ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
The role of satellite cells...
The role of Notch and Wnt in...
Physiological stimuli and muscle...
The interaction of physiological...
Conclusion
References

Int J Biol Sci 2010; 6(3):268-281. doi:10.7150/ijbs.6.268 This issue Cite
Review
Notch and Wnt Signaling, Physiological Stimuli and Postnatal Myogenesis
Susan Tsivitse ![]()
Department of Kinesiology, Exercise Physiology Laboratory, University North Carolina - Charlotte, Charlotte, NC 28223, USA
Abstract
Adult skeletal muscle stem cells, termed satellite cells are imperative to muscle regeneration. Much work has been performed on satellite cell identification and the subsequent activation of the myogenic response but the regulation of satellite cells including its activation is not well elucidated. The purpose of this review article is to synthesize what the literature reveals in regards to the current understanding of satellite cells including their contribution to muscle repair and growth following physiological stimuli. In addition, this review article will describe the recent findings on the roles of the classic developmental signaling pathways, Notch and Wnt, to the myogenic response in various muscle injury models. This purpose of this summary is to bring awareness of the impact that muscle contraction models have on the local and systemic environment of adult muscle stem cells which will be beneficial for comprehending and treatment development for muscle -associated ailments and other organ diseases.
Keywords: Notch and Wnt, muscle repair and growth, physiological stimuli
Introduction
Since the identification of satellite cells on skeletal muscle fibers in 1961(1), the field of satellite cell biology has progressed immensely to its recent characterization as an adult stem cell and its implications in regenerative medicine. Although portrayal of postnatal myogenesis predominates the satellite cell biology field, what is less known is/are the factor(s) responsible for satellite cell activation and the myogenic response. In addition, there is no confirmation on the overall importance of satellite cells to muscle repair and growth following physiological stimuli. In recent years, Notch and Wnt signaling have been determined to be critical for postnatal myogenesis but limited muscle regeneration models have been utilized to test these pathways (2-6). Employing a variety of muscle injury and growth techniques will result in a definitive understanding of satellite cell contribution to muscle repair and hypertrophy. With its systemic effects, physiological stimuli are an ideal muscle model as relates to investigating the myogenic properties of muscle regeneration and growth including satellite cell activation. Although the amount of muscle injury induced may not be as profound as artificial models, contraction-induced muscle injury and muscle overloading exercises are identified to up-regulate myogenic proteins. This review will summarize the literature to address the interest on the effects of physiological stimuli on Notch and Wnt signaling pathways during postnatal myogenesis. We will discuss the current knowledge on satellite cell regulation as well as the recent developments of the importance of Notch and Wnt signaling to muscle repair. We will also examine the usefulness of physiological stimuli to studying the regulation of satellite cell activation and muscle regeneration. Furthermore, we will enlighten the reader with what is currently known on the effects of physiological stimuli and muscle repair as relates to Notch and Wnt signaling.
The role of satellite cells during muscle regeneration
Skeletal muscle has the remarkable capability to completely repair within a few weeks of an insult (7). Immediately following exposure to overt skeletal muscle injury (injurious contractions, chemical, freeze and crush), muscle degeneration occurs including ultrastructural damage to the contractile apparatus such as z-line streaming, A-band disruption, and altered sarcomere length (8, 9). In addition, disruptions to the sarcolemma and increased cellular permeability renders the muscle fiber to be susceptible to the autogenetic response via activation of the non-lysosomal calpain system which further disrupts the contractile apparatus (10, 11).
In the hours to days following the initial mechanical injury, the acute inflammatory response is initiated within the injured muscle fiber and is characterized by vasodilation of the microcirculation, increased permeability of proteins accumulation and infiltration of phagocytes (neutrophils, monocytes and macrophages) (12). Chemotactic signals derived from the muscle itself, resident muscle macrophages or the circulatory system direct the inflammatory cells to the injured site within the muscle fiber in which they function to destroy the necrotic tissue and prepare the site for muscle regeneration (13, 14).
The muscle regeneration process in postnatal skeletal muscle is mostly attributed to the small population of myogenic stem cells termed satellite cells. Located beneath the myofiber basal lamina, quiescent satellite cells comprise a small percentage of myonuclei (~5% at two months of age) (15). Quiescent satellite cells are mitotically arrested at G0 state and are characterized with small cytoplasm, condensed chromatin and flatten shape (15). Satellite cells have often been identified to be Pax 7+, CD34+, CD45- and Sca1- (16). However, there are several markers identified to be associated with quiescent satellite cells such as CD34, α7-integrin, Pax-7, ABGC2, MCadherin, Syndecan 3/4, c-met, Cdh15, Foxk1 (formerly myocyte nuclear factor), Pax3, Sox8, Sox15, Vcam1, CXCR4, β1 integrin, and SM/C-2.6 antibody, calcitonin receptor, Caveolin-1and sphingomyelin (15, 17-26). It is improbable to identify a single marker discriminating quiescent satellite cells from proliferating myogenic precursor cells since many of the aforementioned markers may only identify specific subpopulations of satellite cells or stage of satellite cell activation.
Following muscle injury, quiescent satellite cells are exposed to signals derived either from the damaged muscle or elsewhere within the injured microenvironment and the cells become activated. Many factors have been suggested to be responsible for activation of satellite cells, such as muscle-derived growth factors including hepatocyte growth factor, fibroblast growth factor 2, 6, cytokines such as interleukin-6, heat stress, and developmental signaling pathways, but the molecular/cell mechanisms leading to awakening of satellite cells and from the unperturbed state remain to be fully identified (2, 7, 27, 28).
Upon activation, satellite cells undergo increased cytoplasmic volume, decreased heterochromatin and they become mitotically active as they transition from G0 to G1 cell cycle phase. Once in the first cycle satellite cells enter an intermediate progenitor stage and are known as myogenic precursor or progenitor cells (15). Within hours following activation, satellite cells up regulate the expression of basic helix-loop-helix transcriptional activators of the myogenic regulatory factor (MRF) family (MyoD, Myf5) and progress to the highly proliferative phase termed myoblasts. MRFs are essential for muscle regeneration. MyoD (-/-) null mice and mice with either double-knockout for MyoD and Myf5 genes or triple mutant of Myf5, Mrf4 and MyoD demonstrate a complete loss of skeletal muscle formation (29-32). In the days post activation, proliferating myoblasts express the cell cycle arrest protein, p21 and late MRFS (Myogenin and MRF4), resulting in withdrawal from the cell cycle and initiation of terminal differentiation to form multinucleated myotubes. Completion of the differentiation program results in fusion of myotubes with each other or with existing myofibers and is often identified with increased expression of muscle specific Myosin Heavy Chain (MHC).
Myogenin has been identified to be critical for myoblast differentiation as demonstrated in myogenin-deficient embryos that have myoblasts but not myofibers due to defective myotube formation (33, 34). MicroRNAs (miRs), which function in post-transcriptional modification have recently been identified to participate in skeletal muscle repair and may play a role in the transition of proliferation to differentiation state of myoblasts (35, 36). Exogenous application of miR1, miR-133 and miR-206 to lacerated rat TA result in enhanced muscle repair including increased expression of MyoD and Pax7 (35). In embryos, miR-27b targets and down-regulates Pax3 thereby promoting differentiation while miR-27b inhibitors result in continual Pax3 expression and proliferation of satellite cells (36). These data suggest that microRNAs may provide a novel strategy for understanding the regulation of skeletal muscle repair particularly as relates to the transition of myoblast proliferation to the differentiation stage.
The importance of Pax 7 as relates to MRFs and postnatal myogenesis is a prevalent study topic in satellite cell biology. Upon activation and proliferation of satellite cells, Pax 7 has been identified to up regulate MyoD through the recruitment of histone methyltransferase complex (37, 38) and as myoblasts progress to the differentiation stage, Pax 7 expression is down regulated (39). However, there is also a small population of activated satellite cells that retain Pax 7 expression and do not express MyoD or Myf5. These cells function to maintain the progenitor pool by returning to a quiescent state instead of committing to myogenic differentiation (38, 39). The importance of Pax 7 to muscle regeneration may end by 21 days of age in rodents (32, 40). Lepper et al. (40) tracked the temporal fate of inactive Pax7 gene expression and reports that in adult mice with mutant Pax 7, myogenesis was able to ensue (40). Therefore, Pax 7 may be critical for satellite cell activation up to a “juvenile age” in which young skeletal muscle is developed and the remaining satellite cells are not needed and go into quiescence.
There is much evidence supporting the notion that satellite cells possess stem cell properties such as their ability to generate non-muscle lineages. Satellite cells migrated from single muscle fibers cultured on Matrigel formed myoblasts and myotubes but also expressed osteogenic and adipogenic protein markers suggesting that satellite cells have the potential to convert to other mesodermal derivatives including osteocytes and adipocytes (41). Myoblasts treated with bone morphogenic protein (BMPs) differentiate into osteocytes and suppress the expression of MRFs such as MyoD (42, 43). In addition, myoblasts convert to adipocytes when cultured with thiazolidinedione or fatty acids (44). C2C12s engineered for expression of CCAAT/enhancer-binding proteins (C/EBPs) (adipogenic protein) or BMPs (osteogenic protein) result in differentiation of each respective fate (45). MyoD (-/-) null mice are not more susceptible to transition to osteogenic or adipogenic fates when treated with BMP or thiazolidinedione respectively relative to control mice suggesting that MyoD is not critical for possession of a myogenic fate (41). In vivo models also show the conversion of muscle to non-myogenic lineages including the presence of adipose tissue and bone formation within muscle diseases such as Duchenne Muscular Dystrophic patients and mdx mice (30, 46-54).
