ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2008; 4(1):29-36. doi:10.7150/ijbs.4.29 This issue Cite
Research Paper
Reduced white fat mass in adult mice bearing a truncated Patched 1
Zili Li, Heng Zhang, Leslie A. Denhard, Lan-Hsin Liu, Huaxin Zhou, Zi-Jian Lan ![]()
Birth Defects Center, Department of Molecular, Cellular and Craniofacial Biology, University of Louisville Health Sciences Center, Louisville, KY 40202, USA
Abstract
Hedgehog (Hh) signaling emerges as a potential pathway contributing to fat formation during postnatal development. In this report, we found that Patched 1 (Ptc1), a negative regulator of Hh signaling, was expressed in the epididymal fat pad of adult mice. Reduced total white fat mass and epididymal adipocyte cell size were observed in naturally occurring spontaneous mesenchymal dysplasia (mes) adult mice (Ptc1mes/mes), which carry a deletion of Ptc1 at the carboxyl-terminal cytoplasmic region. Increased expression of truncated Ptc1, Ptc2 and Gli1, the indicators of ectopic activation of Hh signaling, was observed in epididymal fat pads of adult Ptc1mes/mes mice. In contrast, expression of peroxisome proliferator-activated receptor gamma, CCAAT/enhancer binding protein alpha, adipocyte P2 and adipsin were reduced in epididymal fat pads of adult Ptc1mes/mes mice. Taken together, our results indicate that deletion of carboxyl-terminal tail of Ptc1 can lead to the reduction of white fat mass during postnatal development.
Keywords: gene expression, mouse, Patched 1, spontaneous mesenchymal dysplasia, white fat mass, body weight
Introduction
Hedgehog (Hh) signaling plays a critical role during embryonic development and tumorigenesis [1, 2]. The canonical Hh signaling cascade is initiated through the binding of secreted Hh proteins to the membrane-bound receptor Patched-1 (Ptc1) or Patched-2 (Ptc2) to relieve patched-mediated repression of the signal transducer smoothened (Smo), leading to the activation of transcription factors Gli1, Gli2 and Gli3 to enhance transcription of downstream targets such as Ptc1, Ptc2 and Gli1 [1-5]. In addition to its critical roles during embryonic development and tumorigenesis, Hh signaling emerges as a potential mediator of fat formation and body weight gain during postnatal development [6, 7]. Injection of adult mice with sonic Hh (Shh)-IgG fusion protein (a Hh agonist) causes a significant increase in fat mass and body weight without affecting food consumption and fluid intake [8]. In agreement with this observation, inactivation of Hh signaling by injection of anti-Hh antibodies in normal adult mice fed with a high-fat diet and in obese leptin-deficient (ob/ob) mice fed with a low-fat diet can significantly reduce body weight gain in these animals [9]. These animal studies suggest that Hh signaling may stimulate fat formation (adipogenesis) and promote body weight gain in adult mice [8, 9]. In contrast to the above animal studies [8, 9], it has been reported that Hh can block adipogenesis in cultured mammalian adipocytic or multipotent mesenchymal cells [10-13]. This is highlighted by the recent study by Suh et al [13]. They found that fat-body-specific activation of Hh signaling inhibited fat formation in Drosophila and that Hh blocked adipogenesis in mammalian cells by regulating antiadipogenic gene expression. In addition, they found that Hh signaling molecules such as Gli1, Gli2 and Gli3 are expressed in white fat tissues (WAT) of adult mice and that expression of these genes are down-regulated in normal mice fed with high fat diet and in ob/ob mice. A model of Hh signaling in inhibiting fat formation has been proposed in which Hh activates Gli transcription factors (Gli1, Gli2 and Gli3), which then activate antiadipogenic gene expression to inhibit the expression of adipogenic genes such as peroxisome proliferator-activated receptor gamma (PPARgamma) [14, 15] and CCAAT/enhancer binding protein alpha (CEBPalpha) [16] in adipose tissues, resulting in inhibition of adipogenesis [6, 13]. According to this model, it is expected that ectopic activation of Hh signaling in adipose tissues will reduce fat formation in adults, which would be contradictory to observations reported by Martin et al [8]. Therefore, whether Hh signaling acts to stimulate or inhibit fat formation during postnatal development remains to be characterized in vivo using animal models such as mutant mice carrying deletions of Hh signaling molecules.
