ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
1. INTRODUCTION
2. MATERIALS AND METHODS
3. RESULTS
4. DISCUSSION
Acknowledgements
Conflicts of interest
References

Int J Biol Sci 2007; 3(7):477-485. doi:10.7150/ijbs.3.477 This issue Cite
Research Paper
Smad3 -signalling and Th2 cytokines in normal mouse airways and in a mouse model of asthma
Minna Anthoni1, Guoying Wang1,2, Marina S. Leino1, Antti I. Lauerma3,4, Harri T. Alenius1, Henrik J. Wolff5,6,7 ![]()
1. Unit of Excellence for Immunotoxicology, Finnish Institute of Occupational Health, Helsinki, Finland
2. Department of Dermatology and Venereology, Qingdao University Medical College Hospital, P.R.China
3. Control of Hypersensitivity Diseases, Finnish Institute of Occupational Health, Helsinki, Finland
4. Skin and Allergy Hospital, Helsinki University Hospital, Helsinki, Finland
5. Team for Biological Mechanisms and Prevention of Work-related Diseases, Finnish Institute of Occupational Health, Helsinki, Finland
6. Department of Pathology, Helsinki University Central Hospital, Finland
7. Department of Pathology, Kymenlaakso Central Hospital, Finland
Abstract
This study investigates the role of Smad3 signalling for the T-helper2 (Th2) cytokine homeostasis in normal lungs and in a mouse model of asthma.
We used mice deficient for Smad3, a central part of the major signal transduction pathway for TGF-β and other related cytokines, and a mouse model for allergic asthma with ovalbumin (OVA) as the antigen.
Compared to wild type mice, naive (unmanipulated) Smad3-/- mice exhibited significantly increased levels of proinflammatory cytokines and IL-4 as well as the Th2 associated transcription factor GATA-3 in the lung tissue and bronchoalveolar lavage (BAL). In the asthma model, mucin secretion and airway hyperresponsiveness (AHR) after allergen exposure was significantly increased in the Smad3-/- mice as compared to wild type (WT) mice. IL-4 levels in Smad3-/- were similar to those encountered in WT mice but IL-13 levels were decreased in the airways of OVA sensitized Smad3-/- mice compared to corresponding WT mice.
The results indicate that a lack of Smad3 dependent signalling in the normal state will lead to an increase in the GATA-3 levels and as a result of this the levels of IL-4 increase. However, the lack of Smad3 also seems to inhibit expression of some cytokines, especially IL-13. Our results also indicate that in the inflammatory state TGF-β or related cytokines functions to counterbalance the effects of IL-4 rather than to critically regulate its expression.
Keywords: Smad3, TGF-β, Asthma.
1. INTRODUCTION
Transforming growth factor -beta (TGF-β) is a central regulator of immunological activity. TGF-β regulates a variety of cellular responses, such as proliferation, differentiation, migration and apoptosis. TGF-β can either stimulate or inhibit immune cell function and opposite effects may be elicited at different concentrations [1]. Total disruption of the TGF-β1 gene has been reported to cause gross developmental abnormalities. These transgenic mice succumb about 20 days after birth to a wasting syndrome with a multifocal inflammatory response and tissue necrosis [2].
All three mammalian isoforms of TGF-β are present in the airways and at least TGF-β1 and TGF-β2 have been shown to be increased in asthmatic airways and cells, with evidence of increased TGF-β signalling [3-5]. The increased expression of TGF-β in asthmatic airways and an increased number of infiltrated eosinophils have been shown to correlate with each other [6]. TGF-β1 polymorphism has been shown to be associated with asthma [7]. TGF-β1 has been shown to reverse airway inflammation and airway hyperresponsiveness (AHR) in experimental asthma models [8, 9]. TGF-β has also been detected in the BAL fluid of a murine model of asthma [10]. In the work by Scherf et al, where decreased expression of TGF-β1 was induced in an asthma model, an exaggerated asthmatic phenotype was seen [11]. A number of different immune cells and a complex array of cytokines are involved in the development of asthmatic inflammation. T-helper 2 (Th2) cells are known to be important in generating an inflammation that characterizes asthma and allergic disease. However, the regulation of inflammatory cascade and immune system signals involved in the generation of the Th2 cell responses and, on the other hand, downregulating the inflammatory responses, is less well understood. In addition to TGF-β1, another member of the TGF superfamily, activin A has been suggested to be involved in the pathogenesis of asthma [12, 13].
In order to initiate signalling, TGF-β binds to TGF-β receptor types I and II. TGF-β signals are delivered from the cytoplasm to the nucleus via TGF-β signal transducers called Smads. Smad3 is a central signal transducer in TGF-β -signalling [14]. Smads are intracellular proteins, transcription factors 42-65 kDa in mass [15, 16]. There are three families of Smads: receptor-activated Smads (Smads 1, 2, 3, 5 and 8), common-partner Smads (Smad 4) and the inhibitory Smads (Smads 6 and 7). Activin A binds to its own specific receptor, distinct from the TGF-β1 receptor, but both receptors share the same intracellular Smad signalling pathway [17]. After activation of type II and type I -receptors, the receptor-activated -Smads become phosphorylated and form heteromeric complexes with common partner -Smads, that translocate into the nucleus. In the nucleus, these complexes control the expression of target genes in a cell type specific manner. In a study using transgenic mice expressing Smad7, Nakao et al. demonstrated that blockade of TGF-β/Smad -signalling in T cells enhanced antigen-induced airway inflammation and airway reactivity [18].
