ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2010; 6(6):546-555. doi:10.7150/ijbs.6.546 This issue Cite
Research Paper
Myocardial deletion of Smad4 using a novel α skeletal muscle actin Cre recombinase transgenic mouse causes misalignment of the cardiac outflow tract
Mohamad Azhar1* ![]() , Pei-Yu Wang2,3*, Tony Frugier2,4, Kyoko Koishi2, Chuxia Deng5, Peter G. Noakes6, Ian S. McLennan2
, Pei-Yu Wang2,3*, Tony Frugier2,4, Kyoko Koishi2, Chuxia Deng5, Peter G. Noakes6, Ian S. McLennan2
1. BIO5 Institute, and Department of Cell Biology and Anatomy, University of Arizona, Tucson, AZ 85724, USA;
2. Department of Anatomy & Structural Biology, Brain Health and Repair Research Centre, University of Otago, New Zealand;
3. Institute of Neuroscience and Research Center for Mind, Brain and Learning, National Cheng-Chi University, Taipei 116, Taiwan;
4. National Trauma Research Institute, The Alfred Hospital and Medicine Department, Monash University, Melbourne, VIC 3004, Australia;
5. GDDB, NIDDK, National Institutes of Health, Bethesda, MD 20892, USA;
6. School of Biomedical Sciences, Queensland Brain Institute, University of Queensland, Queensland 4072. Australia.
* Equal contribution
Abstract
SMAD4 acts as the converging point for TGFβ and BMP signaling in heart development. Here, we investigated the role of SMAD4 in heart development using a novel α skeletal muscle actin Cre recombinase (MuCre) transgenic mouse strain. Lineage tracing using MuCre/ROSA26LacZ reporter mice indicated strong Cre-recombinase expression in developing and adult heart and skeletal muscles. In heart development, significant MuCre expression was noted at E11.5 in the atrial, ventricular, outflow tract and atrioventricular canal myocardium, but not in the endocardial cushions. MuCre-driven conditional deletion of Smad4 in mice caused double outlet right ventricle (DORV), ventricular septal defect (VSD), impaired trabeculation and thinning of ventricular myocardium, and mid-gestational embryonic lethality. In conclusion, MuCre mice effectively delete genes in both heart and skeletal muscles, thus enabling the discovery that myocardial Smad4 deletion causes misalignment of the outflow tract and DORV.
Keywords: heart, myogenesis, transforming growth factor beta, SMAD, Marfan syndrome.
Introduction
Congenital cardiac malformations are the most common life-threatening birth defects [1]. Most major malformations are detected at birth, but less severe defects can go undetected, leading to cardiac disease later in life [2]. In mice, the development of the heart begins with the formation of the cardiac tube, which soon undergoes dextral looping and other morphogenic events to form the heart chambers [3]. During this process, the ventricles and atria become partially separated by the formation of atrioventricular cushions. The valves and septa arise from these cushions, through inductive influences from the underlying myocardium [4-6].
Transforming growth factor β (TGFβ) and bone morphogenic protein (BMP) ligands and/or their receptors are expressed by myocardium, endocardium, cushion mesenchyme, epicardium, cardiac neural crest cells and second heart field cardiac progenitor cells [7-9]. Mice with gene targeted mutation in TGFβ or BMP pathways exhibit multiple cardiovascular developmental defects, including valvuloseptal septal defects [10-20]. These malformations are consistent with myocardial TGFβ2 and BMP2/4 acting on the overlying cushion to promote an epithelial to mesenchymal transition of a distinct group of endocardial cells [21,22].
The TGFβs and BMPs are members of the TGFβ superfamily of proteins, which signal through a complex consisting of type I and II receptors. Signal transduction is mainly through the SMAD pathway [23], although other pathways such as PI3K-Akt and Ras-Raf have been implicated in some physiological events [24]. There are 8 SMADs, which can be sub-divided into three functional groups. The receptor-mediated SMADs (SMAD1, 2, 3, 5, 8) are directly phosphorylated by the TGFβ receptors, and then translocated to the nucleus, after binding to the single co-mediator SMAD, SMAD4 [24]. The inhibitory SMADs (SMAD6, 7) reduce signaling by preventing activation of the receptor mediated-SMADs [23]. SMAD6 appears to be specific for BMP signaling, whereas SMAD7 can reduce either BMP or TGFβ based signaling pathways [25-27].
The role of the single co-mediator SMAD, Smad4, in the myocardium is unclear due to variations in the timing and efficiency of previous myocardial-specific deletions of Smad4 [28-30]. Smad4 deficient embryos die shortly after implantation [31], but the broad actions of SMAD4 can be investigated through cell-specific deletions [32-35]. We report here that a broad myocardial conditional deletion of Smad4 produces a double-outlet right ventricle (DORV), as well as other cardiac malformations. This suggests that myocardial-SMAD4 has a more extensive role in cardiac morphogenesis than currently reported. The novel alpha skeletal muscle actin Cre transgenic mouse (MuCre) used in this study produces early, rapid and robust cleavage of floxed genes, making it a valuable tool for the study of both cardiac and skeletal muscle formation.
Materials and Methods
Animals
The University of Otago's Animal Ethics Committee approved all experiments. Mice were bred and maintained in M.I.C.E.™ cages and their food sterilized by gamma irradiation. The room had a 14h white light / 10h dark-sodium light phase [35]. Floxed-Smad4 (Smad4flox/flox) [36] and ROSA26LacZ [37] mice were bred at the University of Otago.