Another unique stem cell characteristic of satellite cells are their ability to self renew (20, 22, 55-59). It has been suggested that a small population of activated satellite cells do not proliferate but assist in maintaining the quiescent satellite cell pool to ensure enough satellite cells are present for repairing of injured skeletal muscle (20, 22, 55-57). Transplantation of genetically-labeled myofibers into mdx mice that were depleted of satellite cells result in replenishment of the satellite cell pool (58). There are multiple mechanisms for self-renewal. Activated satellite cells have been demonstrated to divide into 2 separate satellite cell populations, one faction that rapidly divides and another group that divides at a slower rate and may have the duty of replenishing the satellite cell pool (59). Self-renewal may also occur through asymmetric division in which a daughter cell does not further divide and maintains stem-cell properties including expression of Sca1, Notch1and Pax7 while the other daughter cell proliferates and progresses down the myogenic lineage by expressing the muscle differentiation marker Desmin (2, 60-65). Notch inhibitor, Numb is asymmetrically expressed on the activated satellite cells and may regulate cell fate choices by promoting progression down the myogenic lineage (2). Self renewal may also occur through symmetrical division in which both daughter cells maintain stem-cell properties (66, 67). Cells that do not express MyoD but continue to express Pax 7 are suggested to be refrained for self-renewal (32).
There is great advancement in postnatal myogenesis characterization as well as the recognition of the stem cell properties of satellite cells including their tactics to maintaining their numbers. The factors responsible for satellite cell regulation are under study and need to be determined. Ascertaining satellite cell regulation will have implications in muscle -associated ailments and disease as well as implications in stem-cell regenerative medicine.
The role of Notch and Wnt in muscle regeneration
Notch and Wnt signaling are well known to play a critical role in determining tissue specificity of stem cells by regulating cell fate decisions during embryonic development (68-72). Since satellite cells are stem cells it is plausible to infer that Notch and Wnt are important for postnatal myogenesis.
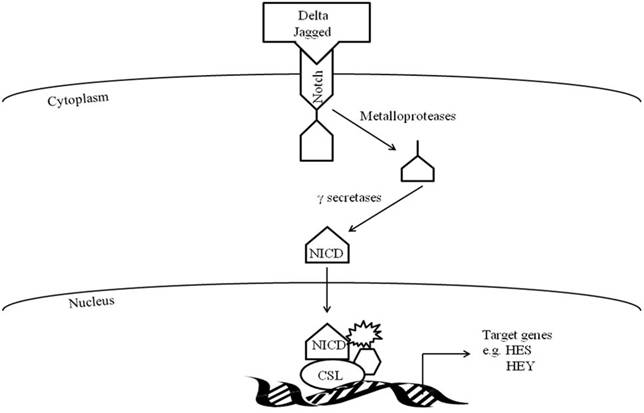
Activation of Notch signaling occurs when the transmembrane Notch receptor interacts with one of its cell-membrane-anchored DSL ligands (Delta/Jagged, Serrate, or Lag2) and undergoes a series of cleavages from metalloproteases and γ-secretases resulting in the intercellular domain of Notch (NICD) translocating to the nucleus (Figure 1). Within the nucleus, the now active NICD interacts with transcriptional repressors of the CSL family (CBF1/RBP-J, Suppressor of Hairless (Su[H]), and Lag-1) and converts them to transcriptional activators (NICD-CSL). With involvement of other proteins, the CSL-NICD complex regulates target genes such as Hey and Hes genes, which encode proteins known to be involved in myogenesis (68).
Within hours to days following muscle injury there is increased expression of Notch signaling components (Delta-1, Notch-1 and active Notch) on activated satellite cells and neighboring muscle fibers(2, 3). Up-regulation of Notch signaling promotes the transition of activated satellite cells to highly proliferative myogenic precursor cells and myoblasts, as well as prevents myoblast differentiation to myotubes (3-6, 73, 74). Myoblast proliferation is decreased and differentiation is promoted when Notch activity is inhibited in myoblasts with Notch antagonist, Numb,γ-secretase inhibitor, or with small-interfering RNA (siRNA) knockdown of Presenilin-1 (2, 4, 75-78). In addition, mutations in Delta-like 1 or CSL result in excessive premature muscle differentiation and defective muscle growth (79, 80).
Multiple mechanisms for how Notch signaling prevents myoblast differentiation have been proposed (81-87). Hes1 or Hey1 may form a complex with MyoD to repress its expression resulting in inhibiting critical proteins for myotube formation such as Mef2D and Myogenin (6, 81-83, 88, 89). However, a recent study by Buas et al. (84) does not show an interaction with Hey1 and MyoD and instead proposes that Hey1 prevents MyoD to bind to Myogenin or Mef2C promoters thereby inhibiting their transcription and ultimately repressing muscle differentiation(84). Another mechanism for Notch preventing differentiation is a potential cooperative effort between Notch and other signaling pathways to allow for sufficient myoblast proliferation prior to differentiation (85, 86). Forkhead Box O 1 transcription factor (FOXO1) prevents muscle differentiation and is suggested to be critical for downstream Notch signaling since siRNA-induced inhibition of FOXO1 represses Notch-induced Hes1 activity in C2C12 cells (85). FOXO1 interacts with CSL of Notch resulting in up regulation of Hes1 promoter activity. In addition, Transforming Growth Factor ß signaling pathway (TGFß) induces Hes1 expression in C2C12 cells which may lead to inhibition of MyoD and progression of muscle differentiation (86, 87).
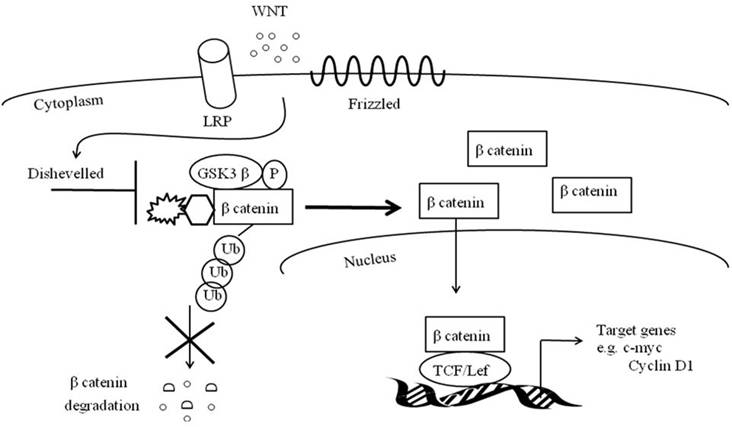
Similar to Notch, canonical Wnt signaling is critical for muscle repair (90-94). The canonical Wnt signaling cascade entails soluble Wnt ligands to interact with Frizzled receptors and low-density lipoprotein receptor-related protein co-receptors (LRP) (Figure 2). This coordination stimulates phosphorylation of Disheveled and inactivates GS3Kß's phosphorylation of ß catenin. With the assistance of Axin, the de-phosphorylated and stable ß- catenin does not undergo ubiquitination and degradation and is instead translocated to the nucleus where it binds to TCF/LEF1 transcription factors (95). TCF/LEF1 transcription factors may directly activate MyF5 and MyoD or may up regulate MRF co-activators such as c-Jun N-terminal kinases (96-98).
Notch signaling pathway.
Canonical Wnt signaling pathway.
There is a preponderance of evidence demonstrating that skeletal muscle injury up regulates Wnt signaling (90-94). mRNA expression of Wnts5a, 5b, 7a, and 7b increases in muscle at four days post cardiotoxin-induced injury (90). At four days post myofiber explantation, there is elevated expression of Wnt3a, Frizzled1 and 2, and Axin2 (93). Using TCF-optimal promoter B-galactosidase mice (TOPGAL mice) there is increased TCF reporter activity in myogenic cells at two and five days post freeze-induced muscle injury (93). Furthermore, differentiating myoblasts express Wnt signaling co-activator BCL9 (94).
While Wnt is critical for muscle development as evident in no dermomyotome formation or Myf5 expression in embryonic Wnt1 and Wnt3a knockout mice, Wnt also contributes postnatal muscle regeneration (90, 93, 98). Activating Wnt signaling by treating myoblasts with lithium chloride (LiCl, which inhibits GSK3ß activity) results in increased MHC and Myogenin expression; and treating myoblasts with Wnt3a results in a ~ 30% increase in Desmin expression (90, 93, 99, 100). Brack et al. (94) reports a ~ 40% increase in muscle regeneration in myogenic cells with forced Wnt expression, further demonstrating that Wnt signaling promotes postnatal muscle repair (94).
