ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2007; 3(6):385-392. doi:10.7150/ijbs.3.385 This issue Cite
Research Paper
Reversal of Systemic Hypertension-Associated Cardiac Remodeling in Chronic Pressure Overload Myocardium by Ciglitazone
Brooke C. Henderson, Utpal Sen, Corey Reynolds, Karni S. Moshal, Alexander Ovechkin, Neetu Tyagi, Ganesh K. Kartha, Walter E. Rodriguez, Suresh C. Tyagi ![]()
Department of Physiology and Biophysics, University of Louisville, Louisville, KY 40202, USA
Abstract
Elevated oxidative stress has been characterized in numerous disorders including systemic hypertension, arterial stiffness, left ventricular hypertrophy (LVH) and heart failure. The peroxisome proliferator activated receptor gamma (PPARγ) ameliorates oxidative stress and LVH. To test the hypothesis that PPARγ decreased LVH and cardiac fibrosis in chronic pressure overload, in part, by increasing SOD, eNOS and elastin and decreasing NOX4, MMP and collagen synthesis and degradation, chronic pressure overload analogous to systemic hypertension was created in C57BL/6J mice by occluding the abdominal aorta above the kidneys (aortic stenosis-AS). The sham surgery was used as controls. Ciglitazone (CZ, a PPARγ agonist, 4 µg/ml) was administered in drinking water. LV function was measured by M-Mode Echocardiography. We found that PPARγ protein levels were increased by CZ. NOX-4 expression was increased by pressure-overload and such an increase was attenuated by CZ. SOD expression was not affected by CZ. Expression of iNOS was induced by pressure-overload, and such an increase was inhibited by CZ. Protein levels for MMP2, MMP-9, MMP-13 were induced and TIMP levels were decreased by pressure-overload. The CZ mitigated these levels. Collagen synthesis was increased and elastin levels were decreased by pressure-overload and CZ ameliorated these changes. Histochemistry showed that CZ inhibited interstitial and perivascular fibrosis. Echocardiography showed that CZ attenuated the systolic and diastolic LV dysfunction induced by pressure-overload. These observations suggested that CZ inhibited pressure-overlaod-induced cardiac remodeling, and inhibition of an induction of NOX4, iNOS, MMP-2/MMP-13 expression and collagen synthesis/degradation may play a role in pressure-overload induced cardiac remodeling.
Keywords: PPARγ, NOX4, MMP, collagen, SOD, TIMP, iNOS, eNOS
1. Introduction
The communication and chemical gradient between the endothelium to myocyte and myocyte to myocyte is very important in maintaining the patency of the organ such as the heart. Extracellular matrix (ECM) connects the myocyte to myocyte, and to the endothelial cells [1]. Studies from our laboratory demonstrated a direct link between diastolic heart failure (DHF), interstitial fibrosis, and left ventricular hypertrophy (LVH). There was a disconnection/uncoupling between cardiomyocytes and endothelial cells by MMP-mediated collagen breaks [2]. Although elevated oxidative stress has been characterized in numerous disorders including systemic hypertension, arterial stiffness, LVH, and DHF, the mechanism and link between oxidative stress and collagen breaks, LVH, DHF and fibrosis is unclear. Peroxinitrite-thiol (ONOO-HS-R) is an intermediate of nitrotyrosine formation. Generation of peroxinitrite leads to the activation of the latent myocardial resident MMPs and causes fibrosis [3]. Also, the formation of nitrotyrosine can be used as a measure of oxidative stress. The peroxisome proliferator activated receptor gamma (PPARγ) ameliorates oxidative stress and LVH [4, 5]. Reduced expression of PPAR causes LVH in a ventricular pressure overload model in mice [6]. In addition, administration of peroxisome proliferator reduces cardiac hypertrophy [7]. We and others have suggested decreased MMP activity by PPARγ induction [8, 9]. Although PPARγ agonists reduce cardiac dysfunction and induce anti-oxidant potential, the mechanism by which PPARγ agonist reduces LVH and cardiac fibrosis is unclear. We hypothesize that PPARγ decreases LVH and cardiac fibrosis in chronic pressure overload, in part, by increasing SOD, eNOS and elastin and decreasing NOX4 (NADPH oxidase subunit), MMP and collagen synthesis and degradation.
