ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
1. Introduction
2. MATERIALS AND METHODS
3. RESULTS
4. DISCUSSION
5. CONCLUSION
Supplementary Material
References

Int J Biol Sci 2008; 4(5):309-317. doi:10.7150/ijbs.4.309 This issue Cite
Research Paper
A combined computational-experimental analyses of selected metabolic enzymes in Pseudomonas species
Deepak Perumal1,2, Chu Sing Lim1,2, Vincent T.K. Chow3, Kishore R. Sakharkar2,4, Meena K. Sakharkar1,2 ![]()
1. Advanced Design and Modeling Lab, Nanyang Technological University, Singapore.
2. Biopharmaceutical Engineering Cluster, BioMedical Engineering Research Centre, Nanyang Technological University, Singapore.
3. Department of Microbiology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore.
4. OmicsVista, Singapore.
Abstract
Comparative genomic analysis has revolutionized our ability to predict the metabolic subsystems that occur in newly sequenced genomes, and to explore the functional roles of the set of genes within each subsystem. These computational predictions can considerably reduce the volume of experimental studies required to assess basic metabolic properties of multiple bacterial species. However, experimental validations are still required to resolve the apparent inconsistencies in the predictions by multiple resources. Here, we present combined computational-experimental analyses on eight completely sequenced Pseudomonas species. Comparative pathway analyses reveal that several pathways within the Pseudomonas species show high plasticity and versatility. Potential bypasses in 11 metabolic pathways were identified. We further confirmed the presence of the enzyme O-acetyl homoserine (thiol) lyase (EC: 2.5.1.49) in P. syringae pv. tomato that revealed inconsistent annotations in KEGG and in the recently published SYSTOMONAS database. These analyses connect and integrate systematic data generation, computational data interpretation, and experimental validation and represent a synergistic and powerful means for conducting biological research.
Keywords: Comparative microbial genomics, metabolic pathways, KEGG, Pseudomonas species, integrative biology, drug discovery.
1. Introduction
The abundance of information provided by completely sequenced genomes offers a starting point for new insights into the multi-level organization of organisms and their evolution [1]. With the availability of an ever increasing number of complete genome sequences and the development of specialized tools for genome comparison, the comparative approach is becoming an extremely powerful strategy for genome analysis [2, 3]. Comparative analysis of metabolic pathways in different genomes yields important information on their evolution, for discovering pharmacological targets and for biotechnological applications[4].
In recent years, several biochemical pathways and metabolic reaction networks have been elucidated and this large pool of biochemical information, together with data from proteomics, metabolomics and genomics have facilitated the design of experiments and the selection of targets for manipulation by metabolic engineers [5, 6]. A comprehensive inventory of the biochemical functions potentially performed by an organism is available in several metabolic databases [7-9]. Comparisons of the differential biochemical pathways by detecting the presence or absence of genes encoding the enzymes that catalyze the reactions, provide a detailed account of the biochemical capacity of the organisms under study. Comparative metabolic pathway analyses also reveal pathway defects, i.e. missing enzymes within the predicted pathways. Thus, comparing the metabolic pathways between different organisms permits the identification of alternative pathways in microorganisms, with the aim of understanding microbial physiology, for designing novel therapeutic products and for antimicrobial drug discovery [10]. Analyzing the role of critical genes or proteins in a metabolic pathway also contributes towards the understanding of cell function and metabolic reactions [11, 12]. Essential microbial enzymes that do not have human homologs may also serve as putative drug targets [13-15].
The genus Pseudomonas is one of the most diverse bacterial genera, being isolated from sources ranging from plants to contaminated soil and water to human clinical samples. Members of Pseudomonas include important plant pathogens as well as biotechnologically ecologically interesting species [16]. Recently, to advance the biology of pseudomonads important in infection and biotechnology, the SYSTOMONAS (SYSTems biology of pseudOMONAS) database was established to provide an integrated bioinformatics platform for a systems biology approach [17]. At least eight Pseudomonas genomes have been hitherto completely sequenced and functionally annotated, i.e. Pseudomonas aeruginosa PAO1, Pseudomonas putida KT2440, Pseudomonas syringae pv.tomato DC3000, Pseudomonas syringae pv.syringae B728a, Pseudomonas syringae pv.phaseolicola 1448A, Pseudomonas fluorescens Pf-5, Pseudomonas fluorescens PfO-1, and Pseudomonas entomophila L48 (Table 1). In this study, we compare the metabolic pathways of the eight Pseudomonas species in order to identify missing enzymes in the metabolic pathways. Though, computational predictions can considerably reduce the volume of experimental studies required to assess basic metabolic properties of multiple bacterial species, experimental validations are required to resolve the problem of infrequently apparent inconsistencies in the predictions by multiple resources. We further carried out polymerase chain reaction (PCR), dot blot hybridization and DNA sequencing for enzymes (selected based on strains available in our lab) to correlate our experimental data with the information in the KEGG and SYSTOMONAS databases. The results of our molecular experiments endorse the fact that complementary advances in computational biology and experimental technologies provide biologists with a markedly improved repertoire of research tools that can help us to better understand the complexities and conundrums of the biology of microorganisms.