Generation of MuCre mice
The MuCre mice carried a transgene consisting of a human alpha skeletal muscle actin promoter [38] and the bacteriophage P1 Cre recombinase gene [39]. The DNA fragments containing the α-skeletal muscle actin promoter and the Cre recombinase gene were isolated from the original clones, integrated into a new cloning vector using standard techniques. The construct was verified by nucleotide sequencing. The transcription unit was then isolated by cleavage of the clone with restriction endonucleases, preparative agarose gel electrophoresis and a DNA isolation kit (QIAGEN). It was then injected into C57BL/6 zygotes (TASQ, University of Queensland, St Lucia, Australia) and the MuCre line was selected from 14 founders, as it had the highest level of Cre recombinase expression. Two different Cre transgenic lines (MuCreA and MuCreB) were generated and characterised. These lines were maintained by breeding transgenic mice with C57BL/6 mice. MuCre refers to the A line, unless otherwise stated. The mice are available from the Riken Bioresource Centre (#RBRC01386, MuCre-A).
The genotype of the mice was determined from ear biopsies, using polymerase chain reaction (PCR), as previously described [40]. The primers used were: Floxed Smad4, 5'-GGGCAGCGTAGCATATAAGA-3' and 5'-TGACCCAAACGTCACCTTCA-3'; ROSA26LacZ, 5'-GTTGCAGTGCACGGCAGATACACTTGCTGA-3' and 5'-GCCACTGGTGTGGGCCATAATTCAATTCGC-3'; MuCre, 5'-GTTGATGCCGGTGAACGTGCAAA-3' and 5'-ATCAGCTACACCAGAGACGGAAA-3'.
The tissue-specificity of Cre expression was examined by quantitative (q) PCR. Total RNA was extracted from the brain, spinal cord, kidney, liver, gastrocnemius muscle and heart using TRIzol reagent, according to the manufacturer's instructions (Invitrogen, Carlsbad, Ca, USA). The total RNA fractions of each sample were DNase treated to remove possible contamination of genomic DNA and then reverse transcribed using SuperScript II Rnase H- reverse transcriptase (Invitrogen) and oligo d(T)15 as primer. Cre cDNA were amplified using primers CreER-F2 (5'-GTT GAT GCC GGT GAA CGT GCA AA-3') and CreER-R1 (5'-ATC AGC TAC ACC AGA GAC GGA AA-3'). GAPDH cDNA were co-amplified using primers GAPDH-forward (5'-CTT CAT TGA CCT CAA CTA-3') and GAPDH-reverse (5'-TTC ACA CCC ATC ACA AAC-3') and used as internal control. Real-time PCR were performed using SYBR Green 1 dye (Invitrogen). Measurements were realized and analyzed by using the ABI Prism 7700 sequence detection system (Applied Biosystems, Foster City, CA, USA).
The characteristics of the MuCre mice were also verified by breeding them with LacZ/alkaline phosphatase (Z-AP) double reporter and ROSA26LacZ reporter mice [37, 41]. In the Z-AP mice, the action of Cre recombinase terminates the production of LacZ and induces alkaline phosphatase (AP) [41]. Successful cleavage is thus indicated by both the loss of LacZ stain and the induction of AP expression.
Embryos were produced at E11.5, E14.5 and E16.5 from time-mated pregnancies, with the date of the vaginal plug designated as E0.5. A proportion of these were stained in situ for LacZ. The dams were killed, the embryos removed and fixed in a solution containing 0.2% glutaraldehyde, 5mM EGTA, 2mM MgCl2 in phosphate-buffered saline (PBS) for 10 minutes at 4°C. The embryos were washed three times in PBS containing 2mM MgCl2 and 1% NP40, and incubated in a solution containing 2.45mM X-Gal, 2mM MgCl2, 20mM K3Fe(CN)6, 20mM K4Fe(CN)6 in PBS for 3 hours. After washing three times in PBS, the embryos were fixed again overnight at 4°C, embedded in Technovit 7100 resin and serially-sectioned at 15µm.
Tissues from embryos and adults were snap frozen in melting isopentane, and sectioned using a cryostat. LacZ staining of the section was as previously described [42]. For alkaline phosphatase (AP) staining, the sections were fixed with 2% paraformaldehyde in 0.1M sodium phosphate buffer, pH 7.4, washed in AP buffer (100mM Tris-HCl pH 9.5, 100mM NaCl and 5mM MgCl2) for 2 minutes and incubated in 318µg/ml nitroblue tetrazolium, 108µg/ml 5-bromo-4-chloro-3-indolyl phosphate, para-toluidium salt and 102µg/ml levamisole in AP buffer for 10 minutes.
The Cre recombinase-ERT contains a tamoxifen-sensitive estrogen receptor domain, which is designed to hold the Cre recombinase in the cytoplasm of the cell, unless tamoxifen is present [39]. This enables temporal control of the cleavage of target sequences, but this strategy is only effective in some mouse lines [43]. The MuCre mice rapidly cleaved target sequences in the absence of tamoxifen, indicating that the Cre recombinase enters the nucleus independent of tamoxifen. The data presented here are therefore from mice that did not receive tamoxifen.