Wnt's main role during muscle regeneration is myoblast differentiation and myotube fusion (99-103). Premature myogenic differentiation occurs in freeze-injured muscle injected with Wnt3a (93). Furthermore, treatment of proliferation C2C12 myoblasts or single fiber cultures with Wnt antagonists or secreted frizzled receptor proteins (sFRPs) repress myotube formation, decreases Myogenin and Mrf4 RNA expression by ~ 50% as well as inhibits Desmin expression (93, 102). Moreover, there is impaired myotube formation and decreased Myogenin and MHC expressions in siRNA-induced inhibition or knock-out mice of Wnt co-activator BCL9 (94). Lastly, there is increased myogenic differentiation in cholesterol depleted- myoblasts identified to promote nuclear translocation of ß-catenin and TCF/LEF activation (103).
Myogenesis consists of a series of orchestrated events that require communication of a variety of signaling pathways when transitioning from the phases of satellite cell activation to myoblast proliferation and differentiation and finally, myotube formation. Notch and Wnt interact during muscle development as well as during postnatal myogenesis and it is plausible that this crosstalk contributes to the stage transitions during adult muscle repair (93, 104). It is suggested that Notch activity presides during myoblast proliferation after which there is a temporal switch to Wnt signaling and subsequent myoblast differentiation and fusion into myotubes (93). Inhibiting Notch (with soluble Jagged ligand or with a γ - secretase inhibitor) or activating Wnt (by inhibiting GSK3ß or adding Wnt3a) decreases Myf5 expression and promotes muscle differentiation providing evidence that Notch signaling needs to be turned off and Wnt turned on for differentiation to ensue (93, 100, 103). When Notch inhibitors are administered during the differentiation phase there is no acceleration of muscle regeneration, further supporting the notion that Notch signaling does not participate in the later stages of muscle repair (93). In addition, during the early stages of muscle repair (24 hours) following cardiotoxin-induced muscle injury, Wnt inhibitor, sFRP2 is expressed in proliferating myoblasts and there is no Wnt activity (105); furthermore, administration of Wnt inhibitor, sFRP3 to single-fiber cultures at early stages of muscle repair (day 2 of culture) has no effect on Wnt inhibition but when sFRP3 is added at later time points (day 3.5 of culture), Wnt expression is inhibited and there is less myoblast differentiation. These data suggest that Wnt activity does not occur during the early proliferative stages of muscle repair (93). Brack et al. (93) recommends that low Wnt signaling is needed for a sufficient quantity of proliferating myoblasts prior to myotube formation (93).
Notch and Wnt orchestrate together for cell fate decisions in other systems but their roles have not been determined to be similar in the skeletal muscle model (106-108). A co-activation of Notch and Wnt promote proliferation of progenitor intestinal epithelial cells (107). When Notch and Wnt are both active in proliferating progenitor cells within the crypt region of the small intestine, the cells continue to proliferate and form the intestinal absorptive cells called enterocytes (107). In addition, administration of Wnt3a to hematopoietic progenitors cultured in presence of Notch ligand, Delta results in increased Hes1 and CD7+ cells (T cells) suggesting that hematopoietic progenitors cells transform into T cells in the presence of Wnt and Notch (108).
Determining if Wnt and Notch regulate each other will aid in delineating the mechanism of orchestration between Notch and Wnt during cell fate determination including postnatal myogenesis. Since Notch appears to dominate during myoblast proliferation and Wnt signaling leads during the differentiation/fusion stages of muscle repair, it is plausible to suggest that Notch signaling is upstream of Wnt. Transfection studies show that the intracellular domain of Notch has binding sites on LEF1 promoter that are different than ß-catenin sites and result in LEF1 transcriptional activation (109). Conversely, it should be noted that there is evidence suggesting Wnt signaling is upstream of Notch (110, 111). The Wnt target genes Brachyury (T-box) and LEF1/TCF1 have binding sites on Notch1 and Delta-like-1 (Dll1) promoter and mice embryos with mutant Tbx6 function (downstream T box target gene) or lacking Brachyury do not express Dll1 (110, 111). Furthermore, in the presence of exogenous Wnt3a and Delta1 there is increased Hes1 and Deltex1 expression in hematopoietic stem cells (108, 112).
Identifying the location for the point of transition between Notch and Wnt signaling would be beneficial in determining the mechanism of Notch and Wnt orchestration for cell fate decisions including postnatal myogenesis. GSK3ß directly binds to Notch 1 intracellular domain (Notch1IC) and facilitates Notch signaling so it seems plausible that GSK3ß may be a critical juncture between Notch and Wnt pathways during postnatal myogenesis (93, 113, 114). While increased Notch signaling corresponds with elevated GSk3ß activity (GSk3ß phosphorylated at tyrosine 216 GSk3ßpY216) at one day post muscle injury, Wnt contradicts GSk3ß activity and is low at that time point (93). However, as the time course of muscle repair progresses from one day to four days, Wnt signaling increases and both GSK3ß and Notch activity declines (93). Inhibiting GSK3ß activity in mononucleated cells of TOPGAL mice results in increased ß-catenin expression and Wnt reporter activity (93). In addition, treating myoblasts with Notch inhibitor γ-secretase, results in decreased GSK3ß activity (93). These data further confirm the corresponding relationship between GSK3ß and Notch and inverse relationship between GSK3ß and Wnt as well as suggests that GSK3ß activity may be pivotal for the transition between Notch and Wnt signaling.
Presenilins, which assist in cleaving full-length Notch to its activated state, have also been suggested to be major players in the coordination of the Notch and Wnt pathways (115, 116). Presenilins have been identified to bind to ß-catenin and possibly participate in ß -catenin ubiquitination(115). In addition, there is increased TCF/LEF1 transcription in Presenilin1 knockout embryonic fibroblasts (116). These date suggest that Presenilins may function as mediator between Notch and Wnt by de-regulating Wnt signaling. Besides GSK3ß and Presenilins, there is evidence that Notch antagonist, Numb may be a pivotal point of transition between Notch and Wnt since forced expression of Numb on erythroid progenitor cells treated with exogenous Wnt5a, results in increased TCF/LEF transcriptional activity (106).
The orchestration of Notch and Wnt is crucial for cell fate decisions of a variety of tissues. However, there are conflicting reports on the mechanisms of their co-regulation during regenerative processes in skeletal muscle and other tissue models. Of particular interest, is understanding Notch and Wnt's role in satellite cell biology such as regulating myogenic proteins during postnatal myogenesis. What is even less known is the effect of different muscle injury models (including physiological stimuli) in regulating Notch, Wnt and myogenic protein expression during adult muscle repair.
Physiological stimuli and muscle regeneration and growth
The current muscle injury models for examining the roles of Notch and Wnt in satellite cell regulation during postnatal myogenesis consist primarily of artificial techniques which do not consider the influences of other biological systems and are not easily applied to muscle function scenarios. Physiological stimuli such as contraction-induced muscle injury (injurious exercise) and growth models (overload hypertrophy, resistance exercises) are functionally relevant and involve the contribution of multiple active biological systems which are important to consider when investigating sources responsible for muscle regenerative processes.
A common injurious exercise model, downhill running (DHR), uses eccentric muscle contractions in which the contracting muscle is forced to lengthen by an external force while producing tension (9, 117-119). DHR in rodents results in decreased isometric force production and elevations in muscle injury makers such as creatine kinase activity and muscle injury criteria identified with Hematoxylin and Eosin staining (118, 120, 121). DHR is also known to stimulate muscle remodeling and myogenesis processes (121-128). DHR induce ~80% up regulation of mitogen-activated protein kinase (MAPK) signaling such as phosphorylation of JNK, p38, and ERK (129). Myogenic markers Myf5, MyoD, Myosin Heavy Chain (MHC), MCadherin, Desmin, and Pax 7 increase expression within the first week following DHR (121-127). There is increased Myf5 transcriptional activity in Myf5LacZ reporter mice at 48 hours to 96 hours post DHR (128). Overall, there is sufficient data to exhibit that DHR induces a myogenic response and may be worthy as a model for studying postnatal myogenesis regulation.
One aspect to consider when using the DHR model to study myogenesis is that the time course of muscle repair with the DHR model may be different than with artificial muscle injury models. There is a delay in the onset of muscle regeneration in the DHR model relative to cardiotoxin injections (CTX) (122). Armand et al. (122) reports that CTX-injured soleus fibers exhibit extensive myofiber damage at 24 hours to 72 hours post injury, while DHR-injured fibers are not apparent until 96 hours to 144 hours post exercise. At 120 hours post-CTX, muscle regeneration dominates while signs of muscle repair are not present until 144 hours to 168 hours post-DHR. Although DHR does not induce as profound of muscle injury as seen in the CTX model, this exercise injury model still induces a myogenic response.
Besides the DHR model, other eccentric contraction models are available to study muscle regeneration (130-139). Eccentric contractions on an isokinetic dynamometer results in muscle remodeling in the hours to days post exercise including increased expression of hepatocyte growth factor (HGF), MyoD, mechano growth factor, neural cell adhesion molecule, Pax7 and insulin-like growth factor 1 (IGF1) (134-138). In situ muscle lengthening contraction injury model is a physiological test that uses a rigorous and precise control of muscle contraction by electrically stimulating motor neurons of associated muscles of interest (130-132, 139). With its ability to quantify muscle function this biological model offers a unique approach relative to other muscle injury protocol. There is a 70%-80% decrease in isometric force production and a 15- 20% increase in injured fibers in mice EDL at three days following exposure to electrically stimulated lengthening contractions (131). Inflammation is also present at three days post injury with reports of a 7.9 fold and 51.2 fold increase in muscle associated neutrophils and macrophages respectively (140). With the profound magnitude of muscle injury present in this physiological stimulus model as well as the presence of myogenic-associated proteins, it seems plausible that one could use this novel muscle injury model to study the regulation of myogenic repair as it relates to recovery of function.