Genome information for the completely sequenced Pseudomonas species. Strains (expts) implies the Pseudomonas strains used for experimental analyses.
| # | Genome | RefSeq | GenBank | Length (Mbp) | GC content | Proteins | Strains (expts) | Reference |
|---|---|---|---|---|---|---|---|---|
| 1 | Pseudomonas aeruginosa PAO1 | NC_002516 | AE004091 | 6.3 | 66.60% | 5568 | ATCC® 15692 | [27] |
| 2 | Pseudomonas entomophila L48 | NC_008027 | CT573326 | 5.9 | 64.20% | 5134 | ||
| 3 | Pseudomonas fluorescens Pf-5 | NC_004129 | CP000076 | 7.1 | 63.30% | 6138 | ||
| 4 | Pseudomonas fluorescens PfO-1 | NC_007492 | CP000094 | 6.4 | 60.50% | 5736 | ||
| 5 | Pseudomonas putida KT2440 | NC_002947 | AE015451 | 6.18 | 61.50% | 5350 | ATCC® 47054; rmo- , mod+ | [28] |
| 6 | Pseudomonas syringae | |||||||
| pv. phaseolicola 1448A | NC_005773 | CP000058 | 5.9 | 58.90% | 4984 | |||
| 7 | Pseudomonas syringae | |||||||
| pv. syringae B728a | NC_007005 | CP000075 | 6.09 | 59.20% | 5089 | |||
| 8 | Pseudomonas syringae pv. tomato str. DC3000 | NC_004578 | AE016853 | 6.4 | 58.40% | 5470 | ATCC® 10862; Derivative of NCPPB1106; Rifr | [29] |
2. MATERIALS AND METHODS
Detection of the presence or absence of enzymes in different Pseudomonas species
Metabolic pathway information was downloaded from KEGG (Kyoto Encyclopedia of Genes and Genomes), a computational representation of metabolic pathway systems [7]. The Enzyme commission (EC) numbers for the eight Pseudomonas species were extracted. Three Pseudomonas strains were selected based on their availability in our lab (Table 1). Genomic DNAs were prepared from P. aeruginosa PAO1, P. putida KT2440 and P. syringae pv. tomato DC3000 as described by Manning [18]. The enzymes showing differential presence or absence were validated by PCR, dot blot hybridization and DNA sequencing as described below.
Polymerase chain reaction (PCR)
The PCR mixture contained 0.2 mM each of dATP, dCTP, dGTP and dTTP, 10 pmol each of forward and reverse primer, 1× PCR buffer [50 mM KCl, 10 mM Tris- HCl (pH 8.3)], 2.5 mM MgCl2, 0.25 U Taq DNA polymerase, 50 ng of template DNA [19]. The reaction mixture was made up to 25 µl with water. Template chromosomal DNA was denatured at 95°C for 4 min, followed by 30 cycles of amplification each of denaturation at 95°C for 30 sec, annealing at 50-60°C (depending on the melting temperature of the primers listed in Table 2) for 30 sec, and extension at 72°C for 1 min followed by a final extension at 72°C for 1 min. Initially, to confirm the presence of genes encoding 6-phosphogluconate dehydrogenase (EC: 1.1.1.44), Xylulose kinase (EC: 2.7.1.17), O-acetyl homoserine (thiol) lyase (EC: 2.5.1.49) in the chromosomes of P. aeruginosa PAO1, P. putida KT2440 and P. syringae pv. tomato DC3000, DNA was amplified using specific primers (Table 2). The resultant PCR products were analyzed by agarose gel electrophoresis. Finally, hybridization analyses were performed using DIG-labeled probes.
The oligonucleotides used in this study for PCR analyses. F- Forward Primer, R- Reverese Primer.
