ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
Acknowledgements
Abbreviations
References

Int J Biol Sci 2009; 5(6):611-620. doi:10.7150/ijbs.5.611 This issue Cite
Research Paper
Nucleotide excision repair and recombination are engaged in repair of trans-4-hydroxy-2-nonenal adducts to DNA bases in Escherichia coli
Beata Janowska1, Marek Komisarski1, Paulina Prorok2, Beata Sokołowska3, Jarosław Kuśmierek1, Celina Janion1, Barbara Tudek1,2 ![]()
1. Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Pawińskiego 5a, 02-106 Warsaw, Poland;
2. Institute of Genetics and Biotechnology, Warsaw University, Pawińskiego 5a, 02-106 Warsaw, Poland;
3. Medical Research Center, Polish Academy of Sciences, Pawinskiego 5, 02-106 Warsaw, Poland
Abstract
One of the major products of lipid peroxidation is trans-4-hydroxy-2-nonenal (HNE). HNE forms highly mutagenic and genotoxic adducts to all DNA bases. Using M13 phage lacZ system, we studied the mutagenesis and repair of HNE treated phage DNA in E. coli wild-type or uvrA, recA, and mutL mutants. These studies revealed that: (i) nucleotide excision and recombination, but not mismatch repair, are engaged in repair of HNE adducts when present in phage DNA replicating in E. coli strains; (ii) in the single uvrA mutant, phage survival was drastically decreased while mutation frequency increased, and recombination events constituted 48 % of all mutations; (iii) in the single recA mutant, the survival and mutation frequency of HNE-modified M13 phage was slightly elevated in comparison to that in the wild-type bacteria. The majority of mutations in recA- strain were G:C → T:A transversions, occurring within the sequence which in recA+ strains underwent RecA-mediated recombination, and the entire sequence was deleted; (iv) in the double uvrA recA mutant, phage survival was the same as in the wild-type although the mutation frequency was higher than in the wild-type and recA single mutant, but lower than in the single uvrA mutant. The majority of mutations found in the latter strain were base substitutions, with G:C → A:T transitions prevailing. These transitions could have resulted from high reactivity of HNE with G and C, and induction of SOS-independent mutations.
Keywords: trans-4-hydroxy-2-nonenal, HNE-DNA adducts, mutations, recombination, NER, M13 phage
1. Introduction
The oxidation of polyunsaturated fatty acids is a free radical process initiated by reactive oxygen species such as the hydroxyl radical. Lipid peroxidation leads to the formation of a large family of alkenals, but the major product of the peroxidation of ω-6 polyunsaturated fatty acids is trans-4-hydroxy-2-nonenal (HNE) [1-5].
HNE occurs in tissues in the range of 0.1 - 3.0 µM, but under conditions of oxidative stress can increase up to 50 µM [4]. It is an important factor regulating cell metabolism as well as a contributor to several pathological processes. HNE is highly cytotoxic and induces apoptosis in neuronal cells. Neuronal apoptosis may occur in stroke [6-8] and in Alzheimer and Huntington diseases [9, 10]. Antioxidants that suppress lipid peroxidation protect against apoptosis induced by oxidative insult, but not that induced by HNE. Glutathione binds HNE and protects neurons against apoptosis induced by oxidative stress and HNE [4]. PC12 cells expressing Bcl2 exhibit elevated levels of glutathione and lowered levels of HNE under oxidative stress [11]. Livers of humans with Wilson's disease and hemochromatosis contain mutations at CpG sites in the coding regions of the p53 tumor suppressor gene. It has been suggested that these mutations may be caused by modification of deoxyguanosine with HNE [3]. Genotoxic effects of HNE were also observed as a consequence of normal cellular metabolism. It was shown that choline-deficient L-amino acid defined diet (CDAA) caused hepatocellular carcinoma (HCC) in rats [12]. Mitochondria isolated from livers of rats fed the CDAA diet produced significantly more H2O2 per NADH reducing equivalents oxidized and contained significantly higher 8-oxodG level in DNA than rats fed a normal diet [13]. In pre-neoplastic liver foci of rats fed the CDAA diet adducts to DNA of the HNE analog, 4-oxo-2-nonenal (ONE), were detected after only three days on this diet [14]. This suggests that the CDAA diet induces oxidative stress associated lipid peroxidation and causes modifications of DNA bases in the early phase of carcinogenesis. It was also shown that a deficiency of glucose 6-phosphate dehydrogenase (G6PD) in the mouse mutant may cause endogenous oxidative stress, which generates trans-4-hydroxy-2-nonenal and causes tissue mutagenesis [15]. The brains of G6PD-deficient males exhibit a considerable accumulation of promutagenic etheno-DNA adducts (13-fold increase in etheno-deoxyadenosine and 5-fold increase in etheno-deoxycytidine), and substantial elevation of somatic mutation rates (3-fold increase in lacZ mutant frequencies, predominated by illegitimate genetic recombinations of lacZ with mouse genomic sequences) [15].
So far, two types of mutagenic lesions arising by fatty acid oxidation have been described, bulky adducts to DNA bases, e.g., HNE-DNA adducts [16, 17], and relatively small etheno-adducts to G, A and C [3, 18]. HNE forms adducts to all four DNA bases, namely, cyclic propano- or etheno-adducts bearing a hexyl- or heptyl- side chain [17]. In the presence of atmospheric oxygen or lipooxygenase, HNE can be transformed into 2,3-epoxy-4-hydroxynonanal (EH), and this compound modifies guanine, adenine and possibly other DNA bases in nucleosides and nucleic acids much more efficiently than HNE [3]. The aldehyde group of propano-type HNE adduct to guanine, and most probably to the other DNA bases, is labile and in dsDNA may react with amino groups of nucleic acid bases and/or proteins, forming DNA-DNA and DNA-protein crosslinks [19].