2. Materials and Methods
Animals
Wild-type C57BL/6J 8 weeks old male mice were obtained from Jackson Laboratories (Bar Harbor, Maine). These animals were kept in animal care facility of University of Louisville, where they were monitored daily. Animal protocols were performed in accordance to the National Institutes of Health guidelines and approved by the IACUC of the University Of Louisville School Of Medicine. All animals were fed rodent chow and housed in cycle of 12 hr light and 12 hr dark conditions. The room was kept at 24°C.
Pressure Overload and Systemic Hypertension Model
To mimic the condition where systemic hypertension imposed load on the heart, abdominal aortic banding above the kidneys was created. The mice were anesthetized with 2, 2, 2 tribromoethanol (TBE, 100 mg/kg of body weight, I. P.) (Sigma-Aldrich, St. Louis, MO). A midline incision was made to expose the abdominal area. The abdominal aorta was constricted (i.e. aortic stenosis-AS) by tightening a 6-0 nylon suture against a 25-gauge blunted needle with the needle being removed after the suture was secure. The sham animals underwent the same procedure without the banding. The incision was closed in layers and the animals were allowed to recover. Animals were weighed and tissues were harvested at 4 weeks for heart, kidneys, lungs and blood. The tissues were either snap frozen in liquid nitrogen or placed in 10% formalin.
Treatment Groups
To induce PPARγ, ciglitazone (CZ, CalBiochem Corp) 4 µg/ml was administered in drinking water for 4 wks. The amount (4 µg/ml) of CZ is soluble in aqueous conditions: the binding constant between ciglitazone and PPARγ is in the micromole range [10]. Based on the assumption that mice have a blood volume of ~2 ml and drink ~5 ml water/day, each will ingest ~2 mg/kg/day CZ. This produced a blood concentration of ~32 µMole/L, which was enough to saturate most binding sites on PPAR. In humans, 100 mg/day PPAR agonist has a potent effect [11]. The mice were treated for 4 weeks and grouped into: 1) Sham, 2) AS; 3) Sham+CZ; 4) AS+CZ.
Measurement of Echocardiography systolic and diastolic function
A Hewlett-Packard Sonos 5500 echocardiographic system equipped with a 15-MHz shallow-focus 15-6L phased-array transducer was used for measurements of LV function on TBE-anesthetized mice. The M-mode selection allowed for the visualization of systole and diastole within the beating heart. Also from M-mode LV wall thickness and diameter were assessed as well as ejection fraction (EDD-ESD/EDD).
Western Blot
Collagen, nitrotyrosine, SOD, NOX4, MMP, and TIMP-4 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). eNOS and iNOS were from BD Biosciences (Franklin Lakes, NJ). Elastin antibody was from Chemicon (Temecula, CA). The LV extracts were prepared in an overnight cacodylic acid extracellular buffer (10mM/L Cacodylic Acid pH 5.0, 8.76g/L NaCl, 5mM/L ZnCl2, 2.22 g/L CaCl2, 0.098g/L NaN3, 100 µL/L Trition X-100). The samples were assessed for protein concentration with a Bio-Rad assay (Philadelphia, PA) using the Bradford Method [20]. 20μg of the protein was mixed with equal volumes of reducing 1x SDS sample buffer. They were electrophoresed on 10% SDS-PAGE gels. The gel was then blotted onto a polyvinylidene difluoride membrane. After being transferred, blots were washed with Tris-buffered saline (TBS) for 5 min at room temperature and incubated in blocking buffer of 5% milk in Tris-buffered saline with 1% tween (TBST) for 1 h at room temperature. The blots were then incubated with the indicated primary antibodies (3% milk in TBST) overnight at 4°C using slow rocking. The blots were washed three times (for 15-min each) with TBST and incubated with HRP-conjugated secondary antibody (3% milk in TBST). After being washed three times (for 15-min each) with TBST, the proteins of interest were detected using an ECL plus kit (Amersham Biosciences, Piscataway, NJ). The membranes were then stripped using 0.2 M NaOH/ 0.1% SDS solution for 30 min at room temperature and reprobed for β-actin. Band densities were normalized to the untreated controls and presented as percentage changes.
Histology
The heart sections were placed in 10% formalin. The sections then underwent a graded alcohol dehydration process. The sections were fixed to a holder using paraplast and sat overnight. Then they were cut into 3 mm sections and stained with Sirius red for collagen (fibrosis) and H & E for cellularity. Optical light microscopy was performed at 40 X magnification.