Assessment of cardiac malformations
The embryos produced by crossing MuCre/Smad4flox/+ and Smad4flox/flox mice were fixed in 4% paraformaldehyde in 0.1M phosphate buffer, pH7.4. The hearts from five MuCre/Smad4flox/flox and six control mice were embedded in wax, serially sectioned at 5µm and stained with hematoxylin and eosin for the histological and morphometric examination.
Results
MuCre mice have cardiac and skeletal muscle expression of Cre recombinase
Muscle-specific Cre recombinase transgenic mouse lines (MuCre) were generated using an α skeletal muscle actin promoter. The endogenous α-skeletal muscle actin promoter is skeletal-muscle specific, but when used in transgenic mice it drives expression in the heart, in a minority of mouse lines [38]. Consistent with this, the hearts of adult MuCre mice had levels of Cre recombinase mRNA that varied from 0.1% (line B) to 40% (line A, MuCrA, hereafter referred to as MuCre) of the level in skeletal muscle. Cre recombinase mRNA was not detected in the brain, spinal cord, liver or kidneys of the MuCre mice. The MuCre mice alone did not exhibit a phenotype.
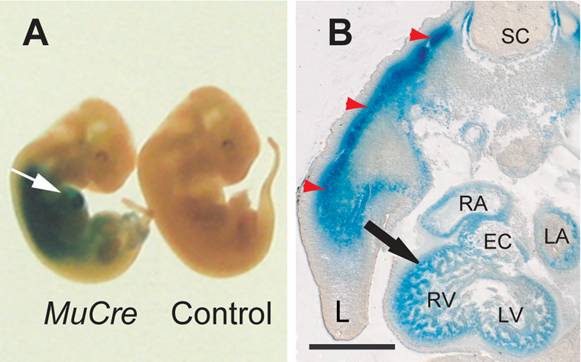
The cells expressing the Cre recombinase were examined by crossing the MuCre mice with a ROSA26LacZ and/or the Z-AP reporter mice, with the observed pattern of stain being consistent with the mRNA data. In whole mount E11.5 MuCre/ROSA26LacZ embryos, the LacZ reporter staining was detected in the developing heart and skeletal muscles (Fig. 1A). Sectioning of the heart revealed strong staining in the myocardium of atria, ventricles and outflow tract (outflow tract) and atrioventricular (AV) canal myocardium, but not in the endocardial cushions (Fig. 1B). Skeletal myotubes were also stained (red arrows in Fig. 1B).
MuCre-driven expression of ROSA26LacZ reporter gene in developing skeletal and cardiac muscle cells. A: Whole mount LacZ staining in E11.5 MuCre/ROSA26LacZ and a littermate non-transgenic control fetuses. The white arrow points to LacZ stain in the heart. B: The MuCre/ROSA26LacZ fetuses were stained for LacZ, embedded in Technovit resin and serially sectioned. The LacZ stain was present in cardiac myocytes (black arrow) and skeletal muscle myotubes (red arrowheads) of the developing limb (L). EC, endocardial cushion; LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle. The scale bar represents 500 µm.
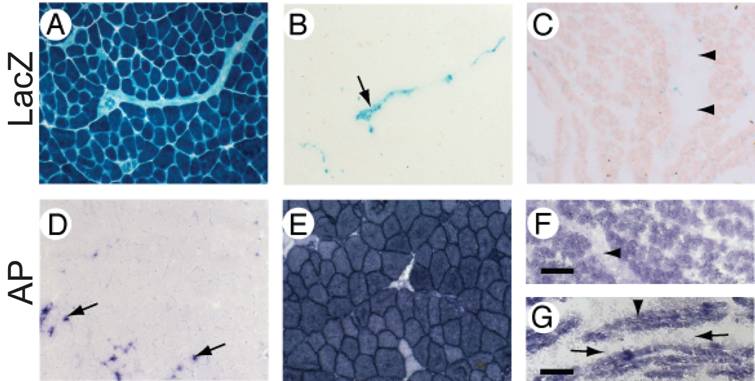
MuCre-driven expression of the Z-AP reporter genes in skeletal muscle myotubes and fibers. With the Z-AP reporter, Cre activity is detected by the expression of alkaline phosphatase (AP) and the cessation of LacZ expression. A, D: Sections of skeletal muscle from an adult non-transgenic Z-AP reporter mouse. All muscle fibres were LacZ-positive and AP-negative (A and D respectively). The expression of AP was restricted to resident macrophages (arrows in D). B, C, E, F, G: Sections of skeletal muscle from adult (B, E), neonatal (C, F) or E16.5 (G) MuCre/Z-AP mice. Activation of Cre was limited to the myogenic cells, which were AP-positive (E, F and G) and LacZ-negative (B and C). The non-myogenic cells in the muscle lack Cre expression. For example, the arrow in (B) points to an intramuscular nerve, whereas the arrowheads in (C, F) point to an intramuscular tendon, which had minimal expression of reporter genes. The AP-positive myotubes in (G) are indicated by an arrowhead, whereas the AP-negative mononucleated cells are indicated by arrows. It is also noteworthy that the AP staining of muscle fibers in (E) is specific because AP staining is absent in the negative control (D). The scale bar = 50µm (A-F) and 30 µm (G).