Muscle overload techniques (heavy resistance exercise, stretch hypertrophy and compensatory hypertrophy) are physiological stimuli capable of investigating the contribution of myogenic properties to growth. Overload-induced muscle hypertrophy is a type of a compensatory hypertrophy model in which muscles are ablated (such as the gastrocnemius, soleus or tibialis anterior) and the remaining muscles (such as the plantaris or extensor digitorum longus) work harder to maintain muscle function (141). This compensation of the remaining muscles results in growth including increased DNA synthesis, muscle mass and force production (141-148). Overloaded rat soleus has a 40% increase in wet weight and ~ 6.76% to 12.74% increase in de novo myofibers (142). The cross sectional area of a single myofiber as well as myonuclei number per millimeter of a myofiber increase at 46% and 44% respectively in an overloaded rat plantaris (149). It is suggested that activation of satellite cells is a source of the new myonuclei rather than sole provision from differentiated myotubes (149, 150). Petrella et al. (151) reports that a 117% increase in satellite cells accompanies the 26% increase in myonuclei following heavy resistance exercises (151). During the early hours and within the first week of compensatory overload there is evidence of satellite cell activation and myogenic response including up-regulated Pax 7, M-Cadherin, MyoD, Desmin, Myogenin, MHC as well as fibroblast growth factors 1 and 7, IGF1 and HGF (142, 145, 147, 148, 152-155).
The contribution of satellite cells to muscle repair and growth is not conclusive. There is evidence supporting the notion that satellite cells are critical to muscle repair, recovery of muscle function as well as important to muscle growth (142, 149, 156-158). Irradiation-induced inhibition of proliferating myogenic cells results in decreased force recovery following injurious eccentric contractions to rodent anterior crural muscles (156, 157). Non-irradiated mice have complete force recovery at three weeks post exposure to an injurious lengthening protocol while irradiated mice exhibit a 35% force deficit (156). In addition, MyoD and Myogenin transcripts are increased at three, seven, 14, and 21 days post lengthening contractions in non-irradiated mice while irradiated mice do not show any MRF up-regulation (156). There is evidence of the importance of satellite cells in the overload muscle growth models (142, 149). Phelan et al. (1997) reports no hypertrophy following four weeks of overloading in irradiated rat soleus (142). In addition, there is no increase in muscle mass, myofibrillar protein, DNA content, or cross sectional area of a single myofiber at three months following overloading in irradiated rat hindlimb muscle (149).
However, it needs to be mentioned that there is data demonstrating the occurrence of muscle hypertrophy without up regulation of myogenic proteins following muscle overload and proponents that propose the critical component for muscle hypertrophy is protein synthesis and not satellite cells (159, 160). Since IGF1 induces muscle hypertrophy by stimulating protein production via signaling pathways such as PI3K/Akt/mTOR/S6K1 (161-164) investigators from this camp manipulate IGF to study the impact of protein synthesis on muscle hypertrophy. There is increased muscle hypertrophy in overloaded mice that over-express muscle IGF1 or Akt (165-167). In addition, irradiated mice that are introduced with viral-mediated IGF1 gene transfer have greater muscle growth than with just irradiation alone (162).
To add complication to the controversy, it is also proposed that both satellite cells and protein synthesis contribute to muscle repair and hypertrophy (162, 168, 169). Barton Davis et al. (162) identified IGF1 receptors on satellite cells and differentiated muscle fibers (162). They suggest that IGF1 could co-activate satellite cells and adult muscle fibers to induce hypertrophy so that in conditions in which irradiation depletes proliferating activated satellite cells the mature muscle fibers could be responsible for the increased protein synthesis (162). Others suggest that the predominating mechanism of either satellite cell activation or increased protein synthesis may be due to the timeline of events following a hypertrophic stimulus (168, 169). Perhaps “early” hypertrophy is due to increased transcription and translation while hypertrophy that occurs at later time points may be due to increased satellite cell activation, resulting in increased myonuclei number (168, 169). More work is needed to delineate the importance of protein synthesis compared to satellite cells to muscle repair and growth.
The interaction of physiological stimuli, Notch and Wnt, and muscle regeneration
There are still many questions in regards to the contribution of satellite cells to muscle repair and growth. Using “global” forms of muscle injury and growth such as contraction-induced muscle injury models and overload-induced hypertrophy models will help identify whether the basis satellite cell activation is intrinsic, muscle-derived, or arise from other systems (nervous, circulatory, inflammatory, etc…). Furthermore, studying the communication of cell signaling pathways important for muscle regeneration (such as Notch, Wnt) as relates to satellite cell activation will aid in delineating the source of satellite cell activation resulting in determining the importance satellite cells to muscle repair and growth. Research on the interaction of physiological stimuli, Notch and Wnt signaling and muscle repair and growth would aid in furthering the understanding of satellite cell regulation. Physiological stimuli up-regulates components of Notch and Wnt signaling (92, 127, 170-175). DHR increases Notch1 and Delta1 expression in myogenic cells at 72 hours through 120 hours post-injury (127). A single injurious bout of resistance exercise increases Notch1 and downstream regulator Hes6 mRNA (170). Overload-induced hypertrophy also increases Notch1 expression in Pax7-positive myogenic cells (171). In vivo exercise decreases GSK3ß activity and ß-catenin phosphorylation which is required for Wnt signaling activation (173, 174). Overload-induced hypertrophy up-regulates total ß-catenin, receptor Frizzled1, Wnt regulator Disheveled1, Wnt downstream target genes cmyc and Cyclin D1 as well as induce a 434% increase in nuclear ß-catenin and promotion of ß-catenin/LEF1 complex formation (172, 175). There is evidence that ß-catenin may be required for muscle hypertrophy since there is no hypertrophy in overloaded mice with conditional inactivation of ß-catenin (92). There is an abundance of evidence stating that physiological stimuli up-regulates Notch and Wnt signaling but more research is needed on the effect of physiological stimuli on the orchestration and regulation of these signaling pathways during muscle repair.
Conclusion
With the development of tools available to identify satellite cells, it is feasible to study the characterization and regulation of postnatal myogenesis. More work is needed to discern the factor(s) responsible for re-awakening these normally quiescent skeletal muscle stem cells as well as to determine the collaboration of the various signaling mechanisms as relates to regulating the myogenic proteins. Notch and Wnt signaling have recently been identified to be critical to muscle regeneration. The revelation of the importance of Notch and Wnt signaling to during muscle repair is recognized in artificial muscle injury models but is largely unknown in physiological stimuli models. Contracting skeletal muscle releases a plethora of signals that induces autocrine and paracrine effects that may influence the regulation of postnatal myogenesis differently than localized muscle injury models. Therefore, understanding the intrinsic and systemic response of contracting muscle will aid in deciphering the regulation of satellite cells and muscle repair as well as understanding the molecular mechanisms responsible for the beneficial protective effects of exercise to cardiovascular disease and cancer.
Conflict of Interest
The author has declared that no conflict of interest exists.