Statistical Analysis
A sample size of n=11 was used in each group. The data was presented as the mean+/-SE. The difference was calculated statistically with the significance level of p < 0.05 using ANOVA followed by Bonferroni post hoc test.
3. Results
PPARγ levels in pressure overload
The levels of PPARγ were decreased in the LV of chronic pressure overload mice. The treatment with ciglitazone (CZ, a PPARγ agonist) mitigated the decrease in aortic stenosis-mediated PPARγ levels in mice with chronic pressure overload (Figure 1).
Levels of oxidative stress
To determine whether the increase in oxidative stress in chronic pressure overload is due to differential expression of Mn-SOD, NADPH oxidase, eNOS, iNOS and nitration of the tyrosine in proteins in the hearts of chronic pressure overload, we measured SOD, NOX4 (NADPH oxidase subunit), eNOS, iNOS and nitrotyrosine. Although it appears that CZ itself increased Mn-SOD expression (vs. sham), there were no change in the levels of SOD with or without AS. Although CZ itself increased NOX4 expression (vs. sham), but decreased the NOX4 expression induced by AS (Figure 2). The levels of eNOS was decreased in aortic stenosis-induced pressure overload mice, there was no effect of CZ on this decrease. The levels of iNOS were increased in pressure overload. The treatment with CZ mitigated this increase in iNOS. The levels of total nitrotyrosine proteins were increased in chronic pressure overload mice. In addition, CZ itself increased nitrotyrosine expression (vs. sham) (Figure 3). These results suggest that pressure overload induces NOX4, and iNOS; but decreases eNOS and had no effect on SOD. The treatment with CZ ameliorated the increase in NOX4 and iNOS but had no decrease in eNOS levels.
Western blot analysis of PPARγ in LV: The LV tissue homogenates from Sham, aortic-stenosis (AS), Sham + CZ and AS+CZ mice were prepared. The levels of PPARγ were measured by immuno-blots using PPARγ antibody. Beta-actin was used as control. The scanned data of the blots were represented by bar graphs. *p<0.05 compared to Sham; ** p<0.05 compared to AS; n=6 in each group.
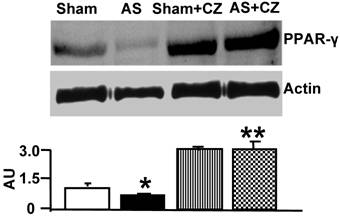
Western blot analysis of Mn-SOD, and NOX4 in LV: The LV tissue homogenates from Sham, aortic-stenosis (AS), Sham + CZ and AS+CZ mice were prepared. The levels of SOD and NOX4 were measured by immuno-blots using respective antibodies. β-actin for equal loading; graphs represent scan data for band intensities; *p<0.05 compared to sham; ** p<0.05 compared to AS; n=6 in each group.
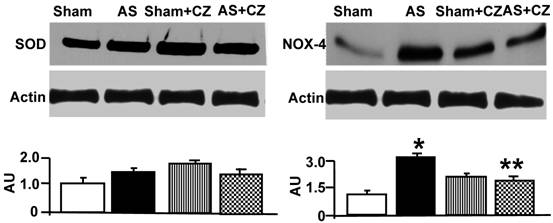
Western blot analysis of eNOS, iNOS and total nitrotyrosine (NO2-T) in LV: The tissue homogenates from Sham, aortic-stenosis (AS), Sham + CZ and AS+CZ mice were prepared. The levels of eNOS, iNOS and protein nitrotyrosine were measured by immuno-blots using respective antibodies. β-actin was used for equal loading; graphs represent scan data for band intensities; *p<0.05 compared to sham; ** p<0.05 compared to AS; n=6 in each group.