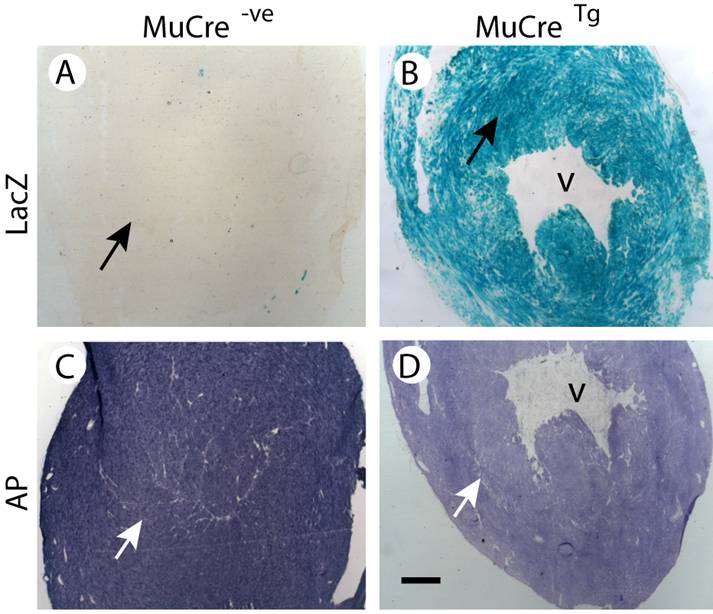
The tissue-specific expression of the MuCre mice was then examined using cryostat sections of MuCre/Z-AP mice, with Cre recombinase expression being detected by the induction of alkaline phosphatase (AP) and the cessation of LacZ expression. In control non-transgenic Z-AP mice all cells were LacZ positive and AP negative (compare Fig 2A with 2D). By contrast, the skeletal muscles of the MuCre/Z-AP mice, the muscle fibers were LacZ negative (Fig. 2B) and AP positive (Fig. 2E), indicating that all muscle fibers had produced high levels of Cre recombinase. The interstitial cells of the muscles, including neural crest-derived Schwann cells (arrow in Fig. 2B) were LacZ positive. The expression of AP was limited to a small population of resident macrophages that naturally produce AP (arrows in Fig. 2D). The complete removal of LacZ and the induction of AP were also observed in all myotubes (immature muscle fibers) of E14.5, E16.5 and newborn MuCre/Z-AP mice (compare Fig. 2C with 2F). The myoblasts and other mononucleated cells were negative for AP (arrows in Fig. 2G). This indicates that the cleavage of target DNA in the myotubes was very rapid, as many of these immature muscle fibers were only a few hours old. All cardiac myocytes of the adult MuCre/Z-AP mice were AP negative (Fig. 3D) and LacZ positive (Fig. 3B). By contrast, no cleavage of the reporter construct was observed in control hearts, with the myocytes being LacZ negative (Fig. 3A). No cleavage of the reporter gene was observed in other tissues, including the brain, spinal cord, liver and intestines. In summary, the MuCre mouse line specifically express Cre recombinase in cardiac and skeletal muscles. The level of expression of the Cre recombinase in both these muscle types is high leading to extremely rapid and robust cleavage of target sequences.
Muscle-specific deletion of Smad4 leads to mid-gestational lethality
MuCre/Smad4+/flox mice were crossed with Smad4flox/flox mice to produce MuCre/Smad4flox/flox (hereafter referred to as myocardial Smad4 conditional knockout) mice. Sixty 10-day-old pups were produced from 14 litters. The data indicated that myocardial Smad4 conditional knockout died in utero (Table 1). The fetuses from 19 time-mated litters were therefore collected. Myocardial Smad4 conditional knockout fetuses were present at the Mendelian ratio (32/125, Table 1), but most of the conditional knockout fetuses older than 13-14 days of gestation were dead in utero (Table 1). The death of the myocardial Smad4 conditional knockout fetuses was independent of whether the MuCre transgene was carried by the dam or by the stud.
Significant mid-gestational embryonic lethality in myocardial Smad4 conditional knockout mice.
| Genotypes | E12.5-13.5 | E14.5 | E18.5 | P10 |
|---|---|---|---|---|
| Smad4flox/+ (control) | 6 (14%) | 12 (35%) | 12 (24%) | 18 (30%) |
| Smad4flox/flox (control) | 13 (30%) | 8 (24%) | 14 (28%) | 16 (27%) |
| MuCre/Smad4flox/+ | 10 (23%) | 6 (18%) | 12 (24%) | 26 (43%) |
| MuCre/Smad4flox/flox | 14 (33%) | 2 [6] (6, 18%) | 2 [8] (4, 16%) | 0 (0%) |
| Number of fetuses/mice | 43 | 34 | 50 | 60 |
| Number of litter | 5 | 5 | 6 | 10 |
MuCre/Smad4flox/+ and non-transgenic Smad4flox/flox mice were mated and their offspring genotyped. The numbers of dead fetuses are indicated in square brackets. Note the significant decrease in the number of live myocardial Smad4 conditional knockout embryos between E13.5 and E14.5 indicates a mid-gestational lethality.