In the model studies HNE is mutagenic, and induces a dose-dependent SOS response in S. typhimurium [20], HPRT mutations in V79, and sister chromatid exchanges in Chinese hamster ovary and in cultured mammalian cells [21, 22].
HNE-dG adducts in DNA were shown to be recognized by bacterial and mammalian nucleotide excision repair (NER) system [23]. However, the consequences of NER deficiency in bacteria and in mammalian cells have not been described extensively. The present paper attempts to remedy this situation.
2. Materials and Methods
2.1.Oligonucleotides and substrates
HNE was synthesized in the form of dimethylacetal derivative according to Chandra and Srivastava [24]. Prior to use, the dimethylacetal of HNE was hydrolyzed to aldehyde by incubation in 0.01 M HCl in a methanol-water 2:1 solution at 37 ºC for 1 h and neutralized with 1.5 M triethylammonium bicarbonate (TEAB) buffer pH 5.5. The BT3 primer used in this study for sequencing of the lacZα gene, 5'-TCG CCA TTC AGG CTG CG-3', was synthesized according to standard procedures using an Applied Biosystems synthesizer (Oligonucleotide Synthesis Laboratory, IBB PAS, Warsaw, Poland). The T7 Sequencing Kit (version 2) and [α-35S]-dATP were from Amersham-Pharmacia Biotech. (Uppsala, Sweden) or ICN (PerkinElmer Life Sci., Boston, MA, USA). dNTPs were from Sigma (St. Louis, MI, USA), X-gal from POCH (Gliwice, Poland) and IPTG from Promega (Madison, WI, USA).
2.2. Bacteria, phages and media
Characteristics of bacterial strains are given in Table 1. Media and plates used for bacterial growth and M13 transfectants were as described before [25].
Bacterial E. coli K12 strains used in this study.
| Strain | Genotype | Phenotype | Source or reference |
|---|---|---|---|
| JM105 | E.coli K12 (supE endA sbc B15 hsd R4 rpsL thi Δ(lac-proAB)/F'lacZ ΔM15) | wild-type for DNA repair | IBB PAS collection |
| BH1100 | as JM105 but recA::Cmr | Recombination and SOS deficient | R. Devoret collection |
| BH430 | as JM105 but uvrA::Tetr | NER deficient | S. Boiteux collection |
| BJ1 | as BH430 but recA::Cmr | NER, recombination and SOS deficient | IBB PAS collection |
| BJ2 | as JM105 but mutL::Kanr | MMR deficient | [Pl(NR 9559) →JM105 Kanr] [45] This work |
| BJ3 | as BH430 but mutL::Kanr | NER, MMR deficient | [Pl(NR 9559) → BH430 Kanr] [45] This work |
To construct new strains bacteria were routinely transformed with the P1vir phage [26].
M13 mp18 contains an operator, promoter and fragments of the N-terminal part of lacZ sequences (lacZα) encoding β-galactosidase. Its C-terminal part (with operator and promoter) encoding the 146 amino acid part of the lacZ gene (lacZβ) is situated on the F' episome of the derivatives of JM105 bacterial host strain. These fragments restore β-galactosidase activity by α-complementation; hence, by sequencing the M13mp18 phage we can detect the forward mutations in the lacZα fragment caused by point or frameshift mutations or by RecA-mediated recombination.
2.3. Modification of phage DNA
M13mp18 phage was grown overnight at 37 ºC in E. coli strain JM 105 (Table 1) in 2YT medium. The phage was collected and its DNA isolated according to Messing [27]. Double stranded M13 phage DNA was incubated with 20, 50 and 100 mM HNE at pH 5.5 for 48 h, at 37 ºC. Control, untreated, phage DNA was prepared in the same way, but HNE was substituted with water. For each HNE concentration, as well as for control DNA, 20 μg of phage DNA in a total volume of 100 μl was used. Subsequently, the DNA was precipitated with ethanol and resuspended in 100 μl of sterile water.
2.4. Preparation of competent cells and transformation
Bacteria were made competent by the CaCl2 method [28]. Transfection was performed according to Sambrook et al. [28]. Phage DNA (100 ng) was used to transfect 100 μl of competent cells. Transfection mixtures were plated on LB solid medium with 3 ml of LB soft agar supplemented with 0.4 mM IPTG and 0.5 mg/ml X-gal. Plates were incubated overnight at 37 ºC and plaques of phage infection centers were scored.
2.5. Collecting lacZα mutants and their sequencing
M13 lacZα mutants lacking α-complementation and exhibiting low or no β-galactosidase activity were recognized as colorless or light blue plaques on LB plates containing IPTG and X-gal. For each bacterial strain used, from 10 000 to 30 000 plaques were analyzed, and 30 - 70 individual mutant phages were isolated only from plates with 100 mM HNE. The frequency of mutations was determined by the ratio of colorless or light blue plaques to all plaques formed after transfection of phage DNA modified with a defined HNE concentration. DNA of mutated phages was isolated and sequenced using the dideoxynucleotide chain termination method [28, 29]. The T7 Sequencing Kit, [α35S]-dATP and a specific 19 nucleotide primer (BT3) annealing at base positions for amino acids 71-78 of the lacZ gene of the M13 "+" strand were used for DNA sequencing. To ensure that mutants were not derived from expansion of clones, eight independent HNE modifications of the phage DNA were performed. Only one transformation was done after each DNA modification, and transfection mixtures were plated onto 4 plates. Usually only one mutant was picked up from each plate (three at maximum).