In this study we used Smad3 deficient transgenic mice to explore the role of Smad3 -signalling for cytokine homeostasis in the airways of naive Smad3-/- mice. In addition, we examined the role of Smad3 -signalling in the regulation of bronchial hyperreactivity and inflammation in a murine model of asthma.
2. MATERIALS AND METHODS
Mice
Smad3 knock-out mice of the C57BL/6 strain were kindly provided by Dr. Chuxia Deng et al and were bred in our facilities. Mice heterozygous for the targeted disruption were intercrossed to produce homozygous offspring. The resulting progeny were screened by PCR to identify Smad3-/- and WT littermates [19]. Animals were used between seven to eleven weeks of age and were age and sex matched within each experiment. The mice were housed in pathogen free facilities. All animal protocols were approved by Health Services of State Provincial Office of Southern Finland.
Induction of Allergic Airway Disease
In this work we used a model for human asthma described by Hamelmann et al [20]. Mice were sensitized intraperitoneally on day 0 and 10 with ovalbumin in alum or PBS emulsified in alum. On days 20-22, the mice were challenged with aerosolized 1% OVA, 20 minutes per day. Airway responsiveness was measured 24 hours after the last airway challenge and specimens were collected. Eight mice per group were challenged in three separate experiments.
AHR
Airway responsiveness was measured in mice 24 h after the final OVA challenge by recording respiratory pressure curves by whole body plethysmography (Buxco Technologies, Troy, NY, USA) in response to nebulized methacholine (MCh) in PBS (Sigma-Aldrich; UK) at increasing concentrations of 3-100 mg/ml. After each nebulization, recordings were taken for five minutes and the results expressed as the increase in enhanced pause (PenH ) compared to baseline (nebulized PBS). Animals were sacrificed by CO2 asphyxiation and lung, serum and bronchoalveolar lavage (BAL) samples were collected for further analysis.
Bronchoalveolar lavage and lung histology
The chest cavity was opened and the lungs were lavaged with PBS via the trachea. The BAL sample was cytocentrifuged (Cytospin, Shandon Ltd, Pittsburgh, PA). The remaining cells were fixed in ethanol (50%). The total amount of cells was determined by counting the cells in a Burker chamber. After cytocentrifuging, the bronchoalveolar lavage cells were stained with May-Grünwald-Giemsa (MGG) stain and the percentage distribution of different types of cells were determined. One part of the left lung was removed for RNA isolation. The right lung was fixed in 10% formalin, embedded in paraffin wax, routinely processed, sectioned 5 μm thick, and stained with H&E and Periodic Acid Schiff (PAS) stains. For the mucin production the results are expressed as the length of basal membrane covered with PAS stained cells / 100 μm stretch of basal membrane from the 3 bronchi in each slide showing the most PAS positivity.
Immunoglobulins
OVA-specific IgE and IgG2a serum levels were measured by ELISA [21]. Bound IgG2a and IgE were detected with biotin-conjugated rat anti-mouse IgG2a and IgE mAb (BD, Pharmingen, San Jose, CA). Streptavidin-HRP (BD, Pharmingen, San Jose, CA) 1/4000 in 1% BSA and peroxidase substrate (Kirkegaard & Perry Laboratories, Gaithersburg, MD) were used and absorbance measured at 405 nm with an ELISA reader (Multiskan MS, Labsystems, Vantaa, Finland).
RT-PCR
One part of the left lung was removed for RNA isolation. Total RNA from lungs was extracted and transcribed into cDNA. Real-time quantitative PCR was performed with an AbiPrism 7700 Sequence Detector System (Applied Biosystems, Foster City, CA, USA). PCR primers and probes were IL-13, IL-10, IL-4, IFN-γ, IL-1β, GATA-3, TGF-β1 and CCL3/MIP-1α (Applied Biosystems) and endogeneous 18S rRNA was used as the housekeeping gene and the target gene expression was expressed as relative units (RU) [22].
Measurement of cytokines by LuminexTM
IL-4, IL-5 and IL-13, cytokines of BAL-samples were measured on a LuminexTM system (Bio-Plex 200) using the Bio-Rad mouse cytokine kit (Bio-Plex cytokine assay) following the manufacturer's instructions. Data were collected with a minimum of 100 beads per analyte using Bio-Plex Manager Software (Bio-Rad Laboratories, Inc, Hercules, CA).
Data Analysis
Data were analyzed with the GraphPadPrism software (GraphPad Software, Inc., San Diego, CA). Data are expressed as mean +/- SEM. Statistical significance was accepted when P<0.05 using the Mann Whitney U Test.