Loss of Smad4 in myocardium causes defects in cardiac morphogenesis
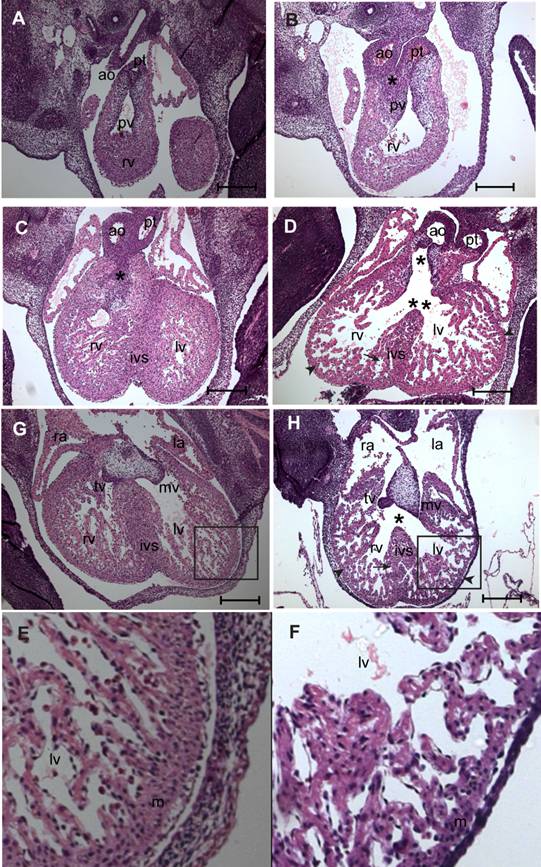
Histological examination by hematoxylin and eosin (H&E) staining was done on serial sections of E11.5-13.5 fetuses of myocardial Smad4 conditional knockout (n = 7) and the littermate MuCre non-transgenic Smad4flox/flox control embryos (n = 5) to determine why the fetuses with myocardial Smad4 deletion die. At E11.5, the transgene was strongly expressed (Fig. 1), but all of the fetuses were still viable (Table 1). The control fetuses had a normal cardiac outflow tract and AV septation and alignment, compact ventricular myocardium, and the endocardial cushions (Fig. 4A,C,E,G). By contrast, myocardial Smad4 conditional knockout fetuses exhibited cardiac defects, including double-outlet right ventricle (DORV) (Fig. 4D,H), ventricular septal defect (VSD) (Fig. 4D,H), decreased density of trabeculae in the ventricles and thinning of the compact layer of the ventricular myocardium (Fig. 4B,D,H,F). DORV was seen with the aortic orifice being in a side-by-side position to the pulmonary orifice (Fig. 4B). In control embryos, tissue between aortic valve and right ventricular outflow tract became myocardial structure (Fig. 4C). By contrast, only a small fibrous ridge separated the aortic valve from the right ventricular outflow tract in myocardial Smad4 conditional knockout fetuses (Fig. 4D). This can be attributed to the fact that the outlet cushions are proximally not fused in the conditional knockout embryos (Fig. 4D). The cardiac phenotype was the same in all embryos examined, indicating that the penetrance of the genetic modification is very high.
In the control E13.5 embryos, the fusion of mesenchyme at the base of the atrial septum and AV cushions nearly completed the inlet septation. However, in the myocardial Smad4 conditional knockout embryos, there was a large perimembranous inlet ventricular septal defect (VSD) due to absence of mesenchyme in the region where outflow tract and the AV cushions should have met (Fig. 4H). The VSD was more closely related to the aortic orifice in the conditional knockout embryo (Fig. 4D). It is also noteworthy that the bifurcated outflow tract is in the right ventricle and not over the ventricular septum (Fig. 4D). The size and the cellularity of endocardial cushions were normal, but their appearance was slightly abnormal (Fig. 4H), which we attributed to the impaired cardiac alignment process in the conditional mutants.
MuCre-driven expression of the Z-AP reporter genes in adult cardiac myocytes. With the Z-AP reporter, Cre activity is detected by the expression of alkaline phosphatase (AP) and the cessation of LacZ expression. A,C: The hearts from control non-transgenic Z-AP reporter mice were LacZ negative and had high levels of AP. B,D: All myocytes in the hearts of MuCre/Z-AP mice were LacZ positive (B) and only had low endogenous AP expression (D). The black (A, B) and the white arrows in (C,D) point to myocytes. V, ventricle. The bar represents 500 µm.
SMAD4 is essential for cardiac morphogenesis. A-F: Light micrographs of serial sections of E13.5 non-transgenic control (A, C, G, E) or MuCre/Smad4flox/flox (B, D, H, F) fetuses stained with haematoxylin and eosin. Asterisk (*) in B shows an abnormal side-by-side alignment of aorta (ao) and pulmonary trunk (pt) leading to the DORV. (D) Asterisk (*) in D indicates missing right ventricular outflow tract septum and a fully developed double-outlet right ventricle (DORV) in the myocardial Smad4 conditional knockout embryo. A clear ventricular septal defect is seen in D (double asterisks) H (asterisk) in the myocardial Smad4 conditional knockout embryo. Note that the DORV is positioned in the right ventricle and not over the ventricular septum (double asterisk in D). Impaired myocardial trabeculation and myocardial hypoplasia in the interventricular septum (arrow in D and H), and thinning of the compact layer of the ventricular myocardial walls (D, H, F) (arrowhead in D & H) is found in the myocardial Smad4 conditional knockout. E-F are 100% zoomed or enlarged images of the boxed regions in G & H, respectively. pv, pulmonary valves; rv, right ventricle; lv, left ventricle; ra, right atrium; la, left atrium; ivs, interventricular septum; mv, mitral valves; tv, tricuspid valves. The scale bars = 200 µm (A-H).