2.6. Statistical Analysis
All data are presented as means ± SD. The STATISTICA (version 5.1) computer software (Stat Soft, Inc., Tulsa) was used for the statistical analysis. All parameters were tested for normal distribution with the tests of Shapiro-Wilk and Kolmogorov-Smirnov with Lillefors correction. The homogeneity of variances was evaluated by the Leven test. Differences among strains were evaluated using ANOVA. Statistical significance was considered at P < 0.05.
3. Results
3.1. Effect of host bacteria on survival and lacZα mutations induced by HNE-treatment of dsDNA of M13 phages
Modification of double stranded M13 phage DNA with HNE resulted in a decrease in phage survival and increase in mutation frequency of the lacZα gene of the M13 phage. As host bacterial strains for phage development we used: JM105 wild-type in DNA repair; BH430 (uvrA), lacking nucleotide excision repair (NER); BH1100 (recA), lacking RecA-recombinational and RecA* coprotease activities; and BJ1 (uvrA recA), lacking both RecA-recombinational and NER mediated repair.
Growth and mutagenesis of HNE-treated M13 mp18 phage differed in different bacterial hosts (Figs. 1, 2; Tables 2, 3). The lowest survival and the highest mutation frequency in the lacZα gene of M13 phage were observed in the BH430 strain deficient in the NER system (Fig. 1). Mutation frequency increased 10-fold, at survival rate of 8.3% when phage DNA was modified with 100 mM HNE. In comparison, in the wild-type E. coli JM105 strain, after the same modification of phage DNA, the mutation frequency increased 3.2-fold, at survival rate of 27.5%. The differences in mutation frequency and survival between the wild-type and NER-deficient strains were statistically significant (P=0.0002). These results confirm the participation of NER in repair of HNE-DNA adducts in bacteria and mammals, as suggested by Feng et al. and Hu et al. [16, 23]. The predominant increase in mutations in uvrA-defective BH430 host-strain was due to recombinantional events, formed by simultaneous deletions of 93- (ΔM15 of F'lacZ) and 54- nucleotides of polylinker fragments, as described in the legend to Fig.3. They constituted 48 % of all mutants, while in the wild-type and recA- strain only 10 % of recombination events were found, and no recombinational repair was found in the double JM105 recA-uvrA- strain (Table 2). Our previous studies on the mutagenic activity of HNE-treated M13mp18 ssDNA showed that HNE treatment inhibits replication and stimulates phage recombination with homologous lacZ gene sequences localized in the F' factor in bacteria [17]. Here we observed that the deficiency of functional RecA protein did not decrease phage survival. In contrast, better survival (P=0.002 at 100 mM HNE), and increased lacZα mutation rate (P=0.03 at 100 mM HNE) of HNE-modified M13 phage was observed in recA- E. coli in comparison to the wild-type bacteria (Tables 2 and 3 and Fig. 1).
Types and frequency (x10-5) of lacZα mutations induced by HNE treatment of dsDNA of M13mp18 phage and allowing it to replicate in E. coli wild-type and mutants defective in NER and/or recA-dependent recombination.
| Type of mutation | Spontaneous mutations | HNE-induced mutations | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| JM105 (wild-type) | JM105 (wild-type) | BH1100 (recA) | BH430 (uvrA) | BJ1 (uvrArecA) | ||||||
| mutation frequency x10-5 | % | mutation frequency x10-5 | % | mutation frequency x10-5 | % | Mutation frequency x10-5 | % | mutation frequency x10-5 | % | |
| Base substitutions | 33.3 | 61.3 | 108 | 80 | 164.6 | 84.2 | 217.2 | 48 | 288 | 94.7 |
| Frameshifts | ||||||||||
| - insertions | nd* | 9.7 | 6.75 | 10 | nd | 5.3 | 18.8 | 4 | 16 | 5.3 |
| - deletions | 6 | 6.75 | 10.3 | nd | nd | |||||
| Recombination deletion of 93+54 nucleotides | 3.5 | 6.5 | 13.5 | 10 | 20.6 | 10.5 | 217.2 | 48 | nd | nd |
| Other deletions | 12 | 22.5 | nd | nd | nd | nd | nd | nd | nd | nd |
| Total | 54.8 | 100 | 135 | 100 | 195.5 | 100 | 453.2 | 100 | 304 | 100 |
*nd - not detected
Types and frequency (x10-5) of lacZα base substitutions mutations induced by HNE treatment of dsDNA of M13mp18 phage and allowing it to replicate in E. coli wild-type and mutants defective in NER and/or recA-dependent recombination.
| Type of mutation | Spontaneous mutations | HNE-induced mutations | |||
|---|---|---|---|---|---|
| JM105 (wild-type) | JM105 (wild-type) | BH1100 (recA) | BH430 (uvrA) | BJ1 (uvrArecA) | |
| A:T→G:C | 3.5 | 6.75 | nd* | 18.1 | 16 |
| A:T→C:G | 2 | 6.75 | nd | nd | nd |
| A:T→T:A | 2 | nd | nd | 18.1 | nd |
| G:C→T:A | 5 | 20.25 | 82.3 | 36.2 | 32 |
| G:C→C:G | 5 | nd | nd | 18.1 | 16 |
| G:C→A:T | 15.8 | 74.25 | 82.3 | 126.7 | 224 |
| Total | 33.3 | 108 | 164.6 | 217.2 | 288 |
*nd - not detected
Survival and mutation frequency in the lacZ gene of M13mp18 phage DNA modified with HNE and transfected into wild-type E. coli and its uvrA and recA mutants.