3. RESULTS
Effects of the Smad3 deletion on normal mouse airways
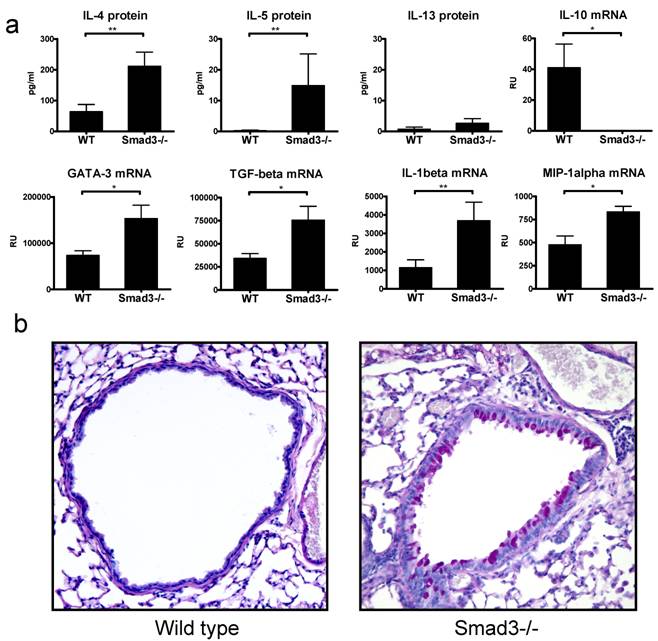
In general there were only minor morphological changes in the lungs of Smad3-/- lungs as compared to WT lungs. H&E stained lung sections revealed a marginal increase in peribronchial lymphocytes (also a few eosinophils and neutrophils were observed) in naive Smad3-/- mice. In addition in some mice (3/8) a patchy area of perivascular inflammation with macrophages was observed. Interestingly, in a few of the naive Smad3-/- animals the numbers of PAS -positive mucin producing cells were increased in the bronchial epithelium, no mucin producing cells could be detected in WT mice (Fig. 1b).
The protein levels of the central Th2 cytokine IL-4 was significantly increased in the BAL of naive Smad3-/- mice as compared to WT controls (Fig. 1a), furthermore the level of IL-5 protein was also increased although the absolute amount of IL-5 was small. The levels of IL-13 were barely above the detection limit. The levels of GATA-3 mRNA, a major Th2 associated transcription factor, were significantly higher in the lung tissue of naive Smad3-/- mice compared to that of WT mice (Fig. 1a). The mRNA expression of TGF-β was significantly increased in the lung of naive Smad3-/- mice compared to WT mice (Fig. 1a). The expression of IL-10 mRNA, another important regulatory cytokine, was significantly decreased in Smad3-/- mice. The expression of the proinflammatory cytokine IL-1β- and the chemokine MIP-1α (CCL3) mRNA was significantly upregulated in naive Smad3-/- mice lungs compared to WT mice. In contrast, no detectable levels of a major Th1 cytokine IFN-γ mRNA were found in the lung tissue of Smad3-/- or WT mice (data not shown).
Cytokines and morphology in the lungs of naive mice. The graphs in figure 1A depict the levels of proteins in BAL fluid (pg/ml) and the levels of mRNA (relative units, RU). Bars and errors represent mean ± SEM. *p<0.05, **p<0.01; n = 8 mice per group. The microscope image in figure 1B shows the mucin production in the lung of a WT mouse and a naive Smad3-/- mouse. Original magnification 200x .
Effects of the Smad3 deletion on mouse airways in the asthma model
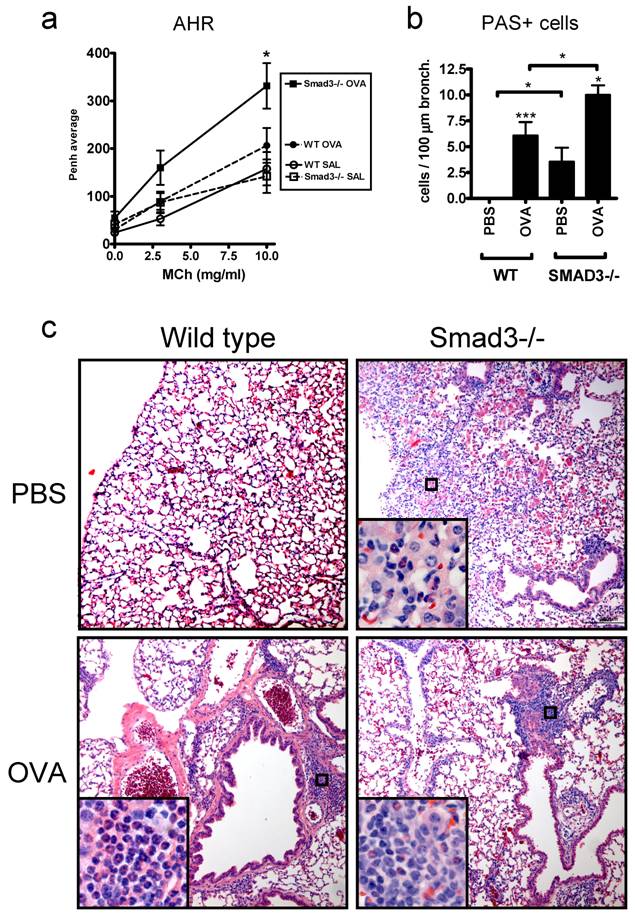
In addition to the naive mice we also examined the effects of the Smad3 deletion in a mouse model of asthma. Airway hyperreactivity to inhaled metacholine, a hallmark of allergic asthma, was significantly increased in OVA sensitized Smad3-/- mice compared to PBS sensitized mice (Fig. 2a). In contrast, no significant differences between OVA and PBS sensitized mice were found in WT mice. Smad3-/- mice exhibited also clear symptoms, including some fatalities, of severe respiratory distress after airway metacholine exposure. In addition to increased bronchial hyperreactitivity, OVA sensitized Smad3-/- mice exhibited increased numbers of PAS -positive mucin producing cells compared to OVA sensitized WT mice (Fig. 2b). PBS treated Smad3-/- mice displayed also significantly increased numbers of PAS-positive cells in the bronchial epithelium compared to WT mice (Fig. 2b). H&E stained lung sections from OVA sensitized Smad3-/- and WT mice showed peribronchiolar and perivascular inflammation consisting of lymphocytes and eosinophils, but no quantitative differences were observed (Fig. 2c). The infiltrate of Smad3-/- mice, but not WT mice, included occasionally neutrophils. Alveolar infiltrates of macrophages, eosinophils and neutrophils were frequently seen in saline treated Smad3-/- mice, but not in WT mice (Fig. 2c).