Matrix remodeling
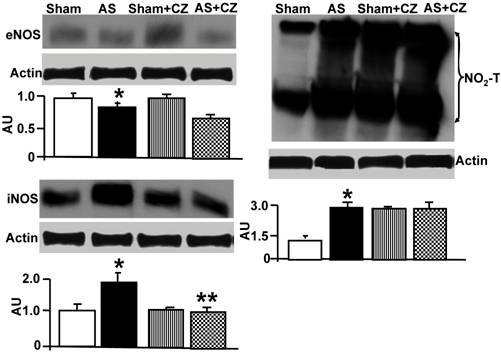
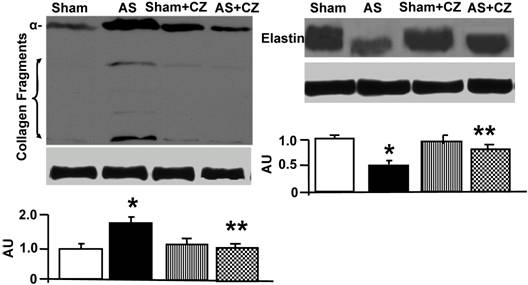
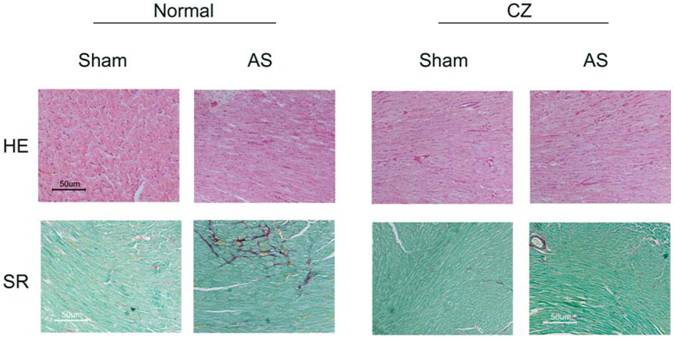
The levels of MMP-2 were increased, and CZ mitigated this increased in AS mice. The CZ itself increased MMP-9 expression (vs. sham), but decreased the MMP-9 expression induced by AS. The levels of MMP-13 were increase in pressure overload, and CZ ameliorated this increase. To determine whether the increase in MMP levels is associated with decrease in cardiac specific TIMP (TIMP-4), we measured the levels of TIMP-4. The levels of TIMP-4 were decreased in pressure overload. The treatment with CZ mitigated this decrease (Figure 4). To determine whether the increase in MMP caused collagen breaks, we measured the collagen alpha-chain expression and collagen fragments. The results suggested that although there was robust increase in collagen synthesis, there was significant degradation (i.e. fragmentation of intact collagen alpha chain were increased in AS) [Figure 5]. To determine where cardiac fibrosis is due to increase in collagen/elastin, we measure elastin levels. The results suggest that there was decrease in elastin expression in chronic pressure overload. The treatment with CZ ameliorated both the collagen degradation and decrease in elastin expression (Figure 5). There was robust interstitial and perivascular fibrosis in the LV of chronic pressure overload. The treatment with CZ mitigated the interstitial and perivascular fibrosis (Figure 6).
Western blot analysis of LV levels of MMP-2, -9, -13 and cardiac specific TIMP-4: The LV tissue homogenates from Sham, aortic-stenosis (AS), Sham + CZ and AS+CZ mice were analyzed for MMP-2, -9, -13 and TIMP-4 using respective antibodies. β-actin was used for equal loading; graphs represent scan data for band intensities; *p<0.05 compared to sham; ** p<0.05 compared to AS; n=6 in each group.
Western blot analysis of collagen (alpha-chain, 100 kDa), collagen fragments, and elastin in LV: The tissue homogenates from Sham, aortic-stenosis (AS), Sham + CZ and AS+CZ mice were prepared. The levels of collagen (alpha-chain), collagen fragments, and elastin were measured by immuno-blots using respective antibodies. β-actin was used for equal loading; graphs represent scan data for band intensities; *p<0.05 compared to sham; ** p<0.05 compared to AS; n=6 in each group.
Representative sections of formalin-fixed tissue section analysis by histological staining with Sirius Red (SR) for collagen and Hematoxin-eosin (HE) for cellularity of hearts from sham, AS, sham+CZ and AS+CZ mice.
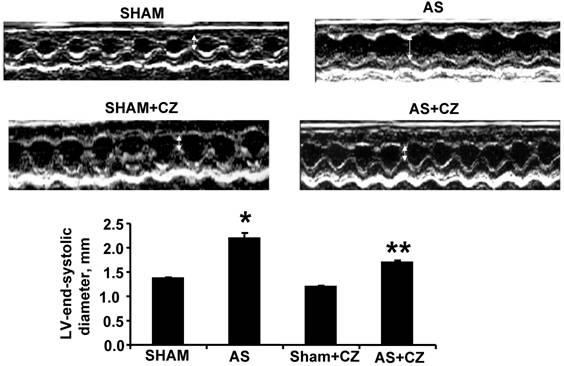
M-mode echocardiography of sham, AS, sham+CZ and AS+CZ hearts. The white arrow indicates the LV end systolic diameter. Note arrow is bigger in AS than other groups. The bar graph represents the LV end systolic diameters of sham, AS, sham+CZ and AS+CZ mice. *p<0.05 compared to sham; ** p<0.05 compared to AS; n=6 in each group.