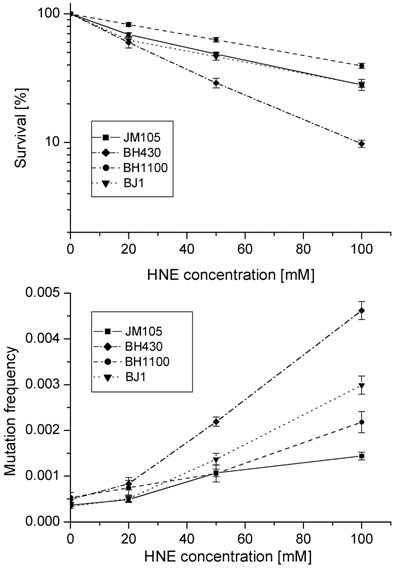
In the double uvrA-recA- defective strain the efficiency of infective centers formation by HNE-modified phage DNA was the same as in the wild-type bacteria; however, the total mutation rate (including base substitutions, frameshifts and recombinations) was higher than in the wild-type strain (P=0.0005 at 100 mM HNE), but still lower than in the uvrA- single mutant (P=0.0003 at 100 mM HNE).
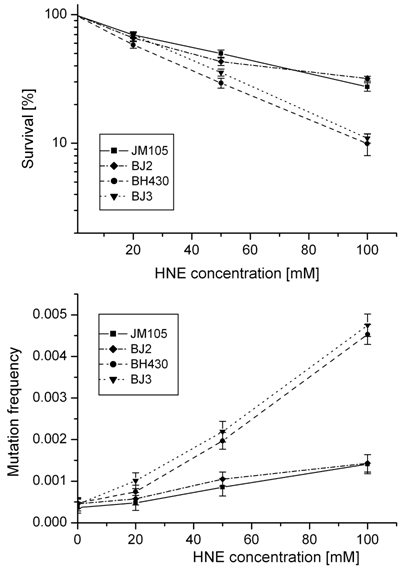
In order to obtain insight into the possible role of mismatch repair system in avoiding the deleterious effects of HNE-DNA adducts, we verified propagation and mutagenesis of HNE modified phage in the mutL- strain. No differences in survival or mutation rate were found between M13 phage transfected into E. coli wild-type or its mutL- derivative, or into E. coli uvrA- or uvrA- mutL- strains (Fig. 2).
Survival and mutation frequency of M13mp18 phage DNA modified with HNE and transfected into wild-type and mutL E. coli and its uvrA and uvrA mutL mutants.
3.2. Spectrum of lacZα gene mutations induced by HNE-treatment of M13mp18 dsDNA
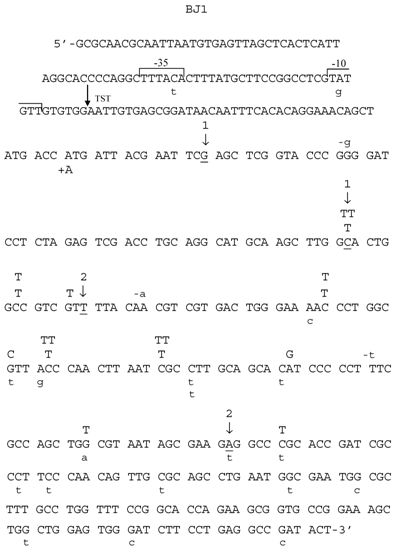
Mutants in lacZα gene obtained after treatment of M13 phage DNA with 100 mM HNE were sequenced, and the results of these analyses are summarized in Tables 2 and 3 and in Figs. 3-6. In HNE-treated M13 phage DNA propagated in wild-type JM105 strain, lacZα mutants arose mainly by base substitutions (80 %), in which G:C → A:T transitions prevailed (69 % of total base substitutions), with 10 % due to frameshifts (-G deletions or +T insertions), and to 10 % recombination events. G:C → A:T transitions were detected as C → T mutations, of which almost all were focused in two mutational hotspots (Fig. 4). The C → T transitions also prevailed in the spectrum of spontaneous mutations, but they were mapped at different loci of the lacZα gene than the HNE-induced mutations, and their efficiency increased 5.4-fold.
Spectrum of HNE-induced point mutations in dsM13mp18 lacZ DNA transfected into the E. coli JM105 strain. The 5'→3' DNA sequence of the lacZ fragment of M13mp18 is shown, from the first nucleotide after lacI termination codon through the coding sequence for aminoacid 65 of the gene. The -10 and -35 promoters, as well as transcription start site (TST) are marked over the sequence. Base substitution mutations are marked in capital letters above the sequence, and frameshifts below, with indication of a subtraction (“-“) or addition (“+”) event. Spontaneous base substitutions are superimposed on the spectrum, and are marked below the sequence in a lower case, while spontaneous frameshift mutations are marked above the sequence, also in a lower case. (1) First and last nucleotide of the 54-nucleotide deletion of the polylinker region; (2) first and last nucleotide of the 93-nucleotide M15 deletion.

Deficiency in DNA repair activity of bacterial hosts remarkably increased the rate of HNE-induced mutations, and completely changed their mutational spectrum.