Ptc1 is a negative regulator of the Hh signaling during embryonic development [1, 4]. It is a Hh receptor protein containing twelve transmembrane domains and intracellular amino- and carboxyl-terminal regions [2, 17]. In the absence of Hh, Ptc1 represses Smo activity. This repression of Smo is relieved once after Hh binds to the extracellular loops within the transmembrane domains of Ptc1, resulting in the activation of Hh signaling downstream target genes to elicit its biological functions [2, 5, 17, 18]. It have been shown that the carboxyl terminus of Ptc1 is important for the inactivation of Hh signaling target genes and the localization and degradation of Ptc1 itself [19, 20]. Deletion of Ptc1 in mice can mimic ectopic Hh signaling and cause embryonic lethality, indicating the essential role of Ptc1 during embryonic development [21]. Although Ptc1 has been shown to be a determinant of body weight in utero [21-23], there is no direct evidence that Ptc1 is also involved in controlling fat formation during postnatal development. The naturally occurring spontaneous mesenchymal dysplasia (mes) mouse line (Ptc1mes/mes), which has a deletion of the Ptc1 protein at the carboxyl-terminal cytoplasmic region and can bypass the embryonic lethality observed in a complete knockout of Ptc1 [22, 24], would be an excellent model to investigate the potential role of Ptc1 in fat formation during postnatal development.
In this communication, we found that Ptc1 was expressed in epididymal white fat tissue (WAT) in adult mice. Loss of the carboxyl-terminus of Ptc1 in adult Ptc1mes/mes mice caused a significant reduction in WAT. The adipocyte size of epididymal WAT was also reduced in Ptc1mes/mes mice. Gene expression analyses showed that truncated Ptc1, Ptc2 and Gli1 were overexpressed in WAT of Ptc1mes/mes mice, indicating ectopic activation of Hh signaling in WAT. Consistent with reduced WAT, expression of adipogenic genes, PPARgamma and CEBPalpha, and the adipocyte marker genes, adipocyte P2 (aP2) [25] and adipsin [26], were reduced in WAT of adult Ptc1mes/mes males. Collectively, our results suggest that Ptc1 play a role in white fat formation during postnatal development. This study supports earlier findings of Suh et al and Spinella-Jaegle et al that Hh signaling inhibits fat formation in cultured cells [10, 13].
Materials and Methods
Animals and genotyping
Ptc1mes/+ mice (B6C3Fe-a/a strain) were purchased from the Jackson Laboratory (Bar Harbor, Maine, USA) and maintained on a 14 hour light:10 hour dark cycle, with free access to normal chow (Lab diet, Catalog #5001, USA) and water, in the vivarium of the University of Louisville. Ptc1+/+, Ptc1mes/+ and Ptc1mes/mes littermates were obtained from the intercross of Ptc1mes/+ mice and genotyped as previously described [22].
Tissue collection and body, tissue and total WAT weight analyses
Mice at various ages were euthanized with carbon dioxide and the body weights of these mice were determined using an electronic balance. Liver, spleen, kidney, lung, heart and total WAT including subcutaneous, abdominal and intra-scapular fat in mice were collected and weighed using a balance. Since it took some time to collect WAT from subcutaneous regions of the whole animal which may damage the WAT histology and cause RNA degradation, WAT from epididymides were quickly harvested for histological and RNA analyses.
Histological analysis of WAT and determination of WAT size
Epididymal WAT was fixed in 4% paraformaldehyde, dehydrated and then embedded in paraffin. The paraffin-embedded tissues were sectioned at 7 μm thickness, stained with hematoxylin and eosin, and then examined under a microscope (Axioskop2, Zeiss, Thornwood, NY, USA). The average size of adipocyte cells from each mouse was determined by analyzing 40 randomly-selected adipocyte cells from 20 sections using the Zeiss Imaging Quantitative Software (Zeiss, Thornwood, NY, USA).
RNA isolation and RT-PCR
RNA was isolated from epididymal WAT of Ptc1mes/mes and control mice using Trizol reagent (Invitrogen, Carlsbad, CA , USA). First strand cDNA was synthesized at 42oC for 60 min using 1 μg of total RNA as template and random hexanucleotides as primers (Promega, Madison, WI, USA). PCR reactions with 1/10 of the cDNA were carried out in a buffer containing 1.5 mM MgCl2 (94oC, 1 min; 52oC, 1 min; 72oC 1 min) for various numbers of PCR cycles (20-38 cycles), which can detect the differential gene expression (within the linear range of PCR products for each gene). The nucleotide sequences of PCR primers are as follows: Ptc1 (Ptc1-F: 5'-tgtctggcatcagtgaggag-3', Ptc1-R: 5'-gacaaggagccagagtccag-3', 190 bp; Ptc1-F2: 5'-gctggaggagaacaagcaac-3', Ptc1-R2: 5'-cattggggtagctggacagt-3', 536 bp), Ptc2 (5'-tgcctctctggagggcttcc-3' and 5'-cagttcctcctgccagtgca-3', 208 bp), Gli1 (5'-actggggtgagttcccttct-3' and 5'-tggcagggctctgactaact-3', 492 bp), Gli2 (5'-ccccctagcatcaatgagaa-3' and 5'-tctgcacggattgtggatta-3', 496 bp), Gli3 (5'-ctacggcgactgagaggaag-3' and 5'- tggcatcaattggtacagga-3', 396 bp), glucocorticoid-induced leucine-zipper protein (Gilz, 5'-tggggcctagtaacaccaag-3' and 5'-aagctgtaaccccacactgg-3', 364 bp), CEBPalpha (5'-atcccagagggactggagtt-3' and 5'-aagtcttagccggaggaagc-3', 373 bp), PARgamma (5'- ccctggcaaagcatttgtat-3' and 5'-aatccttggccctctgagat-3', 403 bp), aP2 (5'- tcacctggaagacagctcct-3' and 5'-tcgactttccatcccacttc-3', 344 bp), adipsin (5'-caagcgatggtatgatgtgc-3' and 5'-atccggtaggatgacactcg-3', 465 bp) and beta-actin (5'-ttgagaccttcaacacccc-3' and 5'-agccagagcagtaatctcc-3', 593 bp). All PCR products were confirmed by DNA sequencing.