| Oligonucleotide number | Sequence | Location/ Description |
|---|---|---|
| ppu44F | 5′-CTCGGTGCACTGGTACAAAA -3′ | P. putida KT2440 (Gene encoded for EC:1.1.1.44) nt 148-167 |
| ppu44R | 5′- GAAGTCCCGACGTCCAGATA -3′ | P. putida KT2440 (Gene encoded for EC:1.1.1.44) Complementary nt 342-362 |
| pst44F | 5′- TCCGACCGAAGACACCATT -3′ | P. syringae pv. tomato DC3000 (Gene encoded for EC:1.1.1.44) nt 213-231 |
| pst44R | 5′- AGCAGCCAGGATGAAACCA -3′ | P. syringae pv. tomato DC3000 (Gene encoded for EC:1.1.1.44) Complementary nt 718-737 |
| pae17F | 5′- TACCTCAACCACTGGCTCA -3′ | P. aeruginosa PAO1 (Gene encoded for EC: 2.7.1.17) nt 558-576 |
| pae17R | 5′- GAAGAACGGCAGCATCAG -3′ | P. aeruginosa PAO1 (Gene encoded for EC: 2.7.1.17) Complementary nt 982-1000 |
| pst17F | 5′- TCGGGCACTGGTTATTTC -3′ | P. syringae pv. tomato DC3000 (Gene encoded for EC: 2.7.1.17) nt 532-549 |
| pst17R | 5′- AGCATCAACACGCCTTCT -3′ | P. syringae pv. tomato DC3000 (Gene encoded for EC: 2.7.1.17) Complementary nt 995-1013 |
| pae49F | 5′- CGTGTTCTGCGAAACCAT -3′ | P. aeruginosa PAO1 (Gene encoded for EC: 2.5.1.49) nt 438-455 |
| pae49R | 5′- GATGAGGAAGGCGTTGAA -3′ | P. aeruginosa PAO1 (Gene encoded for EC: 2.5.1.49) Complementary nt 822-840 |
| ppu49F | 5′- AAGCGGTATTCTGCGAGT -3′ | P. putida KT2440 (Gene encoded for EC: 2.5.1.49) nt 434-451 |
| ppu49R | 5′- ATCAGGAAGGCGTTGAAC -3′ | P. putida KT2440 (Gene encoded for EC: 2.5.1.49) Complementary nt 821-839 |
Preparation of DNA probes and DNA hybridization
Isolated specific DNA fragments were labeled with Dioxygenin (DIG) using a DNA labeling kit (Roche Diagnostics, Germany). Approximately 10 ng to 3 µg of purified DNA fragments were heated at 95°C for 10 min and chilled immediately in ice. Two µl of hexanucleotide mixture, 2 µl of 10× dNTP labeling mixture and 1 µl of Klenow enzyme 1 U/µl were added to the DNA mixture. The reaction mixture was then incubated at 37°C overnight followed by heating at 65°C for 2 min or adding of 2 μl of 0.2 mM EDTA, pH 8.0 to stop the reaction. The efficiency of the labeled DNA was determined according to the manufacturer's instructions. The probe was prepared by denaturing 20 ml of hybridization solution [50% (v/v) formamide, 1% (w/v) SDS, 6× SSC, 1× Denhardt's solution, 100 µg/ml herring sperm DNA and 100 μg/ml heparin] containing 19 μl labeled DNA at 95°C for at least 10 min.
Dot blot hybridization
About 3-5 μl of chromosomal DNA was applied to the positively-charged nylon membrane (Hybond-N+, Amersham Pharmacia Biotech, UK). The transferred DNA was fixed to the membrane by placing on 3MM Whatman No.1 filter paper presoaked with 0.4 N NaOH for 20 min. The membranes were prehybridized with 20 ml of prehybridization solution at 42°C for at least 1 hr in HB-1000 hybridizer (UVP Laboratory, Upland, CA, USA). The prehybridization solution was decanted, the preheated hybridization solution containing the appropriate probe was added to the membrane, and then hybridized at 42°C overnight. The probe solution was then decanted and stored at –20°C for reuse. Immunodetection was performed according to the manufacturer's protocol (Roche Diagnostics), and results were interpreted from the color reaction.
DNA and protein sequence analyses
For DNA sequencing, DNA templates were purified using the Qiagen gel extraction kit (Roche, Valencia, USA). Approximately 100 ng of purified PCR product was sequenced in both directions [20] using the ABI PRISM dye terminator cycle sequencing kit (Perkin-Elmer). The DNA sequence data were analyzed using the DNAMAN program (Lynnon Biosoft®) and BLAST [21].
3. RESULTS
Missing enzymes and metabolic pathways
Our analysis revealed the differential presence or absence of genes in the eight genomes for 11 metabolic pathways (Table 3). Some of the missing enzymes involved in the 11 metabolic pathways were also reported as absent in the Pseudomonas Genome Database (PGD) [22].
“Missing” enzymes and their metabolic pathways in the completely sequenced eight Pseudomonas species. Comparing the metabolic pathways between eight Pseudomonas species revealed 11 pathways with missing enzymes. Alternative pathways or bypasses were found in these pathways to replace the metabolic reactions of these missing enzymes. KEGG (Kyoto Encyclopedia of Genes and Genomes) was used to extract the metabolic pathway information. Enzymes with their EC numbers are tabulated with symbols (+/-) showing the presence or absence of these enzymes in eight Pseudomonas species.