AHR, mucin production and histology of the lungs in WT and Smad3-/- mice after OVA or saline exposure. Graph A shows the AHR response to metacholine (MCh). In graph B, the mucin production after OVA or saline exposure is shown. The results are shown as mean ± SEM. *p<0.05, **p<0.01, ***p<0.001; n = 11 - 19 mice per group. The microscopic images in C show the hematoxylin and eosin (H&E)-stained lung sections of WT (right side) and Smad3-/- mice (left side) after saline (top) or OVA exposure (bottom). Original magnification of the main pictures: x 100 and the inserts x 400.
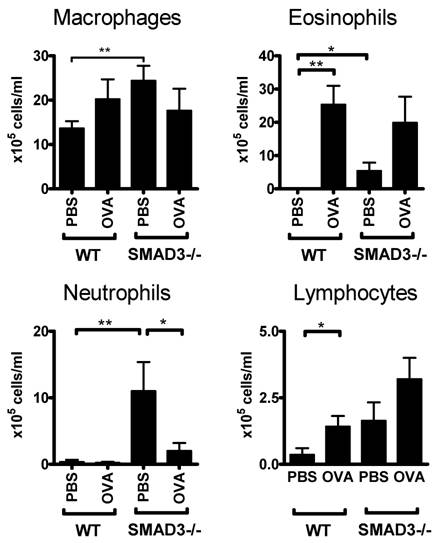
We also examined the distribution of cell types in the BAL fluid of these mice. The number of eosinophils was significantly increased in the BAL fluid of OVA-sensitized WT and Smad3-/- mice compared to their PBS treated controls (Fig 3). There was also an increase in the numbers of eosinophils and neutrophils in PBS exposed Smad3-/- mice when compared to saline treated WT mice (Fig 3). Moreover the cell counts of PBS sensitized mice were higher in Smad3-/- mice compared to those of WT mice.
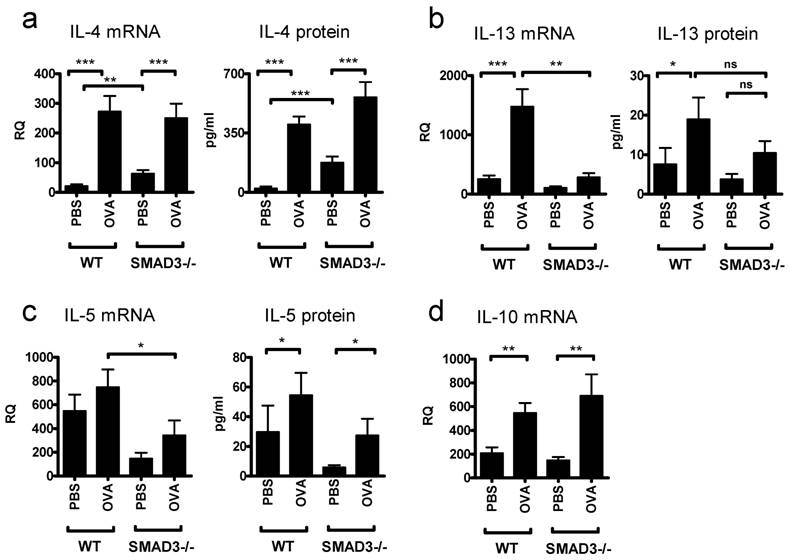
OVA sensitizing caused the levels of IL-4 mRNA and IL-4 protein to rise significantly and to comparable levels in both Smad3-/- and WT mice (Fig. 4a). However, PBS treated Smad3-/- mice exhibited increased amounts of IL-4 mRNA and protein compared to PBS treated WT mice. The expression of mRNA for another major Th2 cytokine, IL-13, was significantly decreased in the lung of OVA sensitized Smad3-/- mice as compared to OVA sensitized WT mice (Fig. 4b). In line with this, no significant differences in the levels of IL-13 were found between OVA and PBS treated Smad3-/- mice (Fig. 4b). However the protein levels of IL-13 were significantly increased in the BAL fluid after OVA sensitization in WT mice compared to PBS treated controls. The lack of Smad3 tended to decrease the levels of IL-5 protein and mRNA in both PBS and OVA exposed mice (Fig. 4c), it appears as if the levels of IL-5 protein in PBS exposed Smad3-/- mice would be lower than in the corresponding naive mice, the difference is however far from achieving significance. OVA evoked significant increases of IL-10 mRNA over the levels seen in PBS treated mice but the presence or absence of Smad3 did not have any effect on IL-10 mRNA. The levels of IL-10 mRNA after either PBS or OVA challenges were, however, much higher than those seen in naive mice (Figs. 1a and 4d). In contrast to the case with naive mice, no significant differences with respect to either the OVA vs PBS or the presence or absence of Smad3, could be observed for the expression of GATA-3 (data not shown).