Food intake measurement
Five adult wild type and Ptc1mes/mes mice at the age of 23-27 weeks were singly housed with free access to water. The food present in a sealed food hooper was recorded weekly for consecutive two weeks using an electronic balance.
Glucose tolerance test
Glucose tolerance test was performed as described previously [23]. Briefly, overnight-fasted 24-26-week-old wild type, heterozygous and homozygous Ptc1mes littermates were injected intraperitoneally with 2 grams/kg body weight of 20% D-glucose. Blood glucose levels at time 0 (immediately before the injection of glucose), 15, 30, 60, 120 and 180 minutes after injection of glucose were determined using a Glucometer Elite glucometer.
Statistical analysis
When applicable, Student's t-test was used to determine the statistical significance of difference between Ptc1mes/mes and control littermates with P value less than 0.05. Data are presented as mean ± SEM of indicated numbers of mice in the figures.
Results
Expression of Hh signaling molecules Ptc1, Ptc2, Gli1, Gli2 and Gli3 in WAT of adult mice
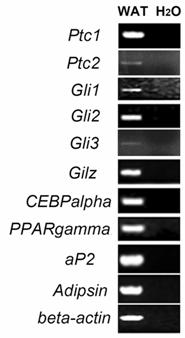
To determine whether Hh signaling molecules such as Ptc1, Ptc2, Gli1, Gli2 and Gli3 are expressed in WAT of adult mice, we performed RT-PCR analyses using specific primers for each gene. As shown in Figure 1, specific PCR products for those genes were detected in RNA samples from epididymal WAT of adult mice. In addition, we also observed the expression of an antiadipogenic gene Gilz [27], adipogenic genes CEBPalpha and PPARgamma, and mature adipocyte marker genes aP2 and adipsin in epididymal WAT (Figure 1).
Expression of Ptc1, Ptc2, Gli1 Gli2 and Gli3 in mouse epididymal WAT. Epididymal WAT RNA from 8-week-old mice was subjected to RT-PCR analyses using specific primers for Ptc1 (Ptc1-F and Ptc1-R), Ptc2, Gli1, Gli2, Gli3, Gilz, PPARgamma, CEBPalpha, aP2 and adipsin. The numbers of PCR reaction cycles are 36 for Ptc1, 38 for Ptc2 and 32 for other tested genes. Water was included as negative controls.
Reduced total WAT and adipocyte cell size in adult Ptc1mes/mes mice
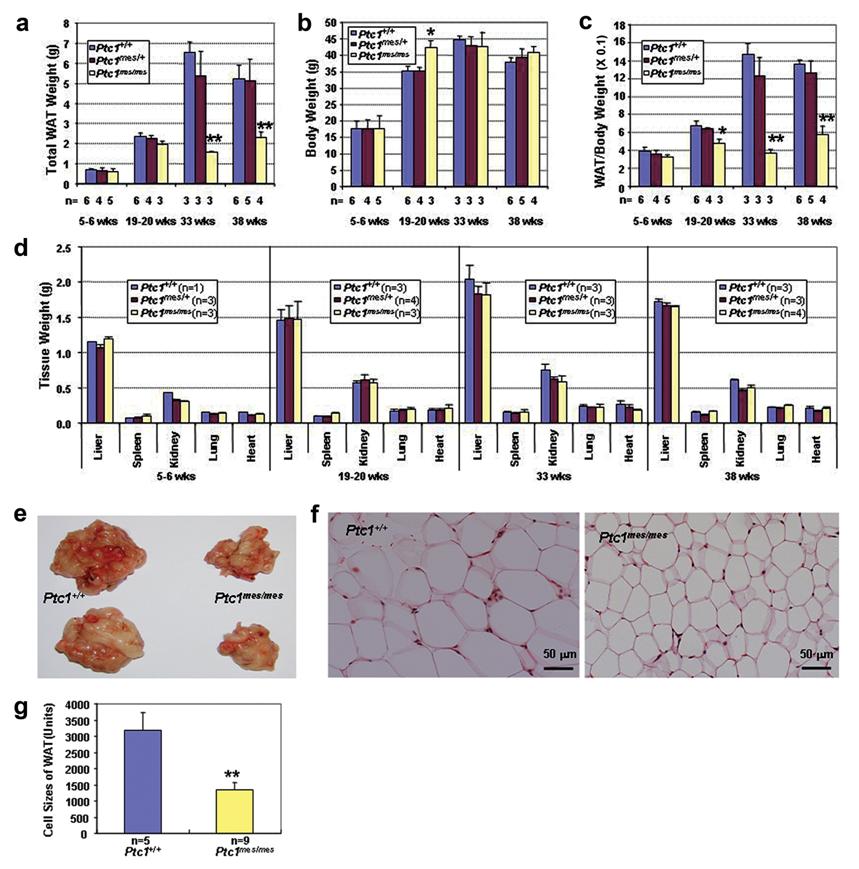
To investigate if there is any defect in WAT during postnatal development, total WAT, body and major organ weights of male Ptc1+/+, Ptc1mes/+ and Ptc1mes/mes littermates at various ages were determined (Figure 2). As shown in Figure 2a, total WAT weights were reduced in Ptc1mes/mes male mice during postnatal development with a modest reduction at the age of 19-20 weeks and the remarkable reduction at the age of 33 or 38 weeks, when compared to their wild type and heterozygous littermates (Figure 2a). No significant changes in body weights were observed between male Ptc1mes/mes and control wild type and heterozygous littermates during postnatal development, except that 19-20-week-old Ptc1mes/mes males were slightly heavier than their controls (Figure 2b). The ratios of total WAT to body weights were also significantly reduced in 19-20-week-old