| Pathways and their enzymes | P.aeruginosa PAO1 | P.entomophila L48 | P.fluorescens Pf-5 | P.fluorescens PfO-1 | P.putida KT2440 | P.syringae pv. syringae B728a | P.syringae pv. phaseolicola 1448A | P.syringae pv. tomato DC3000 |
|---|---|---|---|---|---|---|---|---|
| pae | pen | pfl | pfo | ppu | psb | psp | pst | |
| Pentose phosphate pathway | ||||||||
| *6-phosphogluconate dehydrogenase (EC: 1.1.1.44) | - | + | + | + | + | + | + | + |
| Pentose & Glucuronate interconversions | ||||||||
| *Xylulose kinase (EC:2.7.1.17) | + | + | + | + | - | + | + | + |
| Fructose & Mannose metabolism | ||||||||
| 2,3 butanediol dehydrogenase (EC:1.1.1.14) | + | - | + | - | + | + | - | + |
| Amino sugars metabolism | ||||||||
| Probable UDP-N-acetyl glucosamine 2-epimerase (EC:5.1.3.14) | + | + | + | - | + | + | + | + |
| Nucleotide sugars metabolism | ||||||||
| #UDP-glucuronate 4-epimerase (EC:5.1.3.6) | - | - | - | - | - | - | - | - |
| Synthesis & degradation of ketone bodies | ||||||||
| Probable coA transferase, subunit A (EC:2.8.3.5) | + | + | + | + | + | + | - | + |
| Hydroxymethylglutaryl-coA synthase (EC:2.3.3.10) | + | + | + | + | + | + | - | + |
| Methionine metabolism | ||||||||
| Cystathionine gamma synthase (EC:2.5.1.48) | - | + | - | + | + | + | - | - |
| *O-acetyl homoserine (thiol) lyase (EC:2.5.1.49) | + | + | + | - | + | + | - | - |
| Arginine & Proline metabolism | ||||||||
| Arginase (EC:3.5.3.1) | - | + | - | + | - | + | + | + |
| Urea cycle & metabolism of amino groups | ||||||||
| Arginase (EC:3.5.3.1) | - | + | - | - | - | + | - | - |
| Riboflavin metabolism | ||||||||
| Isocitrate dehydrogenase kinase/phosphatase (EC:3.1.3.-) | + | + | + | + | + | - | + | - |
| Vitamin B6 metabolism | ||||||||
| D-erythrose 4-P dehydrogenase (EC:1.2.1.72) | + | - | + | + | + | + | + | + |
| Erythronate 4-P dehydrogenase (EC:1.1.1.290) | + | - | + | + | + | + | + | + |
* indicates the enzymes selected for PCR and dot blot hybridization analyses. Three Pseudomonas strains (Pseudomonas aeruginosa, Pseudomonas putida and Pseudomonas syringae pv. tomato) were selected based on their availability in our lab. Enzymes present in at least two of above three genomes were chosen for PCR and Dot blot experimental analyses.
# The enzyme UDP-glucuronate-4-epimerase (EC: 5.1.3.6) catalyzes the conversion of UDP-D glucuronate to UDP-D galacturonate which acts as precursor for the synthesis of pectins. None of the Pseudomonas species show the presence of this enzyme. Glycolysis serves as bypass for all these species producing the end product UDP-D galacturonate.
PCR and dot blot hybridization analyses
Among the 11 metabolic pathways described above, three pathways, i.e. pentose phosphate pathway, pentose and glucuronate interconversions and methionine metabolism were selected to confirm the presence or absence of enzymes in three genomes by PCR and dot-blot hybridization experiments (Table 1). The resultant 215-bp PCR product of the gene fragment encoding EC: 1.1.1.44 (ppu44) from P. putida KT2440, as well as the 525-bp PCR product (pst44) for P. syringae pv. tomato DC3000 were observed after gel electrophoresis. For the gene fragments encoding EC: 2.7.1.17, the resultant PCR products of 443-bp (pae17) and 482-bp (pst17) were amplified from P. aeruginosa PAO1 and P. syringae pv. tomato DC3000, respectively. For the gene fragments encoding EC: 2.5.1.49, the resultant PCR products of 403-bp (pae49) and 406-bp (ppu49) were amplified from P. aeruginosa PAO1 and P. putida KT2440, respectively (Figure 1). In order to confirm the presence or absence of genes encoding EC1.1.1.44 in the chromosomes of P. aeruginosa, P. putida and P. syringae pv. tomato, PCR was performed using ppu44 and pst44 primers. Similarly, PCR was also performed for the genes encoding EC 2.7.1.17 (using pae17 and pst17 primers) and EC 2.5.1.49 (using pae49 and ppu49 primers) (Figures 2 and 3).
PCR products of gene fragments of ppu44, pst44, pae17, pst17, pae49, ppu49 of P. aeruginosa PAO1, P. putida KT2440 and P. syringae pv. tomato DC3000. PCR products amplified from genomic DNA of P. aeruginosa, P. putida and P. syringae pv. tomato using the corresponding specific primers. The expected sizes of PCR products of gene fragments are 215-bp ppu44, 525-bp pst44, 443-bp pae17, 482-bp pst17, 403-bp pae49, 406-bp ppu49. The 100-bp DNA size markers are depicted on the left.
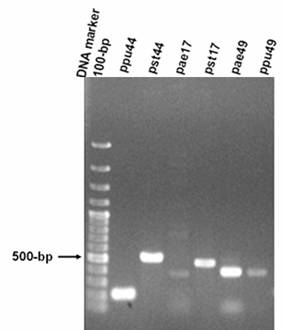
The PCR analyses showed that the gene encoding EC 1.1.1.44 was present in the chromosomes of P. putida, P. syringae pv. tomato and P. aeruginosa. The products of PCR using pae49 primers showed the presence of the gene encoding EC 2.5.1.49 in P. aeruginosa and P. syringae pv. tomato. The products of PCR using ppu49 primers demonstrated the presence of the gene only in P. putida. In addition, the gene encoding EC 2.7.1.17 was found to be present in P. aeruginosa and P. syringae pv. tomato by PCR (Figures 1-3).