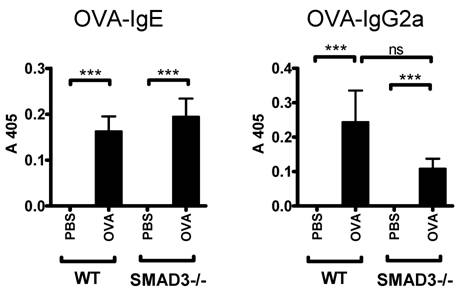
OVA-specific IgE levels (surrogate marker for Th2 type antibody response) were comparably increased both in Smad3-/- and WT mice sensitized with OVA (Fig. 5). In addition, the levels of OVA-specific IgG2a (surrogate marker for Th1 type antibody response) were increased after OVA sensitization in both mice groups. OVA-specific IgG2a levels were decreased in OVA sensitized Smad3-/- mice compared to their WT controls but the difference did not reach statistical significance (Fig. 5).
Cell composition of the bronchoalveolar lavage fluid. The figure shows the proportion of different types of cells in WT and Smad3-/- mice after airway exposure to OVA or saline. * p < 0.05, ** p < 0.01; n = 11 - 14 mice per group. The cell concentrations (106 cells/liter BAL fluid ) were WT PBS 56,4 ±1,5; WT OVA 109,1±21.1; Smad3-/- PBS 149±2,6; Smad3-/- OVA 151,0± 3,9.
Cytokines in the lungs of mice in the asthma model. The graphs depict the levels protein in BAL fluid and the levels of mRNA. after airway exposure to OVA or saline. Bars and errors represent mean ± SEM. *p<0.05, **p<0.01, ***p<0.001; n = 13 - 19 mice per PBS and OVA group.
Mouse serum levels of OVA-specific IgE and IgG2a. The figure shows immunoglobulin levels in WT and Smad3-/- mice after airway exposure to OVA or saline. The data are shown as mean±SEM, *p<0.05, ***p<0.001; n = 13 - 20 mice per group.
4. DISCUSSION
The role of Smad3 -signalling in the regulation of lung homeostasis and in the pathophysiology of respiratory diseases such as allergic asthma is poorly understood. It has been shown, however, that TGF-β is present in the airways and that the levels of certain TGF-β isoforms have been shown to be elevated in asthmatic airways. Another member of the TGF superfamily, activin A, has also been suggested to be involved in the pathogenesis of asthma [12, 13]. Since Smad3 is a critical mediator of TGF-β -signalling and activin A signalling, we utilized Smad3 deficient mice to explore the role of Smad3 -signalling in the regulation of lung functions both in the healthy situation as well as in the diseased state.
Lung histology from naive Smad3-/- mice revealed that the lack of TGF-β -signalling caused a marginal infiltrate of lymphocytes and occasionally modest increases in mucin production in the bronchial epithelium. These changes were accompanied by significant increases in the expression of Th2 cytokine IL-4 and GATA-3, an important transcription factor regulating IL-4 expression. Similarly IL-5 was also significantly increased although the absolute level remained low. The levels of IL-13 were almost nonexistent. It has been shown that GATA-3 expression is suppressed by TGF-β [23]. Thus, the increase of GATA-3 and IL-4 could be viewed to result from the release from TGF-β suppression of GATA-3. In addition to the increase in Th2 cytokines, we found a significant elevation in the expression of proinflammatory cytokines and chemokines (IL-1β and MIP-1α, respectively), the latter being indicative of macrophage activation. In line with this finding, a tendency towards activation of macrophages with regard to matrix metalloproteinases has been observed by Bonniaud et al. in Smad3 knockout mice [24]. The expression levels of a regulatory cytokine, IL-10, were completely abolished in naive Smad3 deficient mice compared to WT mice. The baseline levels of IL-10 in the lungs of WT mice are, however, quite low and it is therefore unclear whether this additional reduction could lead to the increased expression of proinflammatory cytokines and IL-4 seen. The reason for the decrease of IL-10 as well as the reasons for the low levels of IL-5 and IL-13 could be that the presence of Smad3 is required for GATA3 regulation [25], this is discussed in association with the asthma model.