References
1. Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol. 1961;9:493-5
2. Conboy I, Rando TA. The regulation of notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Developmental Cell. 2002;3:397-409
3. Conboy I, Conboy MJ, Smythe GM, Rando TA. Notch-mediated restoration of regenerative potential aged muscle. Science. 2003;302:1575-7
4. Kitzmann M, Bonnieu A, Duret C, Vernus B, Barro M, Laodj-Chevivesse D, Verdi JM, Carnac G. Inhibition of notch signaling induces myotube hypertrophy by recruiting a subpopulation of reserve cells. Journal of Cellular Physiology. 2006;208:538-48
5. Kuroda K, Tani S, Tamura K, Minoguchi S, Kurooka H, Honjo T. Delta-induced notch signaling mediated by RBP-J inhibits MyoD expression and myogenesis. The Journal of Biological Chemistry. 1999;274(11):7238-44
6. Buas M, Kabak S, Kadesch T. Inhibition of myogenesis by Notch: evidence for multiple pathways. J Cell Physiol. 2009;218(1):84-93
7. Hawke T, Garry DJ. Myogenic satellite cells: physiology to molecular biology. J Appl Physiol. 2001;91:534-51
8. Macpherson P, Dennis RG, Faulkner JA. Sarcomere dynamics and contraction-induced injury to maximally activate single muscle fibers from soleus muscles of rat. J Physiol. 1997;500:523-33
9. Fridén J, Lieber RL. Eccentric exercise-induced injuries to contractile and cytoskeletal muscle fibre components. Acta Physiol Scand. 2001;171(3):321-6
10. Belcastro A, Shewchuk LD, Raj DA. Exercise-induced muscle injury: a calpain hypothesis. Mol Cell Biochem. 1998;179(1-2):135-45
11. Duan C, Delp MD, Hayes DA, Delp PD, Armstrong RB. Rat skeletal muscle mitochondrial [Ca2+] and injury from downhill walking. J Appl Physiol. 1990;68:1241-51
12. Smith L. Acute inflammation: the underlying mechanism in delayed onset muscle soreness?. Med Sci Sports Exerc. 1991;23:542-51
13. Frenette J, St-Pierre M, Côté CH, Mylona E, Pizza FX. Muscle impairment occurs rapidly and precedes inflammatory cell accumulation after mechanical loading. Am J Physiol Regul Integr Comp Physiol. 2002;282(2):R351-7
14. Papadimitriou J, Robertson TA, Mitchell CA, Grounds MD. The process of new plasmalemma formation in focally injured skeletal muscle fibers. J Struct Biol. 1990;103(2):124-34
15. Holterman C, Rudnicki MA. Molecular regulation of satellite cell function. Seminars in Cell & Developmental Biology. 2005;16:575-584
16. Montarras D, Morgan J, Collins C, Relaix F, Zaffran S, Cumano A, Partridge T, Buckingham M. Direct isolation of satellite cells for skeletal muscle regeneration. Science. 2005;309(5743):2064-7
17. Cornelison D, Wilcox-Adelman SA, Goetinck PF, Rauvala H, Rapraeger AC, Olwin BB. Essential and separable roles for Syndecan-3 and Syndecan-4 in skeletal muscle development and regeneration. Genes Dev. 2004;18(18):2231-6
18. Tanaka K, Hall JK, Troy AA, Cornelison DD, Majka SM, Olwin BB. Syndecan-4-expressing muscle progenitor cells in the SP engraft as satellite cells during muscle regeneration. Cell Stem Cell. 2009;4(3):217-25
19. Sacco A, Doyonnas R, Kraft P, Vitorovic S, Blau HM. Self-renewal and expansion of single transplanted muscle stem cells. Nature. 2008;456(7221):502-6
20. Shi X, Garry DJ. Muscle stem cells in development, regeneration, and disease. Genes & Development. 2006;20:1692-1708
21. Beauchamp J, Heslop L, Yu DSW, Tajbakhsh S, Kelly RG, Wernig A, Buckinham ME, Partridge TA, Zammit PS. Expression of CD34 and Myf5 defines the majority of quiescent adult skeletal muscle satellite cells. The Journal of Cell Biology. 2000;151(6):1221-33
22. Wagers A, Conboy IM. Cellular and molecular signatures of muscle regeneration: current concepts and controversies in adult myogenesis. Cell. 2005;122:659-67
23. Cornelison D, Wold BJ. Single-cell analysis of regulatory gene expression in quiescent and activated mouse skeletal muscle satellite cells. Developmental Biology. 1997;191:270-83
24. Fukada S, Higuchi S, Segawa M, Koda K, Yamamoto Y, Tsujikawa K, Kohama Y, Uezumi A, Imamura M, Miyagoe-Suzuki Y, Takeda S, Yamamoto H. Purification and cell-surface marker characterization of quiescent satellite cells from murine skeletal muscle by a novel monoclonal antibody. Exp Cell Res. 2004;296(2):245-55
25. Sherwood R, Christensen JL, Conboy IM, Conboy MJ, Rando TA, Weissman IL, Wagers AJ. Isolation of adult mouse myogenic progenitors: functional heterogeneity of cells within and engrafting skeletal muscle. Cell. 2004;119:543-54
26. Gnocchi V, White RB, Ono Y, Ellis JA, Zammit PS. Further characterisation of the molecular signature of quiescent and activated mouse muscle satellite cells. PLoS One. 2009;4(4):e5205
27. Pedersen B. Edward F. Adolph distinguished lecture: muscle as an endocrine organ: IL-6 and other myokines. J Appl Physiol. 2009;107(4):1006-14
28. Oishi Y, Hayashida M, Tsukiashi S, Taniguchi K, Kami K, Roy RR, Ohira Y. Heat stress increases myonuclear number and fiber size via satellite cell activation in rat regenerating soleus fibers. J Appl Physiol. 2009;107(5):1612-21
29. Yablonka-Reuveni Z, Rudnicki MA, Rivera AJ, Primig M, Anderson JE, Natanson P. The transition from proliferation to differentiation is delayed in satellite cells from mice lacking MyoD. Dev Biol. 1999;210(2):440-55
30. Charge S, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev. 2004;84:209-38
31. Rudnicki M, Schnegelsberg PN, Stead RH, Braun T, Arnold HH, Jaenisch R. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell. 1993;75(7):1351-9
32. Tajbakhsh S. Skeletal muscle stem cells in developmental versus regenerative myogenesis. J Intern Med. 2009;266(4):372-89
33. Hasty P, Bradley A, Morris JH, Edmondson DG, Venuti JM, Olson EN, Klein WH. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature. 1993;364(6437):501-6
34. Nabeshima Y, Hanaoka K, Hayasaka M, Esumi E, Li S, Nonaka I, Nabeshima Y. Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature. 1993;364(6437):532-5
35. Nakasa T, Ishikawa M, Shi M, Shibuya H, Adachi N, Ochi M. Acceleration of muscle regeneration by local injection of muscle-specific microRNAs in rat skeletal muscle injury model. J Cell Mol Med. 2009 [Epub ahead of print]
36. Crist C, Montarras D, Pallafacchina G, Rocancourt D, Cumano A, Conway SJ, Buckingham M. Muscle stem cell behavior is modified by microRNA-27 regulation of Pax3 expression. Proc Natl Acad Sci U S A. 2009;11(106):13383-7
37. Rudnicki M, Le Grand F, McKinnell I, Kuang S. The molecular regulation of muscle stem cell function. Cold Spring Harb Symp Quant Biol. 2008;73:323-31
38. Relaix F, Marcelle C. Muscle stem cells. Curr Opin Cell Biol. 2009;21(6):748-53
39. Zammit P, Relaix F, Nagata Y, Ruiz AP, Collins CA, Partridge TA, Beauchamp JR. Pax7 and myogenic progression in skeletal muscle satellite cells. J Cell Sci. 2006;119:1824-32
40. Lepper C, Conway SJ, Fan CM. Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature. 2009;460(7255):627-31
41. Asakura A, M Komaki, MA Rudnicki. Muscle satellite cells are multipotential stem cells that exhibit myogenic, osteogenic, and adipogenic differentiation. Differentiation. 2001;68:245-53
42. Katagiri T, Yamaguchi A, Komaki M, Abe E, Takahashi N, Ikeda T, Rosen V, Wozney JM, Fujisawa-Sehara A, Suda T. Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J Cell Biol. 1994;127:1755-66
43. Yamaguchi A, Katagiri T, Ikeda T, Wozney JM, Rosen V, Wang EA, Kahn AJ, Suda T, Yoshiki S. Recombinant human bone morphogenetic protein-2 stimulates osteoblastic maturation and inhibits myogenic differentiation in vitro. J Cell Biol. 1991;113(3):681-7
44. Teboul L, Gaillard D, Staccini L, Inadera H, Amri EZ, Grimaldi PA. Thiazolidinediones and fatty acids convert myogenic cells into adipose-like cells. J Biol Chem. 1995;270(47):28183-7
45. Fux C, Mitta B, Kramer BP, Fussenegger M. Dual-regulated expression of C/EBP-alpha and BMP-2 enables differential differentiation of C2C12 cells into adipocytes and osteoblasts. Nucleic Acids Res. 2004;32(1):e1
46. Shore E, Glaser DL, Gannon FH. Osteogenic induction in hereditary disorders of heterotopic ossification. Clin Orthop Relat Res. 2000;374:303-16
47. Dulor J, Cambon B, Vigneron P, Reyne Y, Nouguès J, Casteilla L, Bacou F. Expression of specific white adipose tissue genes in denervation-induced skeletal muscle fatty degeneration. FEBS Lett. 1998;439(1-2):89-92