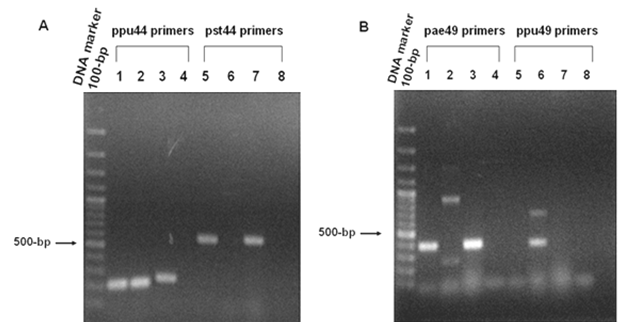
PCR analyses of (A) 6-phosphogluconate dehydrogenase (EC: 1.1.1.44) and (B) O-acetyl homoserine (thiol) lyase (EC: 2.5.1.49). Panel A. PCR products amplified from genomic DNA of P. aeruginosa PAO1, P. putida KT2440 and P. syringae pv. tomato DC3000 using ppu44 primers (Lanes 1-4) and pst44 primers (Lanes 5-8). The 215-bp PCR products of the ppu44 gene fragment were observed in lane 1 (P. aeruginosa), lane 2 (P. putida) and lane 3 (P. syringae pv. tomato). The 525-bp pst44 amplified fragments were seen in lane 5 (P. aeruginosa) and lane 7 (P. syringae pv. tomato). No bands were observed in lane 6 (P. putida), lanes 4 and 8 (negative control). Panel B. PCR products amplified from genomic DNA of P. aeruginosa PAO1, P. putida KT2440 and P. syringae pv. tomato DC3000 using pae49 primers (Lanes 1-4) and ppu49 primers (Lanes 5-8). The 403-bp PCR products of the pae49 gene fragment were observed in lane 1 (P. aeruginosa) and lane 3 (P. syringae pv. tomato). No bands were seen in lane 2 (P. putida) and lane 4 (negative control). The 406-bp ppu49 gene fragment was seen in lane 6 (P. putida). No bands were observed in lane 5 (P. aeruginosa), lane 7 (P. syringae pv. tomato) and lane 8 (negative control).
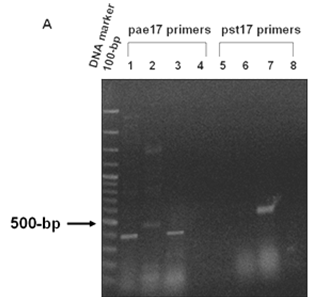
PCR analysis of (A) Xylulose kinase (EC: 2.7.1.7). Panel A. PCR products amplified from genomic DNA of P. aeruginosa PAO1, P. putida KT2440 and P. syringae pv. tomato DC3000 using pae17 primers (Lanes 1-4) and pst17 primers (Lanes 5-8). The 443-bp PCR products of the pae17 gene fragment were observed in lane 1 (P. aeruginosa) and lane 3 (P. syringae pv. tomato). Non-specific bands (~ 500 bp) were seen in lane 2 (P. putida). The 482-bp pst17 gene fragment was seen in lane 7 (P. syringae pv. tomato). No bands were observed in lane 5 (P. aeruginosa), 6 (P. putida), lanes 4 and 8 (negative controls).
Dot blot hybridization
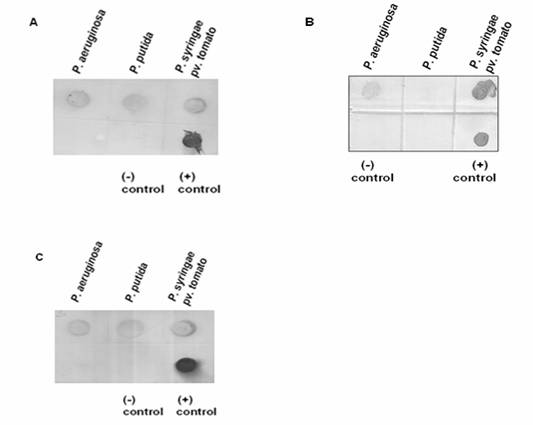
Hybridization analyses were performed using DIG-labeled probes, i.e. 215-bp ppu44, 482-bp pst17 and 403-bp pae49 (Figure 4). The strong hybridization signal with the 215-nucleotide ppu44 probe confirmed that the gene encoding EC 1.1.1.44 is present in P. putida, P. syringae pv. tomato as well as in P. aeruginosa (Figure 4A). This hybridization result supports the data obtained by PCR (Figure 2A). The positive hybridization signals with the 482-nucleotide pst17 probe supported the presence of the gene encoding EC 2.7.1.17 in P. aeruginosa and P. syringae pv. tomato, but showed its absence in P. putida (Figure 4B). The hybridization signals with the 403-nucleotide pae49 probe confirmed that the gene encoding EC 2.5.1.49 is present in P. aeruginosa, P. putida and P. syringae pv. tomato (Figure 4C), in agreement with the PCR analysis (Figure 2B). The data are summarized in Table 4.