Discussion
This study provides clear evidence that muscle-specific deletion of Smad4 leads to defective alignment but not septation of the aortic and pulmonary orifices, resulting in DORV. Interestingly, cardiac phenotypes of myocardial Smad4 conditional knockout mice shares many similarities to the cardiac phenotypes observed in Tgfb2-/-, and TGFβ receptors and Bmp2, Bmp4 and BMP receptors conditional knockout mice [10,12-20,45]. Both TGFβ and BMP ligands play important roles in cardiac morphogenesis by properly recruiting second-heart field progenitors to the outflow tract and to the ventricular myocardium to the developing heart [45,46]. The data suggest a unique requirement of myocardium-produced SMAD4 in the outflow tract alignment since endothelial or cardiac neural crest cell-specific deletion of Smad4 leads to defects in cushion formation or outflow tract septation but not in misalignment of the outflow tract [47,48].
The cardiac phenotype of the MuCre/Smad4flox/flox mice is more extensive than previous studies of myocyte-specific deletion of Smad4, which have used different promoters to drive the Cre recombinase [28-30]. The Smad4flox/flox mice used in these studies are from a common lineage [36], suggesting that the difference in the phenotype relates to the characteristics of the promoters. Alternatively, the phenotypes of mice with TGFβs mutations are influenced by their genetic background [49-51] and by environmental factors [52], and SMAD4 biology could thus be subject to similar regulation.
The development of the heart can proceed normally with attenuated growth factor signaling, as evidenced by the normality of MuCre/Smad4flox/+ mice (this study) and mice with single gene haploinsufficiency for Tgfb2, Bmp2 or Bmp4 [10,12,13,53]. The success of conditional genetic manipulation is thus dependent on early and rapid removal of both alleles of the target gene. Similarly, it may be important for a high percentage of cells to be affected, as a minority of normal cells may be sufficient to drive some morphogenetic events. The MuCre mice effectively achieve these criteria, as evidenced by the speed and completion of removal of floxed genes in the ROSA26LacZ and Z-AP reporter mice, and by the severity of the phenotype of MuCre/Smad4flox/flox mice.
The cardiac malformations in MuCre/Smad4flox/flox mice are concordant with other recent studies of myocyte-specific deletion of Smad4, which have used αSMACre and cTntCre mice [29,30], except for DORV, which is unique to the MuCre/Smad4flox/flox mice. The inductive events that underlie DORV precede those that underlie the other malformations, and presence of DORV in the MuCre/Smad4flox/flox may be because the MuCre mice drive early and possibly more robust removal of floxed genes as compared to the αSMACre driver mouse lines. Although cTnt-Cre mice were effective in robustly deleting Smad4 in early myocardial progenitors at E7.5, it was not possible to determine DORV in cTnt-Cre/Smad4flox/flox mice due to their death before E12.5. Many studies suggested that an interaction between cardiac neural crest cells and second-heart field-derived myocardium play an important role in outflow tract septation and alignment [45]. Since the current study shows that outflow tract is correctly septated but improperly aligned, it is possible that a defective interaction between cardiac neural crest and second-heart field-derived outflow tract myocardium is involved in the formation of DORV in myocardial Smad4 conditional knockout embryos.
Alpha-myosin heavy chain αMHCCre/Smad4flox/flox mice exhibit normal heart development, which is in marked contrast to the MuCre, αSMACre, cTntCre equivalents. αMHCCre mice are commonly used to study cardiac biology, and have proven utility for the study of adult function [41,54]. Their use for the study of cardiac development has yielded conflicting results and the expression of Cre in αMHCCre mice may be insufficiently strong or not early to affect all aspects of myocardial development [28,30,55]. While it is difficult to draw any parallel between MuCre and αMHCCre expression, it is clear that MuCre mice effectively delete genes in myocardium during early cardiac development and in the adulthood.
The MuCre mice were initially developed to study skeletal muscle formation, and they provide rapid and complete removal of floxed genes from myotubes. The MuCre mice may therefore be useful for the study of skeletal myogenesis, provided the target gene is not expressed in the heart. The dual skeletal and cardiac effects of MuCre mice are less problematical for the study of the heart, as embryos develop to term in the complete absence of skeletal muscle [44].
In summary, we report the generation of a new line of α-skeletal-actin Cre-recombinase transgenic mice, which should facilitate the study of cardiac and skeletal myogenesis. The value of these mice is evidenced by their use to provide new insight into the role of Smad4 in outflow tract morphogenesis processes during heart development.
Acknowledgements
The skilled technical assistance of Mrs. Batchelor is gratefully acknowledged. Dr Edna C. Hardeman and Prof. Pierre Chambon are thanked for, respectively, gifting the α-skeletal actin promoter and Cre recombinase-ERT gene. This work was supported by the Marsden Fund Royal Society of New Zealand ( I.S.M.), The Arizona Biomedical Research Commission (M.A.), The Steven M. Gootter Foundation (M.A.), The Stephen Michael Schneider Investigator Award for Pediatric Cardiovascular Research (M.A.), The William J. "Billy" Gieszl Endowment for Heart Research (M. A.), and R01 HL92508 (M.A.), and NHMRC Australia (PGN).