or older Ptc1mes/mes males (Figure 2c). In contrast, there were no significant changes in gross weights of other tested organs including liver, spleen, kidney, lung and heart (Figure 2d). The representative results of reduced total WAT of 33-week-old Ptc1mes/mes male mice were shown in Figure 2e. Histological analyses showed that epididymal fat cells of these Ptc1mes/mes mice were smaller than those of Ptc1+/+ mice (Figure 2f). Quantitative analyses showed that the sizes of epididymal WAT cells in 33-week-old Ptc1mes/mes mice were significantly reduced (P < 0.01), when compared to those of Ptc1+/+ littermates (Figure 2g).
Reduced total WAT and adipocyte cell sizes in adult male Ptc1mes/mes mice, when compared to control Ptc1+/+ littermates. a-d. Total WAT weights (a), body weights (b), the ratios of total WAT to body weights (c) and major organ weights (d) of Ptc1+/+, Ptc1mes/+ and Ptc1mes/mes male mice during postnatal development. e. Representative photos showing reduced total WAT in Ptc1mes/mes mice. Total WAT of two wild type and their Ptc1mes/mes littermates at the age of 33 weeks are shown. f. Histology of WAT from 33-week-old Ptc1+/+ and Ptc1mes/mes littermates. Note the small adipocyte cell size in WAT of Ptc1mes/mes mice. g. Quantitative analyses showing the reduced cell size of WAT in 33-week-old Ptc1mes/mes mice, when compared to their wild type littermates. In a-d and g, data are presented as mean ± SEM of the indicated numbers of mice in the bar graphs. *P < 0.05, **P < 0.01, when compared to Ptc1+/+ and/or Ptc1mes/+ littermates.
Enhanced expression of Hh signaling target genes (amino region of Ptc1, Ptc2, and Gli1) and reduced expression of adipogenic genes PPARgamma and CEBPalpha as well as adipocyte marker genes aP2 and adipsin in epididymal WAT of adult Ptc1mes/mes mice
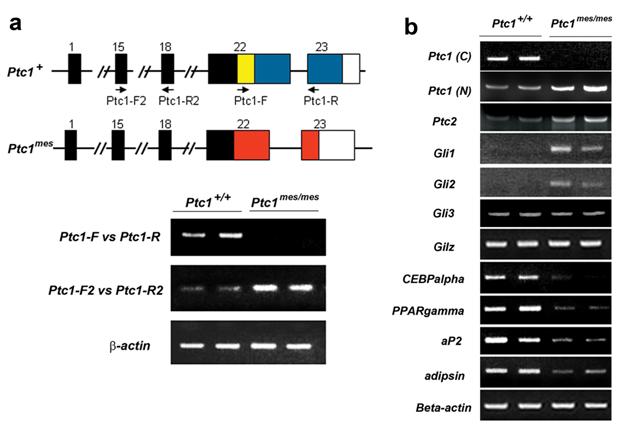
It has been reported that Ptc1mes mutant mice carry a deletion of a 32 bp DNA fragment in the exon 22 of Ptc1 allele [22] (Figure 3a). To confirm this, we isolated RNA from epididymal WAT of 8-10-week-old wild type and Ptc1mes/mes mice and performed RT-PCR analyses. As shown in Figure 3a, a positive DNA band covering exons 22 and 23 (using Ptc1-F and Ptc1-R primers) was detected in wild type, but not Ptc1mes/mes mice. These results confirmed the absence of the carboxyl-terminus of Ptc1 mRNA in epididymal WAT of Ptc1mes/mes mice. Since Ptc1 is a known downstream target gene of Hh signaling, RT-PCR analyses were also performed to determine whether expression of the amino region of the Ptc1 gene will be upregulated in epididymal WAT of Ptc1mes/mes mice. As shown Figure 3a, positive DNA bands covering exons 15 and 18 of Ptc1 (using Ptc1-F2 and Ptc1-R2 primers) were detected in wild type mice and even more pronounced in Ptc1mes/mes mice. Similar Ptc1 expression patterns were observed in epididymal WAT from 33-week-old mice (Figure 3b), indicating that the amino-terminal Ptc1 mRNA is overexpressed in WAT of Ptc1mes/mes mice.