LV function
The M-mode Echocardiographic measurements revealed that there was increase in LV end-systolic and diastolic diameters in AS mice (Table 1 and Figure 7). The treatment with CZ mitigated this increase in LV size. These data suggested significant systolic and diastolic dysfunction in AS-induced pressure overload mice.
Gravimetric and Echocardiography parameters at 4 weeks: Body weight (BW), heart weights (HW), and lung weight of sham and aortic stenosis (AS) mice treated with and without CZ. Left ventricular end diastolic diameter (LVEDd), Left ventricular end systolic diameter (LVESd), fractional shortening (FS), and ejection fraction (EF).
| Sham | AS | Sham+CZ | AS+CZ | |
|---|---|---|---|---|
| n=11 | n=11 | n=11 | n=11 | |
| Body weight, g | 27±1 | 28±2 | 27±1 | 26±2 |
| Heart weight, g | 0.11±0.01 | 0.21±0.02* | 0.11±0.01 | 0.16±0.02** |
| Lung weight, g | 0.18±0.02 | 0.36±0.02* | 0.19±0.01 | 0.24±0.03** |
| HW/BW (mg/g) | 3.7±0.4 | 7.2±0.7* | 4.0±0.4 | 6.0±0.5** |
| LVEDd (mm) | 2.79±0.06 | 3.76±0.04* | 2.84+0.05 | 3.29+0.03 |
| LVESd (mm) | 1.36±0.03 | 2.2±0.1* | 1.19±0.03 | 1.69±0.04** |
| EF % | 90±2 | 67±2* | 92±2 | 83±2** |
| FS % | 60±2 | 43±1* | 59±1.5 | 51±1** |
* p<0.05 compared with sham; ** p< 0.05 compared with AS.
4. Discussion
To determine the role of collagen degradation in cardiac hypertrophy, we previously [2] examined the effect of pressure overload on extracellular matrix activity, composition, cardiac function and remodeling in collagen mutant transgenic animals. We showed that pressure overload caused an increase in oxidative stress and expression/activity of MMP-2/9, leading to myocardial hypertrophy and heart failure [2]. In the present study we extended the notion that oxidative stress MMP activation with collagen degradation is a pre-requisite for LVH and heart failure. The AS-induced increase in MMP and decrease in TIMP was mitigated by PPARγ. There was increase in collagen synthesis and degradation and decrease in elastin content in AS-induced cardiac failure, however, the treatment with PPARγ normalized the levels of elastin and collagen degradation.
Although we showed that CZ decreased NOX4 levels in the LV tissues of AS-model, the effects of CZ on other NOX family protein expression is unclear. However, it would be important to examine whether CZ decreased NADPH oxidase activity in the LV tissues. Others have suggested induction of SOD and reduction of NOX4 by PPARγ agonist [12-14], Here we demonstrated a differential expression of SOD and NOX4 in chronic pressure overload myocardium. The levels of SOD did not affected but the levels of NOX4 were ameliorated by ciglitazone in aortic stenosis model of systemic hypertension. This study suggested that PPARγ effected only the formation of super oxide, but had no effect on the sequestration of super oxide free radicals.
Although generation of peroxynitrite leads to the activation of the latent myocardial resident MMPs and causes fibrosis, in pressure overload model, levels of eNOS were decreased and those of iNOS and nitrotyrosine were increased. CZ only ameliorated the iNOS but have no effect on eNOS or nitrotyrosine levels. Our data suggested that CZ decreased MMP-2/-13 levels, even though it was not able to ameliorate oxidative marker, nitrotyrosine levels. It may suggest that CZ mediated anti-oxidative properties caused decrease in MMP activation. In addition, we observed that CZ did not ameliorate the increase in nitrotyrosine levels in this model. The differential expression of eNOS and iNOS in pressure overload may suggest that the location of NO is more important than the amount of NO. The pressure overload decreased eNOS and increased the iNOS. The generation of NO in the ECM milieu oxidized the TIMPs by nitrosylation and caused the activation of latent resident MMPs [3]. There was robust increase in total nitrotyrosine in AS as compared to sham. However, PPARγ has no ameliorative effects on eNOS but mitigated the induction of iNOS in AS mice. This may suggest that iNOS is influenced by the levels of PPARγ and the PPARγ directly influenced the expression of iNOS in pressure overload myocardium. PPARγ has no effect on total nitrosylation. This differential effects of PPARγ on eNOS, iNOS and nitrotyrosine suggest that iNOS is influenced by PPARγ however the levels of nitrosylation are independent on PPARγ levels.