Our results suggest that Smad3 signalling is important in the suppression of the proinflammatory as well as the IL-4 response in the airways in the healthy condition, it should be noted that the suppressed features mentioned are also associated with asthma.
When studying the lungs of mice exposed to allergen in the asthma model we found that there was a pronounced increase in the amount of mucin in both PBS and OVA treated Smad3-/- mice compared to WT mice. Our results also revealed that airway hyperreactivity to inhaled metacholine, a classical hallmark of allergic asthma, was significantly increased in OVA sensitized Smad3-/- mice. In contrast, no significant differences between OVA and PBS sensitized mice were found in WT mice. These results probably underestimate the actual degree of AHR since C57BL/6 mice (strain used in the present study) are more resistant to bronchoconstrictors than other mouse strains, and differences between mice might be difficult to observe using whole body plethysmography [24]. Indeed, we were able to observe signs of respiratory distress in Smad3-/- mice after allergen exposure but no symptoms were visible in WT mice. Taken together, we found that the lack of Smad3-/- signalling led to an exaggerated asthmatic phenotype in allergic asthma model. The exaggerated asthmatic phenotype seen in this work resembles that seen in the work of Nakao et al [18] where expression of the inhibitory Smad7 was used to disturb signalling.
Naive Smad3 deficient mice demonstrated elevated levels of IL-4 protein in their lungs. In line with this, the levels of IL-4 mRNA and protein in the airways of PBS treated Smad3-/- mice were significantly increased compared to PBS treated WT mice. However, the IL-4 levels were equivalently increased in OVA sensitized Smad3-/- and WT mice. These results suggest that cytokines belonging to the TGF superfamily play an important role in the regulation of IL-4 production in the normal state but their significance is less critical during inflammation induced in this case by immunization with OVA allergen and/or adjuvant. In contrast to IL-4 expression, the levels of IL-13 (another important Th2 type cytokine) were significantly decreased in OVA sensitized lungs of Smad3-/- mice compared to WT mice. It has been previously reported that GATA-3 interacts extensively and apparently directly with Smad3 to regulate GATA target genes, including IL-10 and IL-5, through GATA3 binding sites in the promoter regions [25]. Thus the low level of increase of IL-5 and the decrease of IL-10 in naive Smad3-/- mice could be caused by an inhibition of GATA-3 function by the lack of Smad3. IL-13 is regulated through a GATA-3 binding promoter region [26], but there are no earlier reports that its expression would be dependent on Smad3 [25]. Our findings, interestingly, suggest a critical dependence of GATA-3 for Smad3 in regulating IL-13 expression. In contrast, no GATA binding exists in the IL-4 promoter and GATA-3 regulates IL-4 production through distal regulatory elements [26-28]. In the asthma model, in contrast to naive mice, there were no significant differences in the amounts of GATA-3 mRNA levels between Smad3-/- and WT. Furthermore, IL-10 levels in the asthma model were much higher than in the naive state and they did not reveal any significant differences between Smad3-/- and WT mice mice, suggesting that there is a difference in the regulation of GATA3 and IL-10 cytokines in the naive state and the inflamed state. In the case of IL-10 it appears that in PBS exposed lungs other regulatory mechanisms override the lack of GATA3 and increase IL-10 mRNA levels substantially as compared to naive mice. Taken together, we think that the unexpectedly low levels or decreases of IL-5, IL-10, and IL-13 in various contexts can best be explained by a need for interactions between GATA3 and Smad3. However, depending on the situation, there are obviously other regulatory mechanisms that strongly affect the expression of these cytokines. Further studies are needed to clarify the molecular mechanisms for GATA3 and Smad3 effects on cytokine expression.
Increased amounts of IL-13 have been previously associated with increased airway hyperreactivity and mucus overproduction. It is interesting that in the present study Smad3 deficient mice with diminished IL-13 production clearly exhibited increased airway hyperreactivity and substantially increased mucus production. This result is in agreement with the finding of Perkins et al [29] demonstrating a role for IL-4 in promoting all the Th2 tissue responses independent of IL-13.