While in HNE-treated M13 phages grown in JM105 wild-type strain G:C → A:T transitions were more abundant than G:C → T:A transversions, in phages grown in BH1100 recA- deficient strain, the number of G:C → T:A and G:C → A:T mutations was equal (Table 3). Moreover, almost all (7 of 8) G:C → T:A transversions detected as G → T mutations, when phages were grown in recA-defective strain, were mapped at codon AGC situated inside the 94 nucleotide ΔM15 fragment, which in the recA-positive cells undergoes deletion by recombination (Fig.5).
Interestingly, in uvrA- recA- bacterial strain in which HNE-treated phage DNA survival was the same as in the wild-type strain, G:C → A:T substitutions still dominated (70% of all mutations), but no hotspot in G → T transversion was observed; moreover the total mutation frequency was decreased in comparison to uvrA single mutant (Table 3, and Figs. 1, 4, 5). A wider spectrum could be observed in uvrA- recA- bacterial host; additionally, the following mutations were found: G:C → T:A, G:C →C:G, A:T → G:C, and frameshift (+A).
Spectrum of HNE-induced point mutations in dsM13mp18 lacZ DNA transfected into the E.coli JM105 recA- strain. Explanations as in Fig. 3. Underlined triplet CGT is a hotspot for G → T transversions in HNE-modified ssDNA of M13mp18 phage [17].

Spectrum of HNE-induced point mutations in dsM13mp18 lacZ DNA transfected into the E. coli JM105 uvrA- strain. Explanations as in Fig. 3.

Spectrum of HNE-induced point mutations in dsM13mp18 lacZ DNA transfected into the E. coli JM105 recA-uvrA- strain. Explanations as in Fig. 3.
4. Discussion
It was previously shown that HNE, one of the major products of fatty acid peroxidation, and 2,3-epoxy-4-hydroxynonanal (EH) are strongly genotoxic for bacterial and mammalian cells [17, 30]. In vitro at pH 7.2, HNE forms adducts to all four deoxynucleosides with the following reactivity: dG ≥ dC > dA >> dT [17]. Upon an oxygen shock HNE may be further oxidized to EH, which reacts with DNA bases more readily than HNE to form mutagenic etheno-G, etheno-C and etheno-A derivatives with long alkyl side chains [2]. In our study we were unable to measure the level of HNE-DNA adducts in M13 DNA. However, a brief estimation of adduct number can be proposed, based on literature data. On average, 1.2 HNE-dG adducts per 1000 bp were quantified in plasmid DNA modified in vitro with 192 mM HNE for 20 h [31]. Thus, at 100 mM HNE approximately 0.6 HNE-dG adducts/1000 bp, which equals to 3.6 per 6 kb M13 phage DNA, may be expected in our study. The same number of HNE-dC adducts is probably formed since in free nucleosides the efficiency of HNE adduction to dG and dC is almost equal, while A and T are at least an order of magnitude less reactive [17].
Lipid peroxidation results also in the formation of mutagenic etheno-adducts to G, C and A without alkyl side chain [30]. These latter lesions are removed by base excision repair (BER) [32] and by AlkB oxidoreductase that is expressed in E. coli as a part of the Ada-system [33]. AlkB homologues exist also in human cells [34].
Many of the lesions in DNA (e.g., bulky adducts, like HNE-DNA adducts, cyclic pyrimidine dimers, pyrimidine-pyrimidone 6-4-photoproducts, 3meA, apurinic sites, and others) arrest DNA synthesis, and require induction of the SOS system to recover replication and induce mutations. In spite of the different types of DNA modifications, induction of the SOS response follows a similar scheme: (i) modified DNA arrests DNA replication and induces formation of ss-DNA; (ii) RecA protein forms with ssDNA a ssDNA/RecA filament, which acquires Rec* coprotease activity that helps LexA protein (SOS suppressor) to undergo autocleavage and more than 50 SOS-regulated proteins to be expressed. Among them are mutagenic DNA polymerases, PolII, PolIV or PolV. All these DNA polymerases may incorporate nucleotides by translesion synthesis (TLS) past the damaged base and induce mutations. Selection of DNA polymerase in an error-prone process depends on the kind of lesion. PolV (UmuD'2C) is the most error prone and the most frequently used [35]. Error-prone DNA polymerases and a similar mechanism of mutagenesis exist also in mammalian cells and constitute the DNA damage response [36].
HNE (at physiological concentrations) is a potent inducer of the SOS response [20]. Moreover, transfection of M13 DNA, in which replication of DNA is arrested, most probably induces the SOS response [37]. However, in this study we observed independence of HNE-induced base substitution mutations of RecA protein expression, since an increased level of mutations was found also in recA- bacterial hosts. Examples of this are hot-spotted HNE-induced G:C → T:A mutations which occur only in HNE-modified phages propagated in bacteria deficient in recA, as well as the high frequency of G:C → A:T transitions in uvrA- recA- double mutant.