CZ decreased MMP-2/-13, but not MMP-9 levels in the LV tissues. This may suggest that MMP-9 activation is also dependent on ROS. It is a paradox that if CZ exerts beneficial effects on LV function in this model via anti-oxidative properties, the MMP should not be inhibited. Studies [15] reported interstitial collagen deposition, collagen content or collagen metabolites in the pathological hearts, here, we reported that the interstitial fibrosis in pressure overload was due in part to increase in MMP and decrease in TIMP. Because LV hypertrophy is associated with marked interstitial fibrosis [15], a decrease in MMP is associated with reduced LV hypertrophy, and may not justify the hypothesis that LV hypertrophy is due to increased MMPs. Also it is possible that increased MMP-9 is a compensatory response to increased collagen production, rather than a cause of LV hypertrophy. However, it is a paradox that MMP activation and cardiac fibrosis track together. This paradox can be explained by considering turnover of the degraded matrix proteins. It is well known that both MMP-2 and -9 are collagenases, as well as excellent elastases. Because turnover of elastin is slower than collagen, therefore, degraded elastin is replaced by oxidatively modified collagen, leading to fibrosis. Furthermore, the activation of MMP transducers proteolytic stress that causes myocyte hypertrophy [16].
Previous studies have shown decrease in MMP-2 activity and TIMP-1 expression in hypertension and associated cardiac fibrosis, stiffness and impaired cardiac diastolic relaxation [17]. The MMP-9 activation and decrease in TIMP-4 expression were linked to cardiac dilatation and heart failure [18]. This leads to robust increase in MMP-9/TIMP-4 ratio causing cardiac systolic myocyte slippage and failure. Although it is well known that there is significant systolic and diastolic dysfunction in AS-induced pressure overload mice. The CZ mitigates the cardiac dysfunction of the mice with AS, in part, by increasing TIMP-4.
Coordination between angiogenesis and cardiac hypertrophy (LVH) is the hallmark of compensatory response to heart failure; however, a disruption in coordination between cardiac hypertrophy and angiogenesis contributes to the transition to heart failure [19]. The angiogenesis, LVH and fibrosis track together, therefore, in the absence of blood supply to the myocardium at risk, the lack of angiogenesis by increase angiostatic factors leads to fibrotic and LVH conditions. Although, it is a paradox that activation of MMP is needed to disruption of the matrix during angiogenesis and to create new blood vessels, or opening the collaterals, however, the peptides generated by MMP activation are angiostatic [20, 21]. Early studies by Folkman et al [20, 21] suggested endostatin and angiostatin generated by MMP action on collagen-18 and plasminogen, respectively, inhibit tumor angiogenesis. In addition, the vasostatin is generated by degradation of calreticulin [22], tumstatin [23], arresten [24] and canstatin [25] by degradation of collagen-4 and restin [26] is generated by degradation of collagen-15. The increase in MMPs (Figure 4) in pressure overload is consistent with the notion that MMPs cause discoordinated matrix degradation leading to decompensatory hypertrophy and heart failure by generating angiostatic factors. This explains however, MMP, TIMP and LVH may co-exist.
Limitations
One of the major concerns is the lack of the evidence indicating that observed effects of CZ is mediated by PPARγ by itself. It is known that CZ is known to be an agonist of PPARγ and Western blot analysis showed that PPARγ protein expression was clearly induced by CZ treatment. Figure 1 showed that PPARγ expression was increased in sham-operated heart and suggested that CZ systemically induced PPARγ. It is important to show whether CZ treatment induces PPARγ expression in others tissues including kidney and lung, and whether expression of PPARalpha or PPARdelta were not affected by CZ. In addition, it is important to show that CZ induces PPARγ at the mRNA levels.