Dot blot hybridization for (A) 6-phosphogluconate dehydrogenase (EC: 1.1.1.44), (B) Xylulose kinase (EC: 2.7.1.7) and (C) O-acetyl homoserine (thiol) lyase (EC: 2.5.1.49). Panel A. Presence or absence of gene encoding EC: 1.1.1.44. Chromosomal DNA samples extracted from P. aeruginosa, P. putida and P. syringae pv. tomato were applied onto a nylon membrane and hybridized with the DIG-labelled 215-bp ppu44 PCR product. The 215-bp ppu44 PCR product from P. putida served as positive control, whereas the 406-bp ppu49 PCR product from P. putida was included as negative control. Strong hybridization signals were detected for the chromosomal DNA of P. aeruginosa, P. putida and P. syringae pv. tomato. Panel B. Presence or absence of the gene encoding EC: 2.7.1.17. Chromosomal DNA samples of P. aeruginosa, P. putida and P. syringae pv. tomato were applied onto a nylon membrane and hybridized with the DIG-labelled 482-bp pst17 PCR product. The 482-bp pst17 PCR product from P. syringae pv. tomato served as positive control, whereas the 406-bp ppu49 PCR product from P. putida was included as negative control. Strong hybridization signals were observed for P. aeruginosa and P. syringae pv. tomato but not for P. putida. Panel C. Presence or absence of the gene encoding EC: 2.5.1.49. Chromosomal DNA samples of P. aeruginosa, P. putida and P. syringae pv. tomato were applied onto a nylon membrane and hybridized with the DIG-labelled 403-bp pae49 PCR product. The 403-bp pae49 PCR product from P. aeruginosa served as positive control, whereas the 482-bp pst17 PCR product from P. syringae pv. tomato was included as negative control. Strong hybridization signals were observed for P. aeruginosa, P. putida and P. syringae pv. tomato.
Results of the PCR and dot-blot hybridization analyses. (+/-) showing the presence or absence of enzymes. pae- Pseudomonas aeruginosa chromosomal DNA, ppu- Pseudomonas putida chromosomal DNA and pst- Pseudomonas syringae pv. tomato chromosomal DNA. NA- Information Not Available.
| Enzymes | Primer/Probe | pae | ppu | pst |
|---|---|---|---|---|
| 6-phosphogluconate dehydrogenase | KEGG database | - | + | + |
| (EC: 1.1.1.44) | SYSTOMONAS | + | + | + |
| PGD | - | + | NA | |
| ppu44 primers | + | + | + | |
| pst44 primers | + | - | + | |
| ppu44 probe | + | + | + | |
| Comment | Probably + | + | + | |
| Xylulose kinase | KEGG database | + | - | + |
| (EC: 2.7.1.17) | SYSTOMONAS | + | NA | + |
| PGD | + | NA | NA | |
| pae17 primers | + | - | + | |
| pst17 primers | - | - | + | |
| pst17 probe | + | - | + | |
| + | - | + | ||
| O-acetyl homoserine (thiol) lyase | KEGG database | + | + | - |
| (EC: 2.5.1.49) | SYSTOMONAS | + | + | NA |
| PGD | NA | NA | NA | |
| pae49 primers | + | - | + | |
| ppu49 primers | - | + | - | |
| pae49 probe | + | + | + | |
| Comment | + | + | Probably + |
DNA sequencing and BLAST analyses
The DNA sequences of the PCR products of P. aeruginosa and P. syringae pv. tomato (EC 1.1.1.44, EC 2.5.1.49) were searched against the genome database (http://www.ncbi.nlm.nih.gov/blast/). The nucleotide sequence alignments for EC 1.1.1.44 displayed 37.70% identity (P. aeruginosa sample sequence with P. syringae pv. tomato genome database sequence), 98.15% identity (P. syringae pv. tomato sample sequence with P. syringae pv. tomato genome database sequence) and 37.77 % identity ( sample sequence of P. aeruginosa with sample sequence of P. syringae pv. tomato determined experimentally)(data not shown). However, it is observed that there is a very high nucleotide sequence homology between pae44 sequence and the genomic sequences of three P. aeruginosa strains (PA01, UCBPP-PA14 and PA7) as well as Pseudomonas phage Pf1. On the basis of the gene prediction from the GenBank the obtained pae44 sequence could be a phage gene instead of 6-phosphogluconate dehydrogenase. Hence the sequencing result for this enzyme correlates with the KEGG database.
The nucleotide sequence alignments for the second enzyme O-acetyl homoserine (thiol) lyase EC 2.5.1.49 exhibited 95.01% identity (P. syringae pv. tomato sample sequence with P. aeruginosa genome database sequence), 97.64% identity (P. aeruginosa sample sequence with P. aeruginosa genome database sequence) and 96.34 % identity ( sample sequence of P. syringae pv. tomato with sample sequence of P. aeruginosa determined experimentally) (Supplementary Sequence Fig A). The protein translation for EC 2.5.1.49 showed higher percentage similarity from the nucleotide sequence alignments (95.01% identity between P. syringae pv. tomato sample sequence and P. aeruginosa genome database sequence, and 97.64% identity between P. aeruginosa sample sequence and P. aeruginosa genome database sequence). The resultant protein sequences were subject to BLAST. The P. syringae pv. tomato sample sequence displayed 99% identity with O-acetyl homoserine sulfhydrylase of the genome database, while the P. aeruginosa sample sequence also exhibited 99% identity with O-acetyl homoserine sulfhydrylase of the genome database. Hence, our results, both from the experimental and DNA sequencing confirm the presence of the enzyme O-acetyl homoserine (thiol) lyase in P. syringae pv. tomato.