To determine whether there is mis-expression of other Hh signaling molecules and WAT marker genes in epididymal WAT, semi-quantitative RT-PCR analyses were performed on RNA samples isolated from 33-week-old Ptc1mes/mes mice. As shown in Figure 3b, expression levels of the Hh signaling downstream target genes Ptc1, Ptc2 and Gli1were increased in epididymal WAT of Ptc1mes/mes mice, when compared to those of Ptc1+/+ mice. Expression of Gli2, but not Gli3, was also increased in epididymal WAT of Ptc1mes/mes mice. In contrast, reduced expression of PPARgamma and CEBPalpha, two known adipogenic genes, were observed in Ptc1mes/mes mice, while there was no obvious change in the expression level of Gilz, an antiadipogenic gene [27], between Ptc1mes/mes and Ptc1+/+ mice (Figure 3b). In addition, expression of WAT marker genes aP2 and adipsin were also reduced in Ptc1mes/mes mice (Figure 3b).
Increased expression of Ptc1, Ptc2, Gli1 and Gli2 and reduced expression of PPARgamma, CEBPalpha, aP2 and adipsin in epididymal WAT of Ptc1mes/mes mice. a. Representative RT-PCR analyses showing the loss of carboxyl-terminal Ptc1 mRNA (using Ptc1-F and Ptc1-R primers, 36 cycles) and increased expression of amino-terminal Ptc1 mRNA (using Ptc1-F2 and Ptc1-R2 primers, 32 cycles) in WAT of two 8-10-week-old Ptc1mes/mes mice. Deletion of 32 bp coding sequence within the exon 22 of Ptc1 in Ptc1mes/mes mice is highlighted by a yellow box. Two sets of RT-PCR primers for Ptc1 mRNA are indicated by arrows. b. Representative semi-quantitative RT-PCR showing the increased expression of amino-terminal Ptc1, Ptc2, Gli1 and Gli2 and the reduced expression of PPARgamma, CEBPalpha, aP2 and adipsin in epididymal WAT of 33-week-old Ptc1mes/mes mice. The numbers of PCR reaction cycles for carboxyl-terminal Ptc1 [Ptc1 (C): using primers Ptc1-F and Ptc1-R], amino-terminal Ptc1 [Ptc1-(N):using primers Ptc1-F2 and Ptc1-R2], Ptc2, Gli1, Gli2, Gli3, Gilz, CEBPalpha, PPARgamma, aP2, adipsin and beta-actin were 36, 38, 38, 26, 26, 38, 36, 28, 28, 20, 20 and 32, respectively. Experiments were repeated twice on independent RNA samples.
No significant changes in food consumption and glucose tolerance in adult Ptc1mes/mes mice
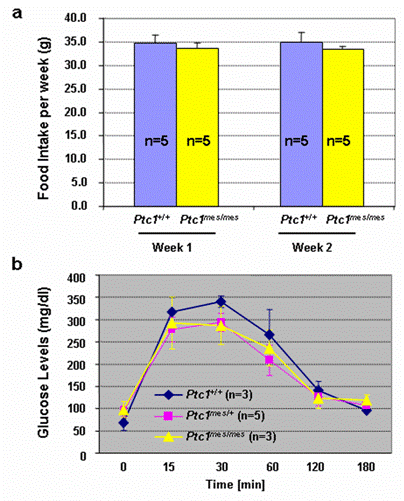
To determine if there is any difference in food consumption by Ptc1mes/mes and their control Ptc1+/+ mice, we performed food intake experiments. As shown in Figure 4a, there was no statistically significant change in food consumption in either adult Ptc1mes/mes or Ptc1+/+ mice during the consecutive two weeks. The average food intake by adult Ptc1mes/mes mice at each tested week was also not significantly different from Ptc1+/+ control mice (Figure 4a). To test if there is any abnormality in serum glucose concentrations in adult Ptc1mes/mes mice, glucose tolerance experiments were performed. As shown in Figure 4b, no statistical significance of difference in serum glucose concentrations among adult Ptc1+/+, Ptc1mes/+ and Ptc1mes/mes mice at each tested time point after the injections of high doses of glucose.