Second, mechanistic analyses of the PPARγ induction by CZ should be performed because thiazolidine derivatives are known to exert their effects by activating PPARγ or by acting as antioxidants independent of PPARγ activation. In vitro experiments to determine whether CZ induces PPARγ expression by PPARγ -dependent or PPARγ -independent mechanisms will be helpful.
Third, we harvested heart 4 weeks after aortic banding. During this period, myocardium responses dynamically, from adaptive phase to maladaptive phase. Thus, in order to show the effect of CZ on cardiac remodeling more precisely, it is important to show the time-course of the gene expression, for example, at day 1, 7, 14 and 28. These studies are in progress.
It is also likely that some synthetic PPAR antagonists, like GW9662, could be helpful in illustrating the mechanism of potential changes in the transcription levels. In Figure 2, we described that CZ increased the expression of NOX4 in the sham-operated group. But CZ reduced the increase of NOX4 expression caused by AS. Similarly, the MMP9 result (Figure 4) has a similar paradox. Although this is not theoretically impossible but is certainly a bizarre finding and can be explained if one assumes that CZ in the absence of injury may stimulate the compensatory responses. There is a study showing that CZ can activate pro-MMP-2 [27]. However, it is in a different cell type, at the enzyme activity level, and may occur under a different set of conditions (like oxidative environment).
Acknowledgements
A part of this study was supported by NIH grants HL-74185, and HL-88012. This work was presented at the Annual Meeting of Experimental Biology, April 28- 2 May, 2007, Washington D. C.
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Henderson BC, Tyagi SC. Oxidative Mechanism and Homeostasis of Proteinase/Antiproteinase in Congestive Heart Failure. J Mol Cell Cardiol. 2006;41:959-962
2. Henderson BC, Tyagi N, Ovechkin A, Kartha GK, Moshal KS, Tyagi SC. Oxidative Remodeling in Pressure Overload Induced Chronic Heart Failure. Euro J Heart Failure. 2007;9(5):450-457
3. Tyagi SC, Rodriguez W, Patel AM, Roberts AM, Falcone JC, Passmore JC, Fleming JT, Joshua IG. Hyperhomocysteinemic Diabetic Cardiomyopathy: Oxidative stress, Remodeling, and Endothelial-Myocyte Uncoupling. J Cardiovasc Pharmacol Therapeutics. 2005;10(1):1-10
4. Lebovitz HE, Manerji MA. Insulin resistance and its treatment by thiazolidinediones. Recent Prog Horm Res. 2001;56:265-294
5. Itoh H, Doi K, Tanaka T, Fukunaga Y, Hosoda K. et al. Hypertension and insulin resistance:role of PPARγ. Clin Exp Pharmacol Physiol. 1999;26:558-560
6. Barger PM, Brandt JM, Leone TC, Weinheimer CJ, Kelly DP. Deactivation of PPAR during cardiac hypertrophic growth. J Clin Invest. 2000;105:1723-1730
7. Yamamoto K, Ohki R, Lee RT. Ikeda U, Shimada K. PPAR activators inhibit cardiac hypertrophy in cardiomyocytes. Circulation. 2001;104:1670-1675
8. Hunt MJ, Tyagi SC. Peroxisome proliferators compete and ameliorate homocysteine-mediated endocardial endothelial cells activation. Am J Physiol. 2002;283:C1073-C1079
9. Marx N, Sukhova G, Murphy C, Libby P, Plutzky J. Macrophages in human atheroma conain PPAR: Differential dependent peroxisomal proliferator activated receptorã expression and reduction of MMP-9 activity through PPAR activation in mononuclear phaocytes in vitro. Am J Pathol. 1998;153:17-23
10. Nolte RT, Wisely GB, Westin S, Cobb JE. et al. Ligand binding and co-activator assembly of the PPARγ. Nature. 1998;395:137-143
11. Evans M, Anderson RA, Graham J, Ellis GR. et al. Ciprofibrate therapy improves endothelial function and reduces postprandial lipemia and oxidative stress in type 2 diabetes mellitus. Circulation. 2000;101:1773-1779