In comparison to spontaneous mutation rate, HNE-treatment of M13 phage DNA increased G:C → A:T transitions frequency from 5.4-fold, when the phage was propagated in the wild-type JM105 strain, to 16-fold, when HNE-damaged phage was propagated in the uvrA- recA- mutant. In recA- defective bacteria the frequency of HNE-induced G:C → A:T transitions was similar to that in the wild-type strain, while in the uvrA- strain the frequency of these mutations increased 9-fold in comparison to the rate of spontaneous mutations. This suggests that the HNE-cytosine adduct may have a strong miscoding potential, and mutations may arise in an SOS-independent manner. Since we used dsDNA of M13 phage for HNE modification and transformation, C → T mutations detected in the spectrum might derive from HNE adduction to C or to G. However, comparison of HNE mutational spectra in dsDNA (Table 3) to those obtained previously for ssDNA of M13 phage [17] shows almost identical specificity of mutations; this strongly suggests that cytosine-HNE adduct(s) is a potent error-prone DNA lesion, inducing C → T transitions [38]. In addition, our previous study [17] as well as literature data suggest that the major mutation induced by HNE-dG adduct(s) is a G → T transversion [38], and G → A transitions are formed to a much lesser extent.
The fact that the frequency of HNE-induced G:C → A:T mutations, detected as C → T transitions, increased twice in the uvrA- strain and three times in the uvrA-recA- double mutant in comparison to that in the wild-type strain suggests that these adducts may be repaired either by NER or by recombination. In the absence of active RecA and NER proteins, HNE-dC adducts may probably be bypassed by replicative DNA polymerase more easily than other HNE DNA adducts in an error-prone mode. Our previous studies suggest that reaction of HNE with dC yields four different products, two containing cyclic rings and two linear ones [17]. In contrast, HNE-dG adducts remain mainly in the form of exocyclic rings in deoxynucleosides and ssDNA. Linear adducts are more flexible than exocyclic rings, and replicative DNA polymerase or DNA polymerase IV present in SOS-uninduced cells [35, 39] might more easily incorporate mismatched nucleotides opposite the lesion, and induce mutations. It is also possible that the bypass of HNE-dC or HNE-dG adducts by SOS DNA polymerases (pol II and pol IV) present in SOS uninduced cells is either error-prone or error-free, and this might explain the better survival and the lower mutation rate of phage DNA in uvrA- recA- bacterial host than in the single uvrA- mutant (Fig. 1).
In recA- E. coli strain, a 16-fold increase of G:C → T:A (detected as G → T in the spectrum, Fig. 4) mutations in comparison to spontaneous mutation frequency was also observed. Comparing the present result with our previous studies performed with ssDNA [17], we observed that this increase was particularly large when the treated DNA was single-stranded (a 73-fold increase), and was not repaired by the NER system, suggesting that the G:C →T:A mutation locus is specifically susceptible to HNE attack. These mutations might arise by a similar mechanism as G:C → A:T transitions (C → T in the spectrum, Figs. 5, 6). Exocyclic ring of HNE-dG adduct opens easily in dsDNA, which creates linear adducts, that subsequently form internal hemiacetals [40]. The contribution of replicative DNA polymerase as well as pol IV and/or Pol II to bypassing such lesions is highly probable.
G → T hotspot in the spectrum occurs in recA-deficient, but not in wild-type bacteria, or in uvrA and recA deficient strains. We hypothesize that modified G triggers DNA recombination, and when recombination is blocked, induces G:C → T:A transversions. This is supported by the fact that in the uvrA deficient strain 48 % of all mutations are recombination events (Table 2). It is worth to note that when HNE treated dsDNA is grown in E. coli recA- strain, the hotspot sequence for G → T mutation in the spectrum, is 5'-G*CTGGCGT-3' (see Fig. 4). This is very similar to chi (χ), a hotspot recombination sequence in E. coli: 5'-GCTGGTGG-3', differing from it in only two nucleotides [41]. It is worth to recall that G → T transversions are frequent mutations at codon 249 (AGG*) of the TP53 gene in human cells [42].
In summary, in this work we established the following: (i) Simultaneous deficiency of uvrA and recA in bacterial host diminishes forward mutations in the lacZα gene, and increases survival of HNE-treated dsDNA M13mp18 phages in comparison to phages propagated in a single uvrA mutant. (ii) By analysis of mutation spectra, we confirmed that HNE-DNA adducts are removed by recA-mediated recombination and uvrA-dependent NER repair. (iii) The most frequent type of mutations in HNE-treated DNA of phages grown in wild-type bacteria were G:C → A:T transitions, whereas in DNA of phages grown in recA- defective bacteria, yields of G:C → T:A mutations greatly increased, and were located in hotspots. (iv) These hotspots differed for dsDNA and ssDNA. In HNE treated dsDNA they occurred in AGC codon, while in ssDNA, in codon CGT, situated two codons downstream (Fig.4). Both loci are situated in the region which is deleted during recombinational repair. (v) HNE-treated DNA is not repaired by dam-directed mismatch repair system of E. coli. Survival and mutagenesis of phages DNA in transformants after treatment of DNA with various concentrations of HNE were nearly the same whether that DNA replicated in E. coli wild-type, mutL or mutL recA mutants. Surprisingly, when damaged phage was grown in the double uvrA- recA- mutant strain of E. coli as a host, G:C → A:T transitions prevailed. This may suggest that an important fraction of HNE-induced point mutations is formed in an SOS-independent manner.
It was noted previously that the defect in UvrA protein decreases the induction of the SOS response by mitomycin C in E. coli recA+ cells [43], most probably by decreasing the formation of ssDNA/RecA protein filaments. On the other hand, overproduction of UvrA protein can ameliorate the ultraviolet sensitivity in E. coli recA- strain [44], which suggests that the UvrA and RecA proteins are competing with each other. However, an exact explanation of how the lack of UvrA and RecA proteins decreases mutations at HNE modified G in DNA and helps in survival of bacteria requires further studies.
Acknowledgements
This work was supported by the grant of the Polish Ministry of Science and Higher Education N303 328834.