No significant change in (a) food intake and (b) glucose tolerance between adult Ptc1mes/mes and their control (Ptc1+/+ or Ptc1mes/+) mice. a. Food consumption by five adult wild type and Ptc1mes/mes mice at the age of 23-27 weeks was recorded weekly for consecutive two weeks. b. Overnight-fasted 24-26-week-old littermates were subjected to glucose tolerance experiments as described in Materials and Methods. Data are presented as mean ± SEM of the indicated numbers of mice in the bar graphs. Student's t-test was performed to determine the significant difference. P value was greater than 0.05 between Ptc1mes/mes and controls (Ptc1+/+ or Ptc1mes/+) at each time point in the figure.
Discussion
In our ongoing study to determine the role of Ptc during ovarian and uterine development in Ptcmes/mes mice, we observed that total white fat masses in adult female Ptcmes/mes mice were less than their wild type littermates (unpublished data). Since female Ptcmes/mes mice are being used to characterize their ovarian and uterine phenotypes, male Ptcmes/mes and their control littermates were used to investigate the potential defects in WAT during postnatal development in this report. We found that Ptc1 was expressed in adult WAT and that loss of the carboxyl-terminus of Ptc1 in adult mice caused a significant reduction in total white fat mass and adipocyte cell size (Figure 1-2). Reduced white fat mass observed in adult Ptc1mes/mes mice can be due to the reduced cell sizes of adipocyte cells (Figure 2) and the reduced adipocyte cell numbers in adult Ptc1mes/mes mice. The latter is indicated by the observation that the reduced levels of adipose cell sizes (40% of controls) are less than the reduced levels of total WAT weights in 33-week-old Ptc1mes/mes (about 75% of controls), even though it is difficult and almost impossible to determine the exact numbers of total adipocyte cells in adult mice. Since we did not observe significant changes in food intake and blood glucose concentrations in adult Ptc1mes/mes mice (Figure 4), the reduced white fat mass (reduced adipocyte cell size and cell numbers) is probably not due to reduced food consumption and aberrant glucose metabolism observed in Ptc1+/- mice [28] as well as other hormonal signaling such as insulin signaling during adipogenesis [29]. Rather, the reduced white fat mass is likely due to the defective Hh signaling during adipogenesis in Ptc1mes/mes mice during postnatal development. By RT-PCR analyses, we found that many Hh signaling molecules are expressed in epididymal WAT (Figure 1), which is consistent with the presence of Gli transcription factors in perigonadal fat pads reported previously [13]. Importantly, we found that truncated Ptc1, Ptc2 and Gli1, the Hh signaling downstream target genes, are overexpressed in epididymal WAT of Ptc1mes/mes mice (Figure 3b), indicating the ectopic activation of Hh signaling in WAT to regulate its downstream gene expression for fat formation. Consistent with reduced white fat mass in Ptc1mes/mes mice, we also observed the reduced expression of adipocyte marker genes aP2 and adipsin. These reduced expression of adipocyte marker genes likely results from the decreased expression of adipogenic genes PPARgamma and CEBPalpha. It has been reported that Shh can inhibit adipocyte differentiation in 3T3-L1 cells, C3H10T1/2 and primary mouse calvaria cells [10, 13]. Overexpression of Gli1 and reduced expression of adipocyte markers, aP2 and/or adipsin, and adipogenic transcription factors, PPARgamma and CEBPalpha, have been observed in Shh-treated 3T3-L1 or C3H10T1/2 cells [10, 13]. Moreover, reduced expression of Gli1 and Gli2 has been reported in ob/ob mice and normal mice fed with a high fat diet to induce obesity [13]. Collectively, our results are consistent with the recent findings from cultured cells [10, 13], indicating that Hh signaling plays a role in inhibiting WAT formation in adult mice.
Since we did not observe overexpression of Gilz, an antiadipogenic gene, which has been reported by Suh et al in cultured cells [13], it is likely that the action of Hh signaling in adult white fat formation is via the inhibition of adipogenic gene transcription, rather than the stimulation of antiadipogenic gene expression. Our gene expression results indicate that Gli1 and Gli2, but not Gli3, may be two major transcription factors involved in adult WAT formation. These Gli transcription factors may inhibit transcription of adipogenic genes PPARgamma and CEBPalpha in adipocyte cells, resulting in reduced WAT formation. Or, they could directly regulate adipocyte gene expression such as aP2 in WAT, given that Gli1 can inhibit aP2 promoter activity in C3H10T1/2 cells [10]. Future studies using molecular approaches such as gel-shift mobility and promoter reporter analyses will determine these possibilities. In addition, further molecular analyses of Hh signaling downstream genes in Hh agonist- or antagonist-treated 3T3-L1 cells over-expressing the amino region of Ptc1 (with a deletion of C-terminal residues of Ptc1) or mouse embryonic fibroblast cells isolated from Ptc1mes/mes embryos will likely provide more details about the mechanism about the action of C-terminal tail of Ptc1 during adipogenesis.