12. Inoue I, Noji S, Awata T, Takahashi K, Nakajima T, Sonoda M, Komoda T, Katayama S. Bezafibrate has an antioxidant effect: PPAR alpha is associated with Cu2+, Zn2+SOD in the liver. Life Sci. 1998;63:135-144
13. Inoue I, Goto S, Mizotani K, Awata T, Mastunaga T, Kawai S, Nakajima T, Hokari S, Komoda T, Katayama S. Lipophilic HMG-CoA reductase inhibitor has an anti-inflammatory effect: reduction of mRNA levels for Il-1beta, Il-6, cyclooxygenase-2, and p22phox by regulation of PPARalpha in primary endothelial cells. Life Sci. 2000;67:863-876
14. Inoue I, Goto SS, Matsunaga T, Nakajima T, Awara T, Hokari S, Komoda T, Katayama S. The ligands/activators for PPAR alpha and gamma increase Cu-Zn SOD and decrease p22phox message expressions in primary endothelial cells. Metabolism. 2001;50:3-11
15. Matsui I, Shimosawa T, Uetake Y, Wang H, Ogura S, Kaneko T, Liu J, Ando K, Fujita T. Protective effect of potassium agonist the hypertensive cardiac dysfunction. Hypertension. 2006;48:225-231
16. Cox MJ, Hawkins UA, Hoit BD, Tyagi SC. Attenuation of oxidative stress and remodeling by cardiac inhibitor of metalloproteinase protein transfer. Circulation. 2004;109(17):2123-2128
17. Lindsay MM, Maxwell P, Dunn FG. TIMP-1: a marker of LV diastolic dysfunction and fibrosis in hypertension. Hypertension. 2002;40:136-141
18. Mujumdar VS, Tyagi SC. Temporal Regulation of ECM Components in Transition from Compensatory Hypertrophy to Decompensatory Heart Failure. J Hypertension. 1999;17:261-270
19. Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115(8):2059-2064
20. O'Reilly MS. Angiostatin: an endogenous inhibitor of angiogenesis and tumor growth. Exs. 1997;79:273-294
21. O'Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88(2):277-285
22. Pike SE, Yao L, Jones KD, Cherney B, Appella E, Sakaguchi K, Nakhasi H, Teruya-Feldstein J, Wirth P, Gupta G, Tosato G. Vasostatin, a calreticulin fragment, inhibits angiogenesis and suppresses tumor growth. J Exp Med. 1998;188(12):2349-2356
23. Maeshima Y, Sudhakar A, Lively JC, Ueki K, Kharbanda S, Kahn CR, Sonenberg N, Hynes RO, Kalluri R. Tumstatin, an endothelial cell specific inhibitor of protein synthesis. Science. 2002;295(5552):140-143
24. Colorado PC, Torre A, Kamphaus G, Maeshima Y, Hopfer H, Takahashi K, Volk R, Zamborsky ED, Herman S, Sarkar PK, Ericksen MB, Dhanabal M, Simons M, Post M, Kufe DW, Weichselbaum RR, Sukhatme VP, Kalluri R. Anti-angiogenic clues from vascular basement membrane collagen. Cancer Res. 2000;60(9):2520-2526
25. Kamphaus GD, Colorado PC, Panka DJ, Hopfer H, Ramchandran R, Torre A, Maeshima Y, Mier JW, Sukhatme VP, Kalluri R. Canstatin, a novel matrix-derived inhibitor of angiogenesis and tumor growth. J Biol Chem. 2000;275(2):1209-1215
26. Ramchandran R, Dhanabal M, Volk R, Waterman MJ, Segal M, Lu H, Knebelmann B, Sukhatme VP. Antiangiogenic activity of restin, NC10 domain of human collagen XV: comparison to endostatin. Biochem Biophys Res Commun. 1999;255(3):735-739
27. Kim KH, Cho YS, Park JM, Yoon SO, Kim KW, Chung AS. Pro-MMP-2 activation by the PPARgamma agonist, ciglitazone, induces cell invasion through the generation of ROS and the activation of ERK. FEBS Lett. 2007;581(17):33303-3310
Author contact
![]() Correspondence to: Suresh C. Tyagi, PhD., Department of Physiology and Biophysics, University of Louisville, 500 South Preston Street. Tel: 502-852-3381; Fax: 502-852-6239
Correspondence to: Suresh C. Tyagi, PhD., Department of Physiology and Biophysics, University of Louisville, 500 South Preston Street. Tel: 502-852-3381; Fax: 502-852-6239
Received 2007-8-2
Accepted 2007-9-5
Published 2007-9-7