Abbreviations
8-oxodG: 8-oxo-deoxyguanosine; CDAA: choline-deficient and L-aminoacid- defined diet; EH: 2,3-epoxy-4-hydroxynonanal; HNE: trans-4-hydroxy-2-nonenal; IPTG: isopropyl-β-D-thiogalactopyranoside; LPO: lipid peroxidation; X-gal: 5-bromo-4-chloro-3-indolyl-β-D-galactoside; ONE: 4-oxo-2-nonenal; G6PD: glucose-6-phosphate dehydrogenase.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Marnett LJ. Oxyradicals and DNA damage. Carcinogenesis. 2000;21:361-370
2. Chung FL, Zhang L. Deoxyguanosine adducts of tert-4-hydroxy-2-nonenal as markers of endogenous DNA lesions. Methods in enzymology. 2002;353:523-536
3. Chung FL, Pan J, Choudhury S, Roy R, Hu W, Tang MS. Formation of trans-4-hydroxy-2-nonenal- and other enal-derived cyclic DNA adducts from omega-3 and omega-6 polyunsaturated fatty acids and their roles in DNA repair and human p53 gene mutation. Mutation research. 2003;531:25-36
4. Poli G, Schaur RJ, Siems WG, Leonarduzzi G. 4-hydroxynonenal: a membrane lipid oxidation product of medicinal interest. Medicinal research reviews. 2008;28:569-631
5. Williams GM, Caldwell J, Armstrong D, Bartsch H, Bevan R, Browne RW, Chipman JK, Iatropoulos MJ, Jeffrey AM, Lunec J, Nair J, Page DL, Reeves BC, Richardson A, Silverstein B, Williams DF. Multicenter study to assess potential hazards from exposure to lipid peroxidation products in soya bean oil from Trilucent breast implants. Regul Toxicol Pharmacol. 2009;53:107-120
6. Linnik MD, Zobrist RH, Hatfield MD. Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke, a journal of cerebral circulation. 1993;24:2002-2008
7. Boortz-Marx RL, Clark HB, Taylor S, Wesa KM, Anderson DC. Sneddon's syndrome with granulomatous leptomeningeal infiltration. Stroke, a journal of cerebral circulation. 1995;26:492-495
8. Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G, Mattson MP. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J Neurosci. 1997;17:5089-5100
9. Lovell MA, Ehmann WD, Mattson MP, Markesbery WR. Elevated 4-hydroxynonenal in ventricular fluid in Alzheimer's disease. Neurobiology of aging. 1997;18:457-461
10. Portera-Cailliau C, Hedreen JC, Price DL, Koliatsos VE. Evidence for apoptotic cell death in Huntington disease and excitotoxic animal models. J Neurosci. 1995;15:3775-3787
11. Gallagher EP, Gardner JL, Barber DS. Several glutathione S-transferase isozymes that protect against oxidative injury are expressed in human liver mitochondria. Biochemical pharmacology. 2006;71:1619-1628
12. Nakae D. Endogenous liver carcinogenesis in the rat. Pathology international. 1999;49:1028-1042
13. Floyd RA, Kotake Y, Hensley K, Nakae D, Konishi Y. Reactive oxygen species in choline deficiency induced carcinogenesis and nitrone inhibition. Molecular and cellular biochemistry. 2002;234-235:195-203
14. Kawai Y, Kato Y, Nakae D, Kusuoka O, Konishi Y, Uchida K, Osawa T. Immunohistochemical detection of a substituted 1,N(2)-ethenodeoxyguanosine adduct by omega-6 polyunsaturated fatty acid hydroperoxides in the liver of rats fed a choline-deficient, L-amino acid-defined diet. Carcinogenesis. 2002;23:485-489
15. Felix K, Rockwood LD, Pretsch W, Nair J, Bartsch H, Bornkamm GW, Janz S. Moderate G6PD deficiency increases mutation rates in the brain of mice. Free radical biology & medicine. 2002;32:663-673
16. Feng Z, Hu W, Amin S, Tang MS. Mutational spectrum and genotoxicity of the major lipid peroxidation product, trans-4-hydroxy-2-nonenal, induced DNA adducts in nucleotide excision repair-proficient and -deficient human cells. Biochemistry. 2003;42:7848-7854
17. Kowalczyk P, Ciesla JM, Komisarski M, Kusmierek JT, Tudek B. Long-chain adducts of trans-4-hydroxy-2-nonenal to DNA bases cause recombination, base substitutions and frameshift mutations in M13 phage. Mutation research. 2004;550:33-48
18. Nair J. Lipid peroxidation-induced etheno-DNA adducts in humans. IARC scientific publications: 55-61. 1999
19. Huang H, Wang H, Qi N, Lloyd RS, Rizzo CJ, Stone MP. The stereochemistry of trans-4-hydroxynonenal-derived exocyclic 1,N2-2'-deoxyguanosine adducts modulates formation of interstrand cross-links in the 5'-CpG-3' sequence. Biochemistry. 2008;47:11457-11472
20. Benamira M, Marnett LJ. The lipid peroxidation product 4-hydroxynonenal is a potent inducer of the SOS response. Mutation research. 1992;293:1-10
21. Cajelli E, Ferraris A, Brambilla G. Mutagenicity of 4-hydroxynonenal in V79 Chinese hamster cells. Mutation research. 1987;190:169-171