We observed that there was a gradual reduction of total WAT in Ptc1mes/mes males during postnatal development with no significant reduction of total WAT weights at the age of 5-6 weeks, modest reduction in 19-20-week-old mice and remarkable reduction in 33-week-old or older mice. This gradual reduction of WAT observed in our studies is contradictory to the previous finding of the increased adipose weights in Shh-treated mice [8]. One possible reason may be due to the differential expression levels of Hh target genes such as Ptc1, Ptc2 and Gli1 in white adipocyte cells in mice during postnatal development. Future gene expression analyses of Ptc1mes/mes mice at different developmental stages and Shh agonist-treated mice will be required to address this possibility.
Increased body weights have been observed in Ptc1 mutant embryos [21-23] and Shh-treated normal mice [8]. Inhibition of body weight gain has also been reported in mice injected with antibodies against Hh proteins [9]. Ectopic activation of Hh signaling in Ptc1mes/mes mice would lead to general stimulation of cell proliferation (other than adipocyte cells), resulting in an increase of body weight. However, we did not observe a significant increase of the body weight in Ptc1mes/mes mice during postnatal development, except those mice at the age of 19-20 weeks after birth (Figure 2). This phenomena is unlikely due to the reduction of food consumption as well as potential aberrant glucose metabolism displayed in Ptc1+/- mice [28], since food consumption and serum glucose levels in adult Ptc1mes/mes mice appears to be comparable with those of control littermates (Figure 4). Considering that the major organ weights including liver, heart, kidney, lung and spleen were not significantly different between Ptc1mes/mes and their control mice (Figure 2d), it is also unlikely the case that the general stimulation of cell proliferation by Hh signaling observed in embryos [22] plays a significant role in body weight gain during postnatal development. Instead, Hh signaling may be active only in a subset of target tissues such as WAT to elicit its biological function in adult Ptc1mes/mes mice. Interestingly, Ptc1mes/mes mice at the age of 33 weeks or older were not lighter than their control littermates, even though there was a significant reduction of total white fat mass (Figure 2). In light of no significant changes in major organ weights in Ptc1mes/mes mice, it is possible that hyperplasia in other tissues and/or cells other than WAT and tested organs exist in Ptc1mes/mes mice, which may offset the reduced WAT weights. In support of this, we did notice that Ptc1mes/mes mice at that age appear to be rounder and bigger than their controls (unpublished data). Considering that activation of Hh signaling can push adipocyte precursor cells toward an osteogenic fate at the expense of adipogenesis [10-13, 30], it is likely that Ptc1mes/mes mice will display aberrant bone formation, which will affect the body weight during postnatal development. Recently, epidermal hyperplasia has been observed in adult Ptc1mes/mes mice [18], which will likely be another factor to offset the reduced WAT weight. Nevertheless, it appears that white fat mass is not a major factor that directly contributes to the body weight gain during postnatal development.
In conclusion, we found that Ptc1 was expressed in the epididymal fat pad of adult mice. Reduced total white fat mass and white adipose cell size were observed in adult Ptc1mes/mes mice. Truncated Ptc1, Ptc2 and Gli1 were overexpressed in WAT of Ptc1mes/mes, suggesting ectopic activation of Hh signaling in WAT. Consistent with reduced WAT, expression of adipogenic genes, PPARgamma and CEBPalpha, and adipocyte marker genes, aP2 and adipsin, were reduced in WAT of adult Ptc1mes/mes mice. Our results suggest that deletion of the carboxyl terminal tail of Ptc1 can lead to a reduction of white fat mass during postnatal development.