48. Pastoret C, Sebille A. mdx mice show progressive weakness and muscle deterioration with age. J Neurol Sci. 1995;129(2):97-105
49. Ross S, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, MacDougald OA. Inhibition of adipogenesis by Wnt signaling. Science. 2000;289(5481):950-3
50. Taylor-Jones J, McGehee RE, Rando TA, Lecka-Czernik B, Lipschitz DA, Peterson CA. Activation of an adipogenic program in adult myoblasts with age. Mech Ageing Dev. 2002;123(6):649-61
51. Sordella R, Jiang W, Chen GC, Curto M, Settleman J. Modulation of Rho GTPase signaling regulates a switch between adipogenesis and myogenesis. Cell. 2003;113(2):147-58
52. Akimoto T, Ushida T, Miyaki S, Akaogi H, Tsuchiya K, Yan Z, Williams RS, Tateishi T. Mechanical stretch inhibits myoblast-to-adipocyte differentiation through Wnt signaling. Biochem Biophys Res Commun. 2005;329(1):381-5
53. Quinn L, Strait-Bodey L, Anderson BG, Argilés JM, Havel PJ. Interleukin-15 stimulates adiponectin secretion by 3T3-L1 adipocytes: evidence for a skeletal muscle-to-fat signaling pathway. Cell Biology International. 2005;29(6):449-57
54. Vertino A, Taylor-Jones JM, Longo KA, Bearden ED, Lane TF, McGehee RE Jr, MacDougald OA, Peterson CA. Wnt10b deficiency promotes coexpression of myogenic and adipogenic programs in myoblasts. Mol Biol Cell. 2005;16(4):2039-48
55. Baroffio A, Hamann M, Bernheim L, Bochaton-Piallat ML, Gabbiani G, Bader CR. Identification of self-renewing myoblasts in the progeny of single human muscle satellite cells. Differentiation. 1996;60(1):47-57
56. Beauchamp J, Morgan JE, Pagel CN, Partridge TA. Dynamics of myoblast transplantation reveal a discrete minority of precursors with stem cell-like properties as the myogenic source. J Cell Biol. 1999;144(6):1113-22
57. Yoshida N, Yoshida S, Koishi K, Masuda K, Nabeshima Y. Cell heterogeneity upon myogenic differentiation: down-regulation of MyoD and Myf-5 generates 'reserve cells'. J Cell Science. 1998;111:769-79
58. Collins C, Olsen I, Zammit PS, Heslop L, Petrie A, Partridge TA, Morgan JE. Stem cell function, self-renewal and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell. 2005;122:289-301
59. Schultz E. Satellite cell proliferative compartments in growing skeletal muscles. Dev Biol. 1996;175(1):84-94
60. Zammit P, Golding JP, Nagata Y, Hudon V, Partridge TA, Beauchamp JR. Muscle satellite cells adopt divergent fates: a mechanism for self-renewal. The Journal of Cell Biology. 2004;166(3):347-57
61. Olguin H, Olwin BB. Pax-7 up-regulation inhibits myogenesis and cell cycle progression in satellite cells: a potential mechanism for self-renewal. Dev Biol. 2004;275:375-88
62. Halevy O, Piestun Y, Allouh MZ, Rosser BW, Rinkevich Y, Reshef R, Rozenboim I, Wleklinski-Lee M, Yablonka-Reuveni Z. Pattern of Pax7 expression during myogenesis in the posthatch chicken establishes a model for satellite cell differentiation and renewal. Dev Dynamics. 2004;231(3):489-502
63. Grefte S, Kuijpers-Jagtman AM, Torensma R, Von den Hoff JW. Grefte S, Kuijpers-Jagtman AM, Torensma R, Von den Hoff JW. Stem Cells Dev. 2007;16(5):857-68
64. Conboy M, Karasov AO, Rando TA. High incidence of non-random template strand segregation and asymmetric fate determination in dividing stem cells and their progeny. PLoS Biol. 2007;5(5):e102
65. Mitchell P, Mills T, O'Connor RS, Kline ER, Graubert T, Dzierzak E, Pavlath GK. Sca-1 negatively regulates proliferation and differentiation of muscle cells. Dev Biol. 2005;283(1):240-52
66. Cosgrove B, Sacco A, Gilbert PM, Blau HM. A home away from home: challenges and opportunities in engineering in vitro muscle satellite cell niches. Differentiation. 2009;78(2-3):185-94
67. Morrison S, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441(7097):1068-74
68. Artavanis-Tsakonas S, Ran MD, Lake RJ. Notch signaling: cell fate control signal integration in development. Science. 1999;284:770-6
69. Baker N. Notch signaling in the nervous system. Pieces still missing from the puzzle. BioEssays. 2000;22:264-73
70. Ehebauer M, Hayward P, Arias AM. Notch, a universal arbiter of cell fate decisions. Science. 2006;314:1414-5
71. Münsterberg A, Kitajewski J, Bumcrot DA, McMahon AP, Lassar AB. Combinatorial signaling by Sonic hedgehog and Wnt family members induces myogenic bHLH gene expression in the somite. Genes Dev. 1995;9(23):2911-22
72. Tajbakhsh S, Borello U, Vivarelli E, Kelly R, Papkoff J, Duprez D, Buckingham M, Cossu G. Differential activation of Myf5 and MyoD by different Wnts in explants of mouse paraxial mesoderm and the later activation of myogenesis in the absence of Myf5. Development. 1998;125(21):4155-62
73. Nofziger D, Miyamoto A, Lyons KM, Weinmaster G. Notch signaling imposes two distinct blocks in the differentiation of C2C12 myoblasts. Development. 1999;126:1689-702
74. Shawber C, Nofziger D, Hsieh JJD, Lindsell C, Bogler O, Hayward D, Weinmaster G. Notch signaling inhibits muscle cell differentiation through a CBF1-independent pathway. Development. 1996;122:3765-73
75. Berezovska O, Jack C, McLean P, Aster JC, Hicks C, Xia W, Wolfe MS, Weinmaster G, Selkoe DJ, Hyman BT. Rapid Notch1 nuclear translocation after ligand binding depends on presenilin-associated gamma-secretase activity. Annals New York Academy of Sciences. 2000;920:223-6
76. Dahlqvist C, ABlokzijl A, Chapman G, Falk A, Dannaeus K, Ibanez CF, Lendahl U. Functional notch signaling is required for BMP4-induced inhibition of myogenic differentiation. Development. 2003;130:6089-99
77. Karlstrom H, Bergman A, Lendahl U, Naslunc J, Lundkvist J. A sensitive and quantitative assay for measuring cleavage of presenilin substrates. The Journal of Biological Chemistry. 2002;277(9):6763-6
78. Ono Y, Gnocchi VF, Zammit PS, Nagatomi R. Presenilin-1 acts via Id1 to regulate the function of muscle satellite cells in a gamma-secretase-independent manner. J Cell Sci. 2009;122:4427-38
79. Schuster-Gossler K, Cordes R, Gossler A. Premature myogenic differentiation and depletion of progenitor cells cause severe muscle hypotrophy in Delta1 mutants. Proc Natl Acad Sci U S A. 2007;104(2):537-42
80. Vasyutina E, Lenhard DC, Wende H, Erdmann B, Epstein JA, Birchmeier C. RBP-J (Rbpsuh) is essential to maintain muscle progenitor cells and to generate satellite cells. Proc Natl Acad Sci U S A. 2007;104(11):4443-8
81. Zhu B, Ramachandran B, Gulick T. Alternative pre-mRNA splicing governs expression of a conserved acidic transactivation domain in myocyte enhancer factor 2 factors of striated muscle and brain. J Biol Chem. 2005;280(31):28749-60
82. Cheng T, Wallace MC, Merlie JP, Olson EN. Separable regulatory elements governing myogenin transcription in mouse embryogenesis. Science. 1993;261(5118):215-8
83. Wang D, Valdez MR, McAnally J, Richardson J, Olson EN. The Mef2c gene is a direct transcriptional target of myogenic bHLH and MEF2 proteins during skeletal muscle development. Development. 2001;128(22):4623-33
84. Buas M, Kabak S, Kadesch T. The Notch effector Hey1 associates with myogenic target genes to repress myogenesis. J Biol Chem. 2010;285(2):1249-58
85. Kitamura T, Kitamura YI, Funahashi Y, Shawber CJ, Castrillon DH, Kollipara R, DePinho RA, Kitajewski J, Accili D. Kitamura T, Kitamura YI, Funahashi Y, Shawber CJ, Castrillon DH, Kollipara R, DePinho RA, Kitajewski J, Accili D. J Clin Invest. 2007;117(9):2477-85
86. Klüppel M, Wrana JL. Turning it up a Notch: cross-talk between TGF beta and Notch signaling. Bioessays. 2005;27(2):115-8
87. Blokzijl A, Dahlqvist C, Reissmann E, Falk A, Moliner A, Lendahl U, Ibáñez CF. Cross-talk between the Notch and TGF-beta signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J Cell Biol. 2003;163(4):723-8
88. Sasai Y, Kageyama R, Tagawa Y, Shigemoto R, Nakanishi S. Two mammalian helix-loop-helix factors structurally related to Drosophila hairy and Enhancer of split. Genes Dev. 1992;6(12B):2620-34
89. Sun J, Kamei CN, Layne MD, Jain MK, Liao JK, Lee ME, Chin MT. Regulation of myogenic terminal differentiation by the hairy-related transcription factor CHF2. J Biol Chem. 2001;276(21):18591-6
90. Polesskaya A, Seale P, Rudnicki MA. Wnt signaling induces the myogenic specification of resident CD45+ adult stem cells during muscle regeneration. Cell. 2003;113:841-52