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Pierpont ME, Basson CT, Benson DW Jr. et al. Genetic basis for congenital heart defects current knowledge a scientific statement from the American Heart Association Congenital Cardiac Defects Committee Council on Cardiovascular Disease in the Young endorsed by the American Academy of Pediatrics. Circulation. 2007;115:3015-3038
2. Markwald RR, Norris RA, Moreno-Rodriguez R, Levine RA. Developmental basis of adult cardiovascular diseases valvular heart diseases. Ann N Y Acad Sci. 2010;1188:177-83
3. Gittenberger-de Groot AC, Bartelings MM, DeRuiter MC, Poelmann RE. Basics of cardiac development for the understanding of congenital heart malformations. Pediatr Res. 2005;57:169-176
4. Person AD, Klewer SE, Runyan RB. Cell biology of cardiac cushion development. Int Rev Cytol. 2005;243:287-335
5. Barnett JV, Desgrosellier JS. Early events in valvulogenesis a signaling perspective. Birth Defects Res C Embryo Today. 2003;69:58-72
6. Combs MD, Yutzey KE. Heart valve development regulatory networks in development and disease. Circ Res. 2009;105:408-421
7. Azhar M, Schultz JE, Grupp I. et al. Transforming growth factor beta in cardiovascular development and function. Cytokine Growth Factor Rev. 2003;14:391-407
8. Delot EC. Control of endocardial cushion and cardiac valve maturation by BMP signaling pathways. Mol Genet Metab. 2003;80:27-35
9. Molin DG, Bartram U, Van der HK. et al. Expression patterns of Tgfbeta1-3 associate with myocardialisation of the outflow tract and the development of the epicardium and the fibrous heart skeleton. Dev Dyn. 2003;227:431-444
10. Azhar M, Runyan RB, Gard C. et al. Ligand-specific function of transforming growth factor beta in epithelial-mesenchymal transition in heart development. Dev Dyn. 2009;238:431-442
11. Compton LA, Potash DA, Brown CB, Barnett JV. Coronary vessel development is dependent on the type III transforming growth factor beta receptor. Circ Res. 2007;101:784-791
12. Bartram U, Molin DG, Wisse LJ. et al. Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping myocardialization endocardial cushion differentiation and apoptosis in Tgfb2 knockout mice. Circulation. 2001;103:2745-2752
13. Uchimura T, Komatsu Y, Tanaka M. et al. Bmp2 and Bmp4 genetically interact to support multiple aspects of mouse development including functional heart development. Genesis. 2009;47:374-384
14. Beppu H, Malhotra R, Beppu Y. et al. BMP type II receptor regulates positioning of outflow tract and remodeling of atrioventricular cushion during cardiogenesis. Dev Biol. 2009;331:167-175
15. Choudhary B, Ito Y, Makita T. et al. Cardiovascular malformations with normal smooth muscle differentiation in neural crest-specific type II TGFbeta receptor (Tgfbr2) mutant mice. Dev Biol. 2006;289:420-429
16. Sridurongrit S, Larsson J, Schwartz R. et al. Signaling via the Tgf-beta type I receptor Alk5 in heart development. Dev Biol. 2008;322:208-218
17. Wang J, Sridurongrit S, Dudas M. et al. Atrioventricular cushion transformation is mediated by ALK2 in the developing mouse heart. Dev Biol. 2005;286:299-310
18. Ma L, Lu MF, Schwartz RJ, Martin JF. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development. 2005;132:5601-5611
19. Jiao K, Langworthy M, Batts L. et al. Tgfbeta signaling is required for atrioventricular cushion mesenchyme remodeling during in vivo cardiac development. Development. 2006;133:4585-4593
20. Jiao K, Kulessa H, Tompkins K. et al. An essential role of Bmp4 in the atrioventricular septation of the mouse heart. Genes Dev. 2003;17:2362-2367
21. Camenisch TD, Molin DG, Person A. et al. Temporal and Distinct TGFbeta Ligand Requirements during Mouse and Avian Endocardial Cushion Morphogenesis. Dev Biol. 2002;248:170-181
22. Sugi Y, Yamamura H, Okagawa H, Markwald RR. Bone morphogenetic protein-2 can mediate myocardial regulation of atrioventricular cushion mesenchymal cell formation in mice. Dev Biol. 2004;269:505-518
23. Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685-700
24. Wharton K, Derynck R. TGFbeta family signaling novel insights in development and disease. Development. 2009;136:3691-3697
25. Tang S, Snider P, Firulli AB, Conway SJ. Trigenic neural crest-restricted Smad7 over-expression results in congenital craniofacial and cardiovascular defects. Dev Biol. 2010;344:233-247
26. Euler-Taimor G, Heger J. The complex pattern of SMAD signaling in the cardiovascular system. Cardiovasc Res. 2006;69:15-25
27. Galvin KM, Donovan MJ, Lynch CA. et al. A role for smad6 in development and homeostasis of the cardiovascular system. Nat Genet. 2000;24:171-174
28. Wang J, Xu N, Feng X. et al. Targeted Disruption of Smad4 in Cardiomyocytes Results in Cardiac Hypertrophy and Heart Failure. Circ Res. 2005;97:821-828