Acknowledgements
We thank Dr. Dennis R. Warner for critical comments of this manuscript. This work was supported by National Institute of Health Center of Biomedical Research Excellence Grant # P20-RR/DE17702.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Briscoe J, Therond P. Hedgehog signaling: from the Drosophila cuticle to anti-cancer drugs. Dev Cell. 2005;8:143-51
2. Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059-87
3. Cohen MM Jr. The hedgehog signaling network. Am J Med Genet A. 2003;123:5-28
4. Lum L, Beachy PA. The Hedgehog response network: sensors, switches, and routers. Science. 2004;304:1755-9
5. Hooper JE, Scott MP. Communicating with Hedgehogs. Nat Rev Mol Cell Biol. 2005;6:306-17
6. Rosen ED. New drugs from fat bugs?. Cell Metab. 2006;3:1-2
7. Gesta S, Tseng YH, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell. 2007;131:242-56
8. Martin PL, Lane J, Pouliot L. et al. Increase in adipose and total body weight, but not in lean body mass, associated with subcutaneous administration of sonic hedgehog-Ig fusion protein to mice. Drug Development Research. 2002;57:107-14
9. Buhman KK, Wang LC, Tang Y. et al. Inhibition of Hedgehog signaling protects adult mice from diet-induced weight gain. J Nutr. 2004;134:2979-84
10. Spinella-Jaegle S, Rawadi G, Kawai S. et al. Sonic hedgehog increases the commitment of pluripotent mesenchymal cells into the osteoblastic lineage and abolishes adipocytic differentiation. J Cell Sci. 2001;114:2085-94
11. Zehentner BK, Leser U, Burtscher H. BMP-2 and sonic hedgehog have contrary effects on adipocyte-like differentiation of C3H10T1/2 cells. DNA Cell Biol. 2000;19:275-81
12. van der Horst G, Farih-Sips H, Lowik CW. et al. Hedgehog stimulates only osteoblastic differentiation of undifferentiated KS483 cells. Bone. 2003;33:899-910
13. Suh JM, Gao X, McKay J. et al. Hedgehog signaling plays a conserved role in inhibiting fat formation. Cell Metab. 2006;3:25-34
14. Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994;79:1147-56
15. Rosen ED, Walkey CJ, Puigserver P. et al. Transcriptional regulation of adipogenesis. Genes Dev. 2000;14:1293-307
16. Cao Z, Umek RM, McKnight SL. Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes Dev. 1991;5:1538-52
17. Stone DM, Hynes M, Armanini M. et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature. 1996;384:129-34
18. Nieuwenhuis E, Barnfield PC, Makino S. et al. Epidermal hyperplasia and expansion of the interfollicular stem cell compartment in mutant mice with a C-terminal truncation of Patched1. Dev Biol. 2007;308:547-60
19. Johnson RL, Milenkovic L, Scott MP. In vivo functions of the patched protein: requirement of the C terminus for target gene inactivation but not Hedgehog sequestration. Mol Cell. 2000;6:467-78
20. Lu X, Liu S, Kornberg TB. The C-terminal tail of the Hedgehog receptor Patched regulates both localization and turnover. Genes Dev. 2006;20:2539-51
21. Goodrich LV, Milenkovic L, Higgins KM. et al. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109-13
22. Makino S, Masuya H, Ishijima J. et al. A spontaneous mouse mutation, mesenchymal dysplasia (mes), is caused by a deletion of the most C-terminal cytoplasmic domain of patched (ptc). Dev Biol. 2001;239:95-106
23. Milenkovic L, Goodrich LV, Higgins KM. et al. Mouse patched1 controls body size determination and limb patterning. Development. 1999;126:4431-40
24. Sweet HO, Bronson RT, Donahue LR. et al. Mesenchymal dysplasia: a recessive mutation on chromosome 13 of the mouse. J Hered. 1996;87:87-95
25. Hunt CR, Ro JH, Dobson DE. et al. Adipocyte P2 gene: developmental expression and homology of 5'-flanking sequences among fat cell-specific genes. Proc Natl Acad Sci U S A. 1986;83:3786-90
26. Min HY, Spiegelman BM. Adipsin, the adipocyte serine protease: gene structure and control of expression by tumor necrosis factor. Nucleic Acids Res. 1986;14:8879-92
27. Shi X, Shi W, Li Q. et al. A glucocorticoid-induced leucine-zipper protein, GILZ, inhibits adipogenesis of mesenchymal cells. EMBO Rep. 2003;4:374-80
28. Hebrok M, Kim SK, St Jacques B. et al. Regulation of pancreas development by hedgehog signaling. Development. 2000;127:4905-13
29. Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. 2006;7:885-96
30. Wu X, Walker J, Zhang J. et al. Purmorphamine induces osteogenesis by activation of the hedgehog signaling pathway. Chem Biol. 2004;11:1229-38
Author contact
![]() Correspondence to: Dr. Zi-Jian Lan, Birth Defects Center, Department of Molecular, Cellular and Craniofacial Biology, University of Louisville Health Sciences Center, Dental Building Room 203B, 501 S. Preston Street, Louisville, KY 40202. Tel: 502-852-4669; Fax: 502-852-4702; Email: z0lan001louisville.edu
Correspondence to: Dr. Zi-Jian Lan, Birth Defects Center, Department of Molecular, Cellular and Craniofacial Biology, University of Louisville Health Sciences Center, Dental Building Room 203B, 501 S. Preston Street, Louisville, KY 40202. Tel: 502-852-4669; Fax: 502-852-4702; Email: z0lan001louisville.edu
Received 2008-1-9
Accepted 2008-1-25
Published 2008-1-26