4. DISCUSSION
Metabolic pathways and networks have been a longstanding focus of biochemical and physiological research. Through the comparison of metabolic pathways, intricate and complex relationships between genes that encode enzymes can be studied, and more sophisticated strategies for diagnosis and treatment of complex diseases may become feasible [23]. Concurrently, metabolic reconstruction encourages the application of comparative genomics to search for missing genes [24]. Most of the missing enzymes can be replaced by alternative reactions that utilize the same substrate or produce the specific product, thereby allowing a given pathway to operate.
Comparative metabolic pathway analyses reveal that several pathways in the Pseudomonas species show high plasticity and versatility. Our results confirm the presence of alternative enzymes and bypass processes for these missing enzymes. Computational predictions from multiple sources of information, though useful, may sometimes be inconsistent and further confound the issue of annotation of genes. In such cases, experimental validations may become essential to resolve and reconcile the apparent inconsistencies. In this report, the confirmation of the presence or absence of three enzymes by molecular techniques validated the presence of the enzyme O-acetyl homoserine (thiol) lyase (EC: 2.5.1.49) in P. syringae pv. tomato. The data for the two enzymes 6-phosphogluconate dehydrogenase (EC 1.1.1.44) and Xylulose kinase (EC 2.7.1.17) conflict with both the KEGG and SYSTOMONAS database whereas the data for the enzyme O-acetyl homoserine (thiol) lyase (EC: 2.5.1.49) are congruent with both KEGG and SYSTOMONAS database (Table 4).
Our results for the first time confirm the presence of DNA sequence encoding the enzyme O-acetyl homoserine (thiol) lyase (EC: 2.5.1.49) in P. syringae pv. tomato. It is also noteworthy that that the mere presence of a gene sequence even when confirmed by experimental procedures does not necessarily ensure that its corresponding enzyme is actually functional. This non-functionality may be attributed to the presence of non-active remnant genes, evolutionary pressures leading to loss of function, inactivating mutations, and the lack of transcription, translation, or post-translational processing [25]. Nonetheless, with the rapid availability of microbial genome sequences, coupled with functional data arising from numerous computational sources, it is essential to characterize the differential occurrence of genes in closely related genomes. Such a monumental effort can be accomplished largely by experimentalists who are able to make effective use of the guidance provided by comparative genome analyses.
5. CONCLUSION
Our experimental results confirmed the presence of the enzyme O-acetyl homoserine (thiol) lyase (EC: 2.5.1.49) in P. syringae pv. tomato. Continued experimental investigation of the metabolic biochemistry of an organism is of importance in (1) to assign pertinent biochemical reactions to the enzymes found in the genome; (2) to validate and scrutinize information already found in the genome; and (3) determination of the presence of reactions or pathways not indicated by current genomic data [26]. The level of metabolic pathway variation(s) in the biosphere remains to be determined.
Despite the relatively small number of carefully studied organisms, significant pathway variations have been observed both between distinct organisms and within organisms with similar life-styles. Our data highlights the importance of isoenzymes, alternative enzymes and alternative pathways/bypasses and built in functional redundancy for the purpose of target identification for novel therapeutic discovery. We concur with the view that erroneous functional assignments should be corrected and disagreements between predictions reconciled, but this is difficult given the size and archival nature of current databases. Computational approaches can help us sift through this mass and help us select cases for validations. We concur with BioCyc's view that there is a critical need to harness the expertise of many scientists to produce biological databases, which accurately reflect the depth and breadth of knowledge that the biomedical research community is producing.