91. Le Grand F, Rudnicki MA. Skeletal muscle satellite cells and adult myogenesis. Curr Opin Cell Biol. 2007;19(6):628-33
92. Armstrong D, Wong VL, Esser KA. Expression of beta-catenin is necessary for physiological growth of adult skeletal muscle. Am J Physiol Cell Physiol. 2006;291(1):C185-8
93. Brack A, Conboy IM, Conboy MJ, Shen J, Rando TA. A temporal switch from Notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell. 2008;2:50-9
94. Brack A, Murphy-Seiler F, Hanifi J, Deka J, Eyckerman S, Keller C, Aguet M, Rando TA. BCL9 is an essential component of canonical Wnt signaling that mediates the differentiation of myogenic progenitors during muscle regeneration. Dev Biol. 2009;335(1):93-105
95. Church V, Francis-West P. Wnt signalling during limb development. Int J Dev Biol. 2002;46(7):927-36
96. Ridgeway A, Petropoulos H, Wilton S, Skerjanc IS. Wnt signaling regulates the function of MyoD and myogenin. J Biol Chem. 2000;275(42):32398-405
97. Li L, Yuan H, Xie W, Mao J, Caruso AM, McMahon A, Sussman DJ, Wu D. Li L, Yuan H, Xie W, Mao J, Caruso AM, McMahon A, Sussman DJ, Wu D. J Biol Chem. 1999;274(1):129-34
98. Cossu G, Borello U. Wnt signaling and the activation of myogenesis in mammals. EMBO J. 1999;18(24):6867-72
99. Rochat A, Fernandez A, Vandromme M, Moles JP, Bouschet T, Carnac G, Lamb NJC. Insulin and Wnt1 pathways cooperate to induce reserve cell activaiton in differentiation and myotube hypertrophy. Mol Biology. 2004;15:4544-55
100. van der Velden J, Langen RC, Kelders MC, Wouters EF, Janssen-Heininger YM, Schols AM. Inhibition of glycogen synthase kinase-3beta activity is sufficient to stimulate myogenic differentiation. Am J Physiol Cell Physiol. 2006;290(2):C453-62
101. Vyas D, Spangenburg EE, Abraha TW, Childs TE, Booth FW. GSK-3beta negatively regulates skeletal myotube hypertrophy. Am J Physiol Cell Physiol. 2002;283(2):C545-C51
102. Descamps A, Arzouk H, Bacou F, Bernardi H, Fedon Y, Gay S, Reyne Y, Rossano B, Levin J. Inibition of myoblast differentiation by Sfrp1 and Sfrp2. Cell Tissu Res. 2008;332:299-306
103. Mermelstein C, Portilho DM, Mendes FA, Costa ML, Abreu JG. Wnt/beta-catenin pathway activation and myogenic differentiation are induced by cholesterol depletion. Differentiation. 2007;75(3):184-92
104. Hurlbut G, Kankel MW, Lake RJ, Artavanis-Tsakonas S. Crossing paths with Notch in the hyper-network. Curr Opin Cell Biol. 2007;19(2):166-75
105. Levin J, El Andalousi RA, Dainat J, Reyne Y, Bacou F. SFRP2 expression in rabbit myogenic progenitor cells and in adult skeletal muscles. J Muscle Res Cell Motil. 2001;22(4):361-9
106. Cheng X, Huber TL, Chen VC, Gadue P, Keller GM. Numb mediates the interaction between Wnt and Notch to modulate primitive erythropoietic specification from the hemangioblast. Development. 2008;135(20):3447-58
107. Nakamura T, Tsuchiya K, Watanabe M. Crosstalk between Wnt and Notch signaling in intestinal epithelial cell fate decision. J Gastroenterol. 2007;42(9):705-10
108. Aoyama K, Delaney C, Varnum-Finney B, Kohn AD, Moon RT, Bernstein ID. The interaction of the Wnt and Notch pathways modulates natural killer versus T cell differentiation. Stem Cells. 2007;25(10):2488-97
109. Ross D, Kadesch T. The notch intracellular domain can function as a coactivator for LEF-1. Mol Cell Biol. 2001;21(22):7537-44
110. Hofmann M, Schuster-Gossler K, Watabe-Rudolph M, Aulehla A, Herrmann BG, Gossler A. WNT signaling, in synergy with T/TBX6, controls Notch signaling by regulating Dll1 expression in the presomitic mesoderm of mouse embryos. Genes Dev. 2004;18(22):2712-7
111. Galceran J, Sustmann C, Hsu SC, Folberth S, Grosschedl R. LEF1-mediated regulation of Delta-like1 links Wnt and Notch signaling in somitogenesis. Genes Dev. 2004;18(22):2718-23
112. Duncan A, Rattis FM, DiMascio LN, Congdon KL, Pazianos G, Zhao C, Yoon K, Cook JM, Willert K, Gaiano N, Reya T. Duncan AW, Rattis FM, DiMascio LN, Congdon KL, Pazianos G, Zhao C, Yoon K, Cook JM, Willert K, Gaiano N, Reya T. Nat Immunol. 2005;6(3):314-22
113. Foltz D, Santiago MC, Berechid BE, Nye JS. Glycogen synthase kinase-3beta modulates notch signaling and stability. Curr Biol. 2002;12(12):1006-11
114. Espinosa L, Inglés-Esteve J, Aguilera C, Bigas A. Phosphorylation by glycogen synthase kinase-3 beta down-regulates Notch activity, a link for Notch and Wnt pathways. J Biol Chem. 2003;278(34):32227-35
115. De Strooper B, Annaert W. Where Notch and Wnt signaling meet. The presenilin hub. J Cell Biol. 2001;152(4):F17-20
116. Soriano S, Kang DE, Fu M, Pestell R, Chevallier N, Zheng H, Koo EH. Presenilin 1 negatively regulates beta-catenin/T cell factor/lymphoid enhancer factor-1 signaling independently of beta-amyloid precursor protein and notch processing. J Cell Biol. 2001;152(4):785-94
117. Armstrong R, Ogilvie RW, Schwane JA. Eccentric exercise-induced injury to rat skeletal muscle. Journal of Applied Physiology. 1983;54(1):80-93
118. Tsivitse S, McLoughlin TJ, Peterson JM, Mylona E, McGregor SJ, Pizza FX. Downhill running in rats: influence on neutrophils, macrophages, and MyoD+ cells in skeletal muscle. Eur J Physiol Occup Physiol. 2003;90:633-8
119. Eston R, Mickleborough J, Baltzopoulos V. Eccentric activation and muscle damage: biomechanical and physiological considerations during downhill running. Br J Sports Med. 1995;29(2):89-94
120. Kyparos A, Matziari C, Albani M, Arsos G, Sotiriadou S, Deligiannis A. A decrease in soleus muscle force generation in rats after downhill running. Can J Appl Physiol. 2001;26(4):323-35
121. Smith H, Plyley MJ, Rodgers CD, McKee NH. Skeletal muscle damage in the rat hindlimb following single or repeated daily bouts of downhill exercise. Int J Sports Med. 1997;18(2):94-100
122. Armand A, Launay T, Gapera BD, Charbonnier F, Gallien CL, Chanoine C. Effects of eccentric treadmill running on mouse soleus: degeneration/regeneration studied with Myf-5 and MyoD probes. Acta Physiol Scand. 2003;179:75-84
123. Darr K, Schultz E. Exercise-induced satellite cell activation in growing and mature skeletal muscle. J Appl Physiol. 1987;63(5):1816-21
124. Smith H, Plyley MJ, Rodgers CD, McKee NH. Expression of developmental myosin and morphological characteristics in adult rat skeletal muscle following exercise-induced injury. Eur J Physiol Occup Physiol. 1999;80(2):84-91
125. Smith H, Maxwell L, Rodgers CD, McKee NH, Plyley MJ. Exercise-enhanced satellite cell proliferation and new myonuclear accretion in rat skeletal muscle. J Appl Physiol. 2001;90:1407-14
126. Tiidus P, Deller M, Liu XL. Oestrogen influence on myogenic satellite cells following downhill running in male rats: a preliminary study. Acta Physiol Scand. 2005;184:67-72
127. Tsivitse S, Peters MG, Stoy AL, Mundy JA, Bowen RS. The effect of downhill running on Notch signaling in regenerating skeletal muscle. Eur J Appl Physiol. 2009;106(5):759-67
128. Parise G, McKinnell IW, Rudnicki MA. Muscle satellite cell and atypical myogenic progenitor response following exercise. Muscle Nerve. 2008;37(5):611-9
129. Boppart M, Burkin DJ, Kaufman SJ. Alpha7-Beta1-Integrin regulates mechanotransduction and prevents skeletal muscle injury. Am J Physiol Cell Physiol. 2006;290:C1660-C1665
130. Faulkner J, Jones DA, Round JM. Injury to skeletal muscles of mice by forced lengthening during contractions. Q J Exp Physiol. 1989;74(5):661-70
131. Koh T, Peterson JM, Pizza FX, Brooks SV. Passive stretches protect skeletal muscle of adult and old mice from lengthening contraction-induced injury. J Gerontol A Biol Sci Med Sci. 2003;58(7):592-7
132. McCully K, Faulkner JA. Injury to skeletal muscle fibers of mice following lengthening contractions. J Appl Physiol. 1985;59(1):119-26
133. Smith C, Stauber F, Waters C, Alway SE, Stauber WT. Transforming growth factor-beta following skeletal muscle strain injury in rats. J Appl Physiol. 2007;102(2):755-61
134. Mahoney D, Safdar A, Parise G, Melov S, Fu M, MacNeil L, Kaczor J, Payne ET, Tarnopolsky MA. Gene expression profiling in human skeletal muscle during recovery from eccentric exercise. Am J Physiol Regul Integr Comp Physiol. 2008;294(6):R1901-10
