. Neurobiol. 2005;76:126-152
22. Devireddy L.R, Green M.R. Transcriptional program of apoptosis induction following interleukin 2 deprivation: identification of RC3, a calcium/calmodulin binding protein, as a novel proapoptotic factor. Mol. Cell Biol. 2003;23:4532-4541
23. Nishida Y, Yoshioka M, St-Amand J. The top 10 most abundant transcripts are sufficient to characterize the organs functional specificity: evidences from the cortex, hypothalamus and pituitary gland. Gene. 2005;344:133-141
24. Yang H.M, Lee P.H, Lim T.M, Sheu F.S. Neurogranin expression in stably transfected N2A cell line affects cytosolic calcium level by nitric oxide stimulation. Brain Res. Mol. Brain. Res. 2004;129:171-178
25. Mayer B. Regulation of nitric oxide synthase and soluble guanylyl cyclase. Cell Biochem. Funct. 1994;12:167-177
26. McDonald L.J, Murad F. Nitric oxide and cyclic GMP signaling. Proc. Soc. Exp. Biol. Med. 1996;211:1-6
27. Garthwaite J, Southam E, Boulton C.L, Nielsen E.B, Schmidt K, Mayer B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol. Pharmacol. 1995;48:184-188
28. Bahler M, Rhoads A. Calmodulin signaling via the IQ motif. FEBS Lett. 2002;513:107-113
29. Holscher C. Nitric oxide, the enigmatic neuronal messenger: its role in synaptic plasticity. Trends Neurosci. 1997;20:298-303
30. Hawkins R.D, Son H, Arancio O. Nitric oxide as a retrograde messenger during long-term potentiation in hippocampus. Prog. Brain Res. 1998;118:155-172
31. Yun HY, Dawson VL, Dawson TM. Nitric oxide in health and disease of the nervous system. Mol. Psychiatry. 1997;2:300-310
32. Christopherson K.S, Hillier B.J, Lim W.A, Bredt D.S. PSD-95 assembles a ternary complex with the N-methyl-D-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J. Biol. Chem. 1999;274:27467-27473
33. Wu J, Huang K.P, Huang F.L. Participation of NMDA-mediated phosphorylation and oxidation of neurogranin in the regulation of Ca2+- and Ca2+/calmodulin-dependent neuronal signaling in the hippocampus. J. Neurochem. 2003;86:1524-1533
34. Huang F.L, Huang K.P, Wu J, Boucheron C. Environmental enrichment enhances neurogranin expression and hippocampal learning and memory but fails to rescue the impairments of neurogranin null mutant mice. J. Neurosci. 2006;26:6230-6237
35. Shughrue P.J, Merchenthaler I. Estrogen prevents the loss of CA1 hippocampal neurons in gerbils after ischemic injury. Neuroscience. 2003;116:851-861
36. Wehrle R, Caroni P, Sotelo C, Dusart I. Role of GAP-43 in mediating the responsiveness of cerebellar and precerebellar neurons to axotomy. Eur. J. Neurosci. 2001;13:857-870
37. Aigner L, Arber S, Kapfhammer J.P, Laux T, Schneider C, Botteri F, Brenner H.R, Caroni P. Overexpression of the neural growth-associated protein GAP-43 induces nerve sprouting in the adult nervous system of transgenic mice. Cell. 1995;83:269-278
38. Miller R.J. Calcium signalling in neurons. Trends Neurosci. 1998;11:415-419
39. Dunlap K, Luebke J.I, Turner T.J. Identification of calcium channels that control neurosecretion. Science. 1994;266:828-831
40. Choi D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1998;1:623-634
41. Trump B.F, Berezesky I.K. The role of cytosolic Ca2+ in cell injury, necrosis and apoptosis. Curr. Opin. Cell Biol. 1992;4:227-232
42. van Dalen J.J, Gerendasy D.D, de Graan P.N, Schrama L.H, Gruol D.L. Calcium dynamics are altered in cortical neurons lacking the calmodulin-binding protein RC3. Eur. J. Neurosci. 2003;18:13-22
43. Cohen R.W, Margulies J.E, Coulter P.M, Watson J.B. Functional consequences of expression of the neuron-specific, protein kinase C substrate RC3 (neurogranin) in Xenopus oocytes. Brain Res. 1993;627:147-152
44. Tsuji Y, Unno N, Menconi M.J, Smith M, Fink M.P. Nitric oxide donors increase cytosolic ionized calcium in cultured human intestinal epithelial cells. Shock. 1996;6:19-24
45. Mishra O.P, ivoria-Papadopoulos M. Nitric oxide-mediated Ca++-influx in neuronal nuclei and cortical synaptosomes of normoxic and hypoxic newborn piglets. Neurosci. Lett. 2002;318:93-97
46. Schröder H. No Nitric Oxide for HO-1 from Sodium Nitroprusside. Mol Pharmacol. 2006;69:1507-1509
47. Kim H.J, Tsoy I, Park M.K, Lee Y.S, Lee J.H, Seo H.G, Chang K.C. Iron Released by Sodium Nitroprusside Contributes to Heme Oxygenase-1 Induction via the cAMP-Protein Kinase A-Mitogen-Activated Protein Kinase Pathway in RAW 264.7 Cells. Mol Pharmacol. 2006;69:1633-1640
48. Gerendasy D. Homeostatic tuning of Ca2+ signal transduction by members of the calpacitin protein family. J. Neurosci. Res. 1999;58:107-119
49. Krucker T, Siggins G.R, McNamara R.K. et al. Targeted disruption of RC3 reveals a calmodulin-based mechanism for regulating metaplasticity in the hippocampus. J. Neurosci. 2002;22:5525-5535
50. Li J, Pak J.H, Huang F.L, Huang K.P. N-methyl-D-aspartate induces neurogranin/RC3 oxidation in rat brain slices. J. Biol. Chem. 1999;274:1294-1300
Author contact
![]() Correspondence to: Dr. Fwu-Shan Sheu, Department of Biological Sciences, National University of Singapore, 14 Science Drive 4, Singapore 117543, Singapore. E-mail address: dbssfsedu.sg
Correspondence to: Dr. Fwu-Shan Sheu, Department of Biological Sciences, National University of Singapore, 14 Science Drive 4, Singapore 117543, Singapore. E-mail address: dbssfsedu.sg
Received 2007-1-11
Accepted 2007-2-23
Published 2007-2-23