Supplementary Material
Sequence Fig A for EC 2.5.1.49
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Baxevanis A.D. Using genomic databases for sequence-based biological discovery. Molecular Medicine. 2003;9:185-192
2. Binnewies T.T, Motro Y, Hallin P.F, Lund O, Dunn D, La T, Hampson D.J, Bellgard M, Wassenaar T.M, Ussery D.W. Ten years of bacterial genome sequencing: comparative-genomics-based discoveries. Funct Integr Genomics. 2006;6:165-185
3. Zhang R, Zhang C.T. The impact of comparative genomics on infectious disease research. Microbes Infect. 2006;8:1613-1622
4. Romero P, Wagg J, Green M.L, Kaiser D, Krummenacker M, Karp P.D. Computational prediction of human metabolic pathways from the complete human genome. Genome biology. 2005;6:R2
5. Cane D.E. BIOSYNTHETIC PATHWAYS:Biosynthesis Meets Bioinformatics. Science. 2000;287:818-819
6. Kern A, Tilley E, Hunter I.S, Legisa M, Glieder A. Engineering primary metabolic pathways of industrial micro-organisms. Journal of Biotechnology. 2007;129:6-29
7. Kanehisa M, Goto S, Kawashima S, Nakaya A. The KEGG databases at GenomeNet. Nucl Acids Res. 2002;30:42-46
8. Karp P.D, Krummenacker M, Paley S, Wagg J. Integrated pathway-genome databases and their role in drug discovery. Trends in Biotechnology. 1999;17:275-281
9. Karp P.D, Riley M, Paley S.M, Pellegrini-Toole A, Krummenacker M. EcoCyc: Encyclopedia of Escherichia coli genes and metabolism. Nucleic Acids Research. 1998;26:50-53
10. Nielsen J, Olsson L. An expanded role for microbial physiology in metabolic engineering and functional genomics: Moving towards systems biology1. FEMS Yeast Research. 2002;2:175-181
11. Nielsen J. Metabolic engineering: Techniques for analysis of targets for genetic manipulations. Biotechnology and Bioengineering. 1998;58:125-132
12. Nielsen J. Metabolic engineering. Applied Microbiology and Biotechnology. 2001;55:263-283
13. Galperin M.Y, Walker D.R, Koonin E.V. Analogous enzymes: Independent inventions in enzyme evolution. Genome Research. 1998;8:779-790
14. Perumal D, Lim C.S, Sakharkar KR, Sakharkar MK. Differential genome analyses of metabolic enzymes in Pseudomonas aeruginosa for drug target identification. In Silico Biology. 2007;07:453-465
15. Sakharkar K.R, Sakharkar M.K, Chow V.T.K. A novel genomics approach for the identification of drug targets in pathogens, with special reference to Pseudomonas aeruginosa. In Silico Biology. 2004;4:355-360
16. Anzai Y, Kim H, Park J.Y, Wakabayashi H, Oyaizu H. Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence. International Journal of Systematic and Evolutionary Microbiology. 2000;50:1563-1589
17. Choi C, Munch R, Leupold S, Klein J, Siegel I, Thielen B, Benkert B, Kucklick M, Schobert M, Barthelmes J, Ebeling C, Haddad I, Scheer M, Grote A, Hiller K, Bunk B, Schreiber K, Retter I, Schomburg D, Jahn D. SYSTOMONAS -- an integrated database for systems biology analysis of Pseudomonas. Nucl Acids Res. 2007;35:D533-537
18. Manning P.A, Heuzenroeder M.W, Yeadon J. Molecular cloning and expression in Escherichia coli K-12 of the O antigens of the Inaba and Ogawa serotypes of the Vibrio cholerae O1 lipopolysaccharides and their potential for vaccine development. Infection and Immunity. 1986;53:272-277
19. Delidow B.C, Peluso J.J, White B.A. Quantitative measurement of mRNAs by polymerase chain reaction. Genetic Analysis Techniques. 1989;6:120-124
20. Sanger F, Nicklen S, Coulson A.R. DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences of the United States of America. 1977;74:5463-5467
21. Altschul S.F, Gish W, Miller W, Myers E.W, Lipman D.J. Basic local alignment search tool. Journal of Molecular Biology. 1990;215:403-410
22. Winsor GL, Lo R, Ho Sui SJ, Ung KSE, Huang S, Cheng D, Ching WKH, Hancock REW, Brinkman FSL. Pseudomonas aeruginosa Genome Database and PseudoCAP: Facilitating community-based, continually updated, genome annotation. Nucleic Acids Research. 2005;33:D338-D343
23. Forst C.V, Schulten K. Phylogenetic analysis of metabolic pathways. Journal of Molecular Evolution. 2001;52:471-489
24. Osterman A, Overbeek R. Missing genes in metabolic pathways: A comparative genomics approach. Current Opinion in Chemical Biology. 2003;7:238-251
25. Cordwell S.J. Microbial genomes and 'missing' enzymes: Redefining biochemical pathways. Archives of Microbiology. 1999;172:269-279
26. Covert M.W, Schilling C.H, Famili I, Edwards J.S, Goryanin I.I, Selkov E, Palsson B.O. Metabolic modeling of microbial strains in silico. Trends in Biochemical Sciences. 2001;26:179-186
27. Cox C.D. Effect of pyochelin on the virulence of Pseudomonas aeruginosa. Infect Immun. 1982;36:17-23
28. Nieto C, Fernandez-Tresguerres E, Sanchez N, Vicente M, Diaz R. Cloning vectors, derived from a naturally occurring plasmid of Pseudomonas savastanoi, specifically tailored for genetic manipulations in Pseudomonas. Gene. 1990;87:145-149
29. Cuppels D.A. Generation and Characterization of Tn5 Insertion Mutations in Pseudomonas syringae pv. tomato. Appl Environ Microbiol. 1986;51:323-327
Author contact
![]() Correspondence to: Meena K. Sakharkar (Ph.D.), Assistant Professor, N3-2C-113B, Nanyang Technological University, Singapore. Tel: 67905836; Email: mmeenaedu.sg
Correspondence to: Meena K. Sakharkar (Ph.D.), Assistant Professor, N3-2C-113B, Nanyang Technological University, Singapore. Tel: 67905836; Email: mmeenaedu.sg
Received 2008-7-14
Accepted 2008-9-6
Published 2008-9-10