29. Song L, Yan W, Chen X. et al. Myocardial smad4 is essential for cardiogenesis in mouse embryos. Circ Res. 2007;101:277-285
30. Qi X, Yang G, Yang L. et al. Essential role of Smad4 in maintaining cardiomyocyte proliferation during murine embryonic heart development. Dev Biol. 2007;311:136-146
31. Sirard C, de la Pompa JL, Elia A. et al. The tumor suppressor gene Smad4/Dpc4 is required for gastrulation and later for anterior development of the mouse embryo. Genes Dev. 1998;12:107-119
32. Wang RH, Li C, Xu X. et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005;2:399-409
33. Xu X, Ehdaie B, Ohara N. et al. Synergistic action of Smad4 and Pten in suppressing pancreatic ductal adenocarcinoma formation in mice. Oncogene. 2010;29:674-686
34. Yang G, Yang X. Smad4-mediated TGF-beta signaling in tumorigenesis. Int J Biol Sci. 2010;6:1-8
35. McLennan IS, Taylor-Jeffs J. The use of sodium lamps to brightly illuminate mouse houses during their dark phases. Lab Anim. 2004;38:1-9
36. Yang X, Li C, Herrera PL, Deng CX. Generation of Smad4/Dpc4 conditional knockout mice. Genesis. 2002;32:80-81
37. Friedrich G, Soriano P. Promoter traps in embryonic stem cells a genetic screen to identify and mutate developmental genes in mice. Genes Dev. 1991;5:1513-1523
38. Brennan KJ, Hardeman EC. Quantitative analysis of the human alpha-skeletal actin gene in transgenic mice. J Biol Chem. 1993;268:719-725
39. Feil R, Brocard J, Mascrez B. et al. Ligand-activated site-specific recombination in mice. Proc Natl Acad Sci USA. 1996;93:10887-10890
40. Frugier T, Koishi K, Matthaei KI, McLennan IS. Transgenic mice carrying a tetracycline-inducible truncated transforming growth factor beta receptor (TbetaRII). Genesis. 2005;42:1-5
41. Lobe CG, Koop KE, Kreppner W. et al. Z/AP a double reporter for cre-mediated recombination. Dev Biol. 1999;208:281-292
42. Wang PY, Protheroe A, Clarkson AN. et al. Mullerian inhibiting substance contributes to sex-linked biases in the brain and behavior. Proc Natl Acad Sci USA. 2009;106:7203-7208
43. Sohal DS, Nghiem M, Crackower MA. et al. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res. 2001;89:20-25
44. Grieshammer U, Lewandoski M, Prevette D. et al. (1998) Muscle-specific cell ablation conditional upon Cre-mediated DNA recombination in transgenic mice leads to massive spinal and cranial motoneuron loss. Dev Biol. 1998;197:234-247
45. Li P, Pashmforoush M, Sucov HM. Retinoic acid regulates differentiation of the secondary heart field and TGFbeta-mediated outflow tract septation. Dev Cell. 2010;18:480-485
46. McCulley DJ, Kang JO, Martin JF, Black BL. (2008) BMP4 is required in the anterior heart field and its derivatives for endocardial cushion remodeling outflow tract septation and semilunar valve development. Dev Dyn. 2008;237:3200-3209
47. Lan Y, Liu B, Yao H. et al. Essential role of endothelial Smad4 in vascular remodeling and integrity. Mol Cell Biol. 2007;27:7683-7692
48. Nie X, Deng CX, Wang Q, Jiao K. Disruption of Smad4 in neural crest cells leads to mid-gestation death with pharyngeal arch craniofacial and cardiac defects. Dev Biol. 2008;316:417-430
49. Kallapur S, Ormsby I, Doetschman T. Strain dependency of TGFbeta1 function during embryogenesis. Mol Reprod Dev. 1999;52:341-349
50. Tang Y, Lee KS, Yang H. et al. Epistatic interactions between modifier genes confer strain-specific redundancy for Tgfb1 in developmental angiogenesis. Genomics. 2005;85:60-70
51. Asano Y, Koishi K, Frugier T, McLennan IS. Mice with disrupted TGFbeta signaling have normal cerebella development but exhibit facial dysmorphogenesis and strain-dependent deficits in their body wall. Cell Mol Neurobiol. 2009;29:621-633
52. Engle SJ, Ormsby I, Pawlowski S. et al. Elimination of colon cancer in germ-free Transforming Growth Factor beta 1-deficient mice. Cancer Res. 2002;62:6362-6366
53. Sanford LP, Ormsby I, Gittenberger-de GA. et al. TGFbeta2 knockout mice have multiple developmental defects that are non- overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659-2670
54. Xu J, Ismat FA, Wang T. et al. Cardiomyocyte-specific loss of neurofibromin promotes cardiac hypertrophy and dysfunction. Circ Res. 2009;105:304-311
55. Gaussin V, Van de PT, Mishina Y. et al. Endocardial cushion and myocardial defects after cardiac myocyte-specific conditional deletion of the bone morphogenetic protein receptor ALK3. Proc Natl Acad Sci USA. 2002;99:2878-2883
Author contact
![]() Corresponding author: Mohamad Azhar, 1656 E Mabel St, PO Box 245217, Tucson, AZ 85724-5217; Phone: 520-626-1861; Fax: 520-626-7600; E-mail: azharmarizona.edu
Corresponding author: Mohamad Azhar, 1656 E Mabel St, PO Box 245217, Tucson, AZ 85724-5217; Phone: 520-626-1861; Fax: 520-626-7600; E-mail: azharmarizona.edu
Received 2010-8-10
Accepted 2010-9-8
Published 2010-9-20