135. McKay B, O'Reilly CE, Phillips SM, Tarnopolsky MA, Parise G. Co-expression of IGF-1 family members with myogenic regulatory factors following acute damaging muscle-lengthening contractions in humans. J Physiol. 2008;586:5549-5560
136. Crameri R, Aagaard P, Qvortrup K, Langberg H, Olesen J, Kjaer M. Myofibre damage in human skeletal muscle: effects of electrical stimulation versus voluntary contraction. J Physiol. 2007;583:365-80
137. O'Reilly C, McKay B, Phillips S, Tarnopolsky M, Parise G. Hepatocyte growth factor (HGF) and the satellite cell response following muscle lengthening contractions in humans. Muscle Nerve. 2008;38(5):1434-42
138. Mikkelsen U, Langberg H, Helmark IC, Skovgaard D, Andersen LL, Kjaer M, Mackey AL. Local NSAID infusion inhibits satellite cell proliferation in human skeletal muscle after eccentric exercise. J Appl Physiol. 2009;107(5):1600-11
139. Pizza F, Peterson JM, Baas JH, Koh TJ. Neutrophils contribute to muscle injury and impair its resolution after lengthening contractions in mice. J Physiol. 2005;562(3):899-913
140. Pizza F, Koh TJ, McGregor SJ, Brooks SV. Muscle inflammatory cells after passive stretches, isometric contractions, and lengthening contractions. J Appl Physiol. 2002;92(5):1873-8
141. Timson B. Evaluation of animal models for the study of exercise-induced muscle enlargement. J Appl Physiol. 1990;69(6):1935-45
142. Phelan J, Gonyea WJ. Effect of radiation on satellite cell activity and protein expression in overloaded mammalian skeletal muscle. Anat Rec. 1997;247(2):179-88
143. Snow M. Satellite cell response in rat soleus muscle undergoing hypertrophy due to surgical ablation of synergists. Anat Rec. 1990;227(4):437-46
144. Tamaki T, Uchiyama Y, Okada Y, Tono K, Nitta M, Hoshi A, Akatsuka A. Multiple stimulations for muscle-nerve-blood vessel unit in compensatory hypertrophied skeletal muscle of rat surgical ablation model. Histochem Cell Biol. 2009;132(1):59-70
145. Marino J, Tausch BJ, Dearth CL, Manacci MV, McLoughlin TJ, Rakyta SJ, Linsenmayer MP, Pizza FX. Beta2-integrins contribute to skeletal muscle hypertrophy in mice. Am J Physiol Cell Physiol. 2008;295(4):C1026-36
146. Noble E, Tang Q, Taylor PB. Protein synthesis in compensatory hypertrophy of rat plantaris. Can J Physiol Pharmacol. 1984;62(9):1178-82
147. Carson J, Booth FW. Myogenin mRNA is elevated during rapid, slow, and maintenance phases of stretch-induced hypertrophy in chicken slow-tonic muscle. Pflugers Arch. 1998;435(6):850-8
148. Winchester P, Davis ME, Alway SE, Gonyea WJ. Satellite cell activation in the stretch-enlarged anterior latissimus dorsi muscle of the adult quail. Am J Physiol. 1991;260:C206-C12
149. Adams G, Caiozzo VJ, Haddad F, Baldwin KM. Cellular and molecular responses to increased skeletal muscle loading after irradiation. Am J Physiol Cell Physiol. 2002;283(4):C1182-95
150. Favier F, Benoit H, Freyssenet D. Cellular and molecular events controlling skeletal muscle mass in response to altered use. Pflugers Arch. 2008;456(3):587-600
151. Petrella J, Kim JS, Mayhew DL, Cross JM, Bamman MM. Potent myofiber hypertrophy during resistance training in humans is associated with satellite cell-mediated myonuclear addition: a cluster analysis. J Appl Physiol. 2008;104(6):1736-42
152. Ishido M, Kami K, Masuhara M. Localization of MyoD, myogenin and cell cycle regulatory factors in hypertrophying rat skeletal muscles. Acta Physiol Scand. 2004;180(3):281-9
153. Ishido M, Uda M, Kasuga N, Masuhara M. The expression patterns of Pax7 in satellite cells during overload-induced rat adult skeletal muscle hypertrophy. Acta Physiologica. 2008;195(4):459-69
154. Adams G, Haddad F, Baldwin KM. Time course of changes in markers of myogenesis in overloaded rat skeletal muscles. J Appl Physiol. 1999;87(5):1705-12
155. Tanaka Y, Yamaguchi A, Fujikawa T, Sakuma K, Morita I, Ishii K. Expression of mRNA for specific fibroblast growth factors associates with that of the myogenic markers MyoD and proliferating cell nuclear antigen in regenerating and overloaded rat plantaris muscle. Acta Physiologica. 2008;194(2):149-59
156. Lovering R, Roche JA, Bloch RJ, De Deyne PG. Recovery of function in skeletal muscle following 2 different contraction-induced injuries. Arch Phys Med Rehabil. 2007;88(5):617-25
157. Rathbone C, Wenke JC, Warren GL, Armstrong RB. Importance of satellite cells in the strength recovery after eccentric contraction-induced muscle injury. Am J Physiol Cell Physiol. 2003;285:R1490-R1495
158. Wozniak A, Kong J, Bock E, Pilipowicz O, Anderson JE. Signaling satellite-cell activation in skeletal muscle: markers, models, stretch, and potential alternate pathways. Muscle & Nerve. 2004;31:283-300
159. McCarthy J, Esser KA. Counterpoint: Satellite cell addition is not obligatory for skeletal muscle hypertrophy. J Appl Physiol. 2007;103(3):1102-3
160. Mozdziak P, Greaser ML, Schultz E. Myogenin, MyoD, and myosin expression after pharmacologically and surgically induced hypertrophy. J Appl Physiol. 1998;84(4):1359-64
161. DeVol D, Rotwein P, Sadow JL, Novakofski J, Bechtel PJ. Activation of insulin-like growth factor gene expression during work-induced skeletal muscle growth. Am J Physiol. 1990;259:E89-E95
162. Barton-Davis E, Shoturma DI, Sweeney HL. Contribution of satellite cells to IGF-I induced hypertrophy of skeletal muscle. Acta Physiol Scand. 1999;167(4):301-5
163. Glass D. Skeletal muscle hypertrophy and atrophy signaling pathways. Int J Biochem Cell Biol. 2005;37(10):1974-84
164. Nader G. Molecular determinants of skeletal muscle mass: getting the "AKT" together. Int J Biochem Cell Biol. 2005;37(10):1985-96
165. Adams G, McCue SA. Localized infusion of IGF-I results in skeletal muscle hypertrophy in rats. J Appl Physiol. 1998;84(5):1716-22
166. Musarò A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, Barton ER, Sweeney HL, Rosenthal N. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet. 2001;27(2):195-200
167. Lai K, Gonzalez M, Poueymirou WT, Kline WO, Na E, Zlotchenko E, Stitt TN, Economides AN, Yancopoulos GD, Glass DJ. Conditional activation of akt in adult skeletal muscle induces rapid hypertrophy. Mol Cell Biol. 2004;24(21):9295-304
168. O'Connor R, Pavlath GK. Point:Counterpoint: Satellite cell addition is/is not obligatory for skeletal muscle hypertrophy. J Appl Physiol. 2007;103(3):1102-3
169. Rehfedt C, Mantilla CB, Sieck GC, Hikida RS, Booth FW, Kadi F, Bodine SC, Lowe DA. In response to Point:Counterpoint: "Satellite cell addition is/is not obligatory for skeletal muscle hypertrophy. Journal of Applied Physiology. 2007;103(3):1104-6
170. Carey K, Farnfield MM, Tarquinio SD, Cameron-Smith D. Impaired expression of notch signaling genes in aged human skeletal muscle. J Gerontoloty Biol Sci A. 2007;62(1):9-17
171. Akiho M, Nakashima H, Sakata M, Yamasa Y, Yamaguchi A, Sakuma K. Expression profile of Notch-1 in mechanically overloaded plantaris muscle of mice. Life Sci. 2009;86(1-2):59-65
172. Armstrong D, Esser KA. Wnt/beta-catenin signaling activates growth-control genes during overload-induced skeletal muscle hypertrophy. Am J Physiol Cell Physiol. 2005;289(4):C853-C859
173. Sakamoto K, Arnolds DE, Ekberg I, Thorell A, Goodyear LJ. Exercise regulates Akt and glycogen synthase kinase-3 activities in human skeletal muscle. Biochem Biophys Res Commun. 2004;319(2):419-25
174. Aschenbach W, Ho RC, Sakamoto K, Fujii N, Li Y, Kim YB, Hirshman MF, Goodyear LJ. Regulation of dishevelled and beta-catenin in rat skeletal muscle: an alternative exercise-induced GSK-3beta signaling pathway. Am J Physiol Endocrinol Metab. 2006;291(1):E152-8
175. Ishido M, Uda M, Masuhara M, Kami K. Alterations of M-cadherin, neural cell adhesion molecule and Beta-catenin expression in satellite cells during overload-induced skeletal muscle hypertrophy. Acta Physiol Scand. 2006;187:407-18
Author contact
![]() Corresponding author: Dr. Susan Tsivitse, Department of Kinesiology, UNC Charlotte, 9201 University City Blvd., Charlotte, NC 28223. Telephone: 704-687-2731; FAX: 704-687-3350; Email: susantsivitseedu
Corresponding author: Dr. Susan Tsivitse, Department of Kinesiology, UNC Charlotte, 9201 University City Blvd., Charlotte, NC 28223. Telephone: 704-687-2731; FAX: 704-687-3350; Email: susantsivitseedu
Received 2010-4-1
Accepted 2010-5-13
Published 2010-5-15