Our results are in many respects contradictory to those described by Le et al [30].They showed that a Smad3 deletion led to a decrease in mucin production and eosinophilia whereas no changes was seen with the AHR. The probable reason for this discrepancy is that in the work of Le et al a different Smad3 deletion was used, i.e that described by Zhu et al in 1998 [31] which has exons 2 deleted on both alleles. In contrast we have used a Smad3 deletion where exons 8 on both alleles were deleted [19]. It is well known that these different Smad3 deletions often give contradictory results. Perhaps the most interesting feature of the Smad3 (exon 2) deletion is a major increase in the incidence of colonic cancer. No increases in colonic cancer has been reported from the Smad3 (exon 8) deletions. As far as we know the reason for these discrepancies have not been fully determined, Yang et al 1999 [19] has discussed this topic to some extent in his article and propose a possible explanation i.e. that some part of the mutant transcript (exons 4-9) could actually be translated in the Smad3 (exon 2) deletion. In contrast there is evidence showing that no part of the mutant transcript in the Smad3 (exon 8) deletion is translated. In addition there are differences in the genetic background of the mice but according to Yang et al this is not likely to explain the discrepancies between the different Smad3 deletions. Thus, it is possible that the explanation for the differences between our results and those of Le et al is that in the latter case a part of Smad3 is translated. If we now consider other models where TGF-β or its receptor has been manipulated the results are in many ways similar to the data concerning the inflammatory state presented here i.e. eosinophilia and AHR is increased and mucin production is increased when TGF β1 levels or effect are decreased [11, 18]. Thus we think that our results are much more likely to reflect a generalizable role for Smad3 signalling in asthma.
TGF-β is not entirely dependent on Smad dependent pathways for its signalling [32, 33] and a total lack of TGF-β will cause a much more severe phenotype than that seen in the Smad3-/- mice [2]. This study showed that a lack of Smad3 in naive mice caused an increase in the central Th2 transcription factor GATA-3 and changes in IL-4 and proinflammatory cytokine levels. Interestingly the lack of Smad3 and presumably TGF-β signalling led to an exaggerated asthmatic phenotype in an asthma model, in line with earlier findings. However, this exaggerated asthmatic phenotype was not accompanied by any increase in IL-4 or other Th2 cytokines, in contrast to earlier findings with other forms of inhibition of TGF-β [11, 18] in asthma models. This indicates that in the inflammatory state, TGF-β or related cytokines function as a counterbalance to IL-4 effects rather than as a regulator of its expression.
Acknowledgements
We wish to thank the late Anita Roberts, PhD, for her interest in this study. We thank Kari Savelius and Virva Paavola for excellent technical assistance. We thank Nummela Sanatoriums Stiftelse, the Finnish Work Environment Fund, Finska Läkaresällskapet, Finnish Government Research Funds (EVO), the Allergy Foundation of the Allergy and Asthma Association, Tampere Tuberculosis Foundation, Finnish Anti-Tuberculosis Association Foundation, the Finnish Society of Allergology and Immunology, the Pulmonary Association Heli, and the Ida Montin Foundation for the financial support. Dr Sampsa Matikainen has given valuable comments on the manuscript. We thank Ewen McDonald for revising the language of this work.
Conflicts of interest
The authors have declared that no conflict of interest exists.
References
1. Letterio JJ, Roberts AB. Regulation of immune responses by TGF-β. Annu Rev Immunol. 1998;16:137-161
2. Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, Annunziata N, Doetschman T. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature. 1992;359:693-699
3. Howell JE, McAnulty RJ. TGF-β: Its role in asthma and therapeutic potential. Current Drug Targets. 2006;7:547-565
4. Adachi T, Motojima S, Hirata A, Fukuda T, Kihara N, Makino S. Detection of transforming growth factor-beta in sputum from patients with bronchial asthma by eosinophil survival assay and enzyme-linked immunosorbent assay. Clin Exp Allergy. 1996;26:557-562
5. Redington AE, Madden J, Frew AJ, Djukanovic R, Roche WR, Holgate ST, Howarth PH. Transforming growth factor-beta 1 in asthma. Measurement in bronchoalveolar lavage fluid. Am J Respir Crit Care Med. 1997;156:642-647
6. Nomura A, Uchida Y, Sakamoto T, Ishii Y, Masuyama K, Morishima Y, Hirano K, Sekisawa K. Increases in collagen type I synthesis in asthma: the role of eosinophils and transforming growth factor -beta. Clin Exp Allergy. 2002;32:860-865
7. Silverman ES, Palmer LJ, Subramaniam V, Hallock A, Mathew S, Vallone J, Faffe DS, Shikanai T, Raby BA, Weiss ST, Shore SA. Transforming growth factor-beta1 promoter polymorphism C-509T is associated with asthma. Am J Respir Crit Care Med. 2004;169:214-219