22. Brambilla G, Sciaba L, Faggin P, Maura A, Marinari UM, Ferro M, Esterbauer H. Cytotoxicity, DNA fragmentation and sister-chromatid exchange in Chinese hamster ovary cells exposed to the lipid peroxidation product 4-hydroxynonenal and homologous aldehydes. Mutation research. 1986;171:169-176
23. Hu W, Feng Z, Eveleigh J, Iyer G, Pan J, Amin S, Chung FL, Tang MS. The major lipid peroxidation product, trans-4-hydroxy-2-nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis. 2002;23:1781-1789
24. Chandra A, Srivastava SK. A synthesis of 4-hydroxy-2-trans-nonenal and 4-(3H) 4-hydroxy-2-trans-nonenal. Lipids. 1997;32:779-782
25. Tudek B, Boiteux S, Laval J. Biological properties of imidazole ring-opened N7-methylguanine in M13mp18 phage DNA. Nucleic acids research. 1992;20:3079-3084
26. Miller JH. Experiments in Molecular Genetics. NY: Cold Spring Harbor Laboratory. 1972
27. Messing J. New M13 vectors for cloning. Methods in enzymology. 1983;101:20-78
28. Sambrook JF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor: Cold Spring Harbor Laboratory Pres. 1989
29. Messing J. In vitro DNA synthesis as a tool to analyze and alter genes. Basic life sciences. 1983;25:9-15
30. Bartsch H, Nair J. Accumulation of lipid peroxidation-derived DNA lesions: potential lead markers for chemoprevention of inflammation-driven malignancies. Mutation research. 2005;591:34-44
31. Choudhury S, Pan J, Amin S, Chung FL, Roy R. Repair kinetics of trans-4-hydroxynonenal-induced cyclic 1,N2-propanodeoxyguanine DNA adducts by human cell nuclear extracts. Biochemistry. 2004;43:7514-7521
32. Gros L, Ishchenko AA, Saparbaev M. Enzymology of repair of etheno-adducts. Mutation research. 2003;531:219-229
33. Delaney JC, Smeester L, Wong C, Frick LE, Taghizadeh K, Wishnok JS, Drennan CL, Samson LD, Essigmann JM. AlkB reverses etheno DNA lesions caused by lipid oxidation in vitro and in vivo. Nature structural & molecular biology. 2005;12:855-860
34. Kurowski MA, Bhagwat AS, Papaj G, Bujnicki JM. Phylogenomic identification of five new human homologs of the DNA repair enzyme AlkB. BMC genomics. 2003;4:48
35. Janion C. Inducible SOS response system of DNA repair and mutagenesis in Escherichia coli. International journal of biological sciences. 2008;4:338-344
36. Budzowska M, Kanaar R. Mechanisms of dealing with DNA damage-induced replication problems. Cell biochemistry and biophysics. 2009;53:17-31
37. Higashitani N, Higashitani A, Horiuchi K. SOS induction in Escherichia coli by single-stranded DNA of mutant filamentous phage: monitoring by cleavage of LexA repressor. Journal of bacteriology. 1995;177:3610-3612
38. Minko IG, Kozekov ID, Harris TM, Rizzo CJ, Lloyd RS, Stone MP. Chemistry and biology of DNA containing 1,N(2)-deoxyguanosine adducts of the alpha,beta-unsaturated aldehydes acrolein, crotonaldehyde, and 4-hydroxynonenal. Chemical research in toxicology. 2009;22:759-778
39. Fuchs RP, Fujii S. Translesion synthesis in Escherichia coli: lessons from the NarI mutation hot spot. DNA repair. 2007;6:1032-1041
40. Huang H, Wang H, Qi N, Kozekova A, Rizzo CJ, Stone MP. Rearrangement of the (6S,8R,11S) and (6R,8S,11R) exocyclic 1,N2-deoxyguanosine adducts of trans-4-hydroxynonenal to N2-deoxyguanosine cyclic hemiacetal adducts when placed complementary to cytosine in duplex DNA. Journal of the American Chemical Society. 2008;130:10898-10906
41. Dixon DA, Kowalczykowski SC. Homologous pairing in vitro stimulated by the recombination hotspot, Chi. Cell. 1991;66:361-371
42. Xing D, Sun L, Cukier RI, Bu Y. Theoretical prediction of the p53 gene mutagenic mechanism induced by trans-4-hydroxy-2-nonenal. The journal of physical chemistry. 2007;111:5362-5371
43. Yamamoto K, Higashikawa T, Ohta K, Oda Y. A loss of uvrA function decreases the induction of the SOS functions recA and umuC by mitomycin C in Escherichia coli. Mutation research. 1985;149:297-302
44. Kiyosawa K, Tanaka M, Matsunaga T, Nikaido O, Yamamoto K. Amplified UvrA protein can ameliorate the ultraviolet sensitivity of an Escherichia coli recA mutant. Mutation research. 2001;487:149-156
45. Fijalkowska IJ, Schaaper RM. Effects of Escherichia coli dnaE antimutator alleles in a proofreading-deficient mutD5 strain. Journal of bacteriology. 1995;177:5979-5986
Author contact
![]() Correspondence to: Barbara Tudek, Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Pawińskiego 5a, 02-106 Warsaw, Poland. Tel: (+4822) 592-3334; fax: (+4822) 592-2190; e-mail: tudekwaw.pl
Correspondence to: Barbara Tudek, Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Pawińskiego 5a, 02-106 Warsaw, Poland. Tel: (+4822) 592-3334; fax: (+4822) 592-2190; e-mail: tudekwaw.pl
Received 2009-6-4
Accepted 2009-9-15
Published 2009-9-23