8. Haneda K, Sano K, Tamura G, Sato T, Habu S, Shirato K. TGF-beta induced by oral tolerance ameliorates experimental tracheal eosinophilia. J Immunol. 1997;159:4484-4490
9. Hansen G, McIntire JJ, Yeung V, Peter BG, Thorbecke GJ, Chen L, DeKruyff RH, Umetsu DT. CD4+ T helper cells engineered to produce latent TGF-β1 reverse allergen-induced airway hyperrreactivity and inflammation. J Clin Invest. 2000;105:61-70
10. Tanaka H, Masuda T, Tokuoka S, Komai M, Nagao K, Takahashi Y, Nagai H. The effect of allergen-induced airway inflammation on airway remodeling in a murine model of allergic asthma. Inflamm Res. 2001;50:616-624
11. Scherf W, Burdach S, Hansen G. Reduced expression of transforming growth factor β1 exacerbates pathology in an experimental asthma model. Eur J Immunol. 2005;35:198-206
12. Cho SH, Yao Z, Wang SW, Alban RF, Barbers RG, French SW, Oh CK. Regulation of activin A expression in mast cells and asthma: its effect on the proliferation of human airway smooth muscle cells. J Immunol. 2003;170:4045-4052
13. Karagiannidis C, Hense G, Martin C, Epstein M, Ruckert B, Mantel PY, Menz G, Uhlig S, Blasser K, Schmidt-Weber CB. Activin A is an acute allergen-responsive cytokine and provides a link to TGF-β-mediated airway remodeling in asthma. J Allergy Clin Immunol. 2006;117:111-118
14. Attisano L, Wrana JL. Signal transduction by the TGF-β superfamily. Science. 2002;296:1646-1647
15. Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753-791
16. Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-beta signal transduction. J Cell Sci. 2001;114:4359-4369
17. Abe M, Minetishi T, Leung PC. Activin receptor signaling. Growth Factors. 2004;22:105-110
18. Nakao A, Miike S, Hatano M, Okumura K, Tokuhisa T, Ra C, Iwamoto I. Blockade of transforming growth factor β/Smad signaling in T cells by overexpression of Smad7 enhances antigen-induced airway inflammation and airway reactivity. J Exp Med. 2000;192:151-158
19. Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, Roberts AB, Deng C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J. 1999;18:1280-1291
20. Hamelmann E, Schwarze J, Takeda K, Oshiba A, Larsen L, Irvin G, Gelfand EW. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am J Respir Crit Care Med. 1997;156:766-775
21. Spergel JM, Mizoguchi E, Brewer JP, Martin TR, Bhan AK, Geha RS. Epicutaneous sensitization with protein antigen induces localized allergic dermatitis and hyperresponsiveness to methacholine after single exposure to aerosolized antigen in mice. J Clin Invest. 1998;101:1614-1622
22. Lehto M, Koivuluhta M, Wang G, Amghaiab I, Majuri M-L, Savolainen K, Turjanmaa K, Wolff H, Reunala T, Lauerma A, Palosuo T, Alenius H. Epicutaneous natural rubber latex sensitization induces T helper 2-type dermatitis and strong prohevein-specific IgE response. J Invest Dermatol. 2003;120:633-640
23. Gorelik L, Fields PE, Flavell RA. Cutting edge: TGF-β inhibits Th type 2 development through inhibition of GATA-3 expression. J Immunol. 2000;165:4773-4777
24. Bonniaud P, Kolb M, Galt T, Robertson J, Robbins C, Stampfli M, Lavery C, Margetts PJ, Roberts AB, Gauldie J. Smad3 null mice develop airspace enlargement and are resistant to TGF-β-mediated pulmonary fibrosis. J Immunol. 2004;173:2099-2108
25. Blokzijl A, ten Dijke P, Ibáñez CF. Physical and functional interaction between GATA-3 and Smad3 allows TGF-β regulation of GATA target genes. Curr Biol. 2002;12:35-45
26. Lavenu-Bombled C, Trainor CD, Makeh I, Romeo P-H, Max-Audit I. Interleukin-13 gene expression is regulated by GATA-3 in T cells. J Biol Chem. 2002;277:18313-18321
27. Zhu J, Min B, Hu-Li J, Watson CJ, Grinberg A, Wang Q, Killeen N, Urban Jr JF, Guo L, Paul WE. Conditional deletion of Gata3 shows its essential function in Th1-Th2 responses. Nature Immunol. 2004;5:1157-1165
28. Kishikawa H, Sun J, Choi A, Miaw S-C, Ho I-C. The cell type-specific expression of the murine IL-13 gene is regulated by GATA-3. J Immunol. 2001;167:4414-4420
29. Perkins C, Wills-Karp M, Finkelman FD. IL-4 induces IL-13-independent allergic airway inflammation. J Allergy Clin Immunol. 2006;118:410-419
30. Le AV, Cho JY, Miller M, McElwain S, Golgotiu K, Broide DH. Inhibition of allergen-induced airway remodeling in Smad 3-deficient mice. J Immunol. 2007;178:7310-7316
31. Zhu Y, Richardson JA, Parada LF, Graff JM. Smad3 mutant mice develop metastatic colorectal cancer. Cell. 1998;94:703-714
32. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature. 2003;425:577-584
33. Moustakas A, Heldin C-H. Non-Smad TGF-β signals. J Cell Sci. 2005;118:3573-3584
Author contact
![]() Correspondence to: Henrik Wolff, Finnish Institute of Occupational Health, Topeliuksenkatu 41aA FI-00250, Helsinki, Finland. FAX: +358 30 474 2021. TEL+358468512261; EMAIL: henrik.wolfffi
Correspondence to: Henrik Wolff, Finnish Institute of Occupational Health, Topeliuksenkatu 41aA FI-00250, Helsinki, Finland. FAX: +358 30 474 2021. TEL+358468512261; EMAIL: henrik.wolfffi
Received 2007-10-29
Accepted 2007-11-22
Published 2007-11-24