ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2011; 7(8):1077-1092. doi:10.7150/ijbs.7.1077 This issue Cite
Research Paper
Cardioprotective Effects of Telmisartan against Heart Failure in Rats Induced By Experimental Autoimmune Myocarditis through the Modulation of Angiotensin-Converting Enzyme-2/Angiotensin 1-7/Mas Receptor Axis
Vijayakumar Sukumaran1, Punniyakoti T. Veeraveedu1,2, Narasimman Gurusamy3, Ken'ichi Yamaguchi4, Arun Prasath Lakshmanan1, Meilei Ma1, Kenji Suzuki5, Makoto Kodama6, Kenichi Watanabe1 ![]()
1. Department of Clinical Pharmacology, Faculty of Pharmaceutical Sciences, Niigata University of Pharmacy and Applied Life Sciences, Niigata, Japan.
2. Department of Single Molecule Imaging, WPI Immunology Frontier Research Center, Osaka University, Osaka, Japan.
3. Department of Anesthesiology and Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, USA.
4. Department of Homeostatic Regulation and Development, Niigata University Graduate School of Medical and Dental Sciences, Niigata, Japan.
5. Department of Gastroenterology and Hepatology, Niigata University Graduate School of Medical and Dental Sciences, Niigata, Japan.
6. First Department of Medicine, Niigata University Graduate School of Medical and Dental Sciences, Niigata, Japan.
Abstract
Angiotensin-converting enzyme-2 (ACE-2) is a homolog of ACE that preferentially forms angiotensin-(ANG)-1-7 from angiotensin II (ANG II). We investigated the cardioprotective effects of telmisartan, a well-known angiotensin receptor blockers (ARBs) against experimental autoimmune myocarditis (EAM). EAM was induced in Lewis rats by immunization with porcine cardiac myosin. The rats were divided into two groups and treated with telmisartan (10 mg/kg/day) or vehicle for 21 days. Myocardial functional parameters were significantly improved by treatment with telmisartan compared with vehicle-treated rats. Telmisartan lowered myocardial protein expressions of NADPH oxidase subunits 3-nitrotyrosine, p47phox, p67 phox, Nox-4 and superoxide production significantly than vehicle-treated rats. In contrast myocardial protein levels of ACE-2, ANG 1-7 mas receptor were upregulated in the telmisartan treated group compared with those of vehicle-treated rats. The myocardial protein expression levels of tumor necrosis factor receptor (TNFR)-associated factor (TRAF)-2, C/EBP homologous protein (CHOP) and glucose-regulated protein (GRP) 78 were decreased in the telmisartan treated rats compared with those of vehicle-treated rats. In addition, telmisartan treatment significantly decreased the protein expression levels of phospho-p38 mitogen-activated protein kinase (MAPK), phospho-JNK, phospho-ERK and phospho (MAPK) activated protein kinase-2 than with those of vehicle-treated rats. Moreover, telmisartan significantly decreased the production of proinflammatory cytokines, myocardial apoptotic markers and caspase-3 positive cells compared with those of vehicle-treated rats. Therefore, we suggest that telmisartan was beneficial protection against heart failure in rats, at least in part by suppressing inflammation, oxidative stress, ER stress as well as signaling pathways through the modulation of ACE2/ANG1-7/Mas receptor axis.
Keywords: Experimental autoimmune myocarditis, endoplasmic reticulum stress, inflammation, telmisartan, oxidative stress, signaling pathways
Introduction
Inflammation and autoimmunity are involved in many cardiac diseases. In humans, acute myocarditis is a potentially lethal disease that frequently precedes the development of dilated cardiomyopathy (DCM). There have been two mechanisms by which myocarditis develop into DCM have been proposed: one is persistent viral infection and the other is progressive autoimmune myocardial injury [1]. Some cases of myocarditis may be mediated by autoimmune response to cardiac antigens. Experimental autoimmune myocarditis (EAM) in rodents may be elicited by immunization of cardiac myosin, and EAM in rats mimics human fulminant myocarditis in the acute phase and human DCM in the chronic phase [2]. EAM is induced by T cell activation and the peak of inflammation is observed in the heart around day 21 after immunization [3-4]. Some evidence supports a role of cellular immune mechanisms in the pathogenesis of myocarditis and subsequent DCM [5].
The renin-angiotensin-aldosterone system (RAAS) plays an important role in the pathogenesis of a variety of clinical conditions, including atherosclerosis, hypertension, left ventricular (LV) hypertrophy, myocardial infarction, and heart failure [6-7]. As a result, the RAAS represents a logical therapeutic target in the management of hypertension, renal disease, and cardiovascular disease. Recently, a new axis of RAAS has been established. In this axis, angiotensin converting enzyme-2 (ACE-2) is a key component of the renin-angiotensin system (RAS) and catalyzes with high efficiency the conversion of angiotensin-II (ANG-II) to angiotensin 1-7 (ANG 1-7) and stimulates the G-protein coupled receptor termed Mas, which has been shown to prevent ANG-II-induced cardiovascular hypertrophy and remodeling associated with blockade of MAPK signaling [8-12]. ACE-2 is reported to be highly expressed in the heart and kidney. ACE-2 is a membrane-associated hydrolase and it also hydrolyzes ANG I to ANG 1-9 [13]. As ANG 1-9 can be converted to ANG 1-7 by ACE or by other peptidases, ACE-2 facilitates ANG 1-7 production by two separate pathways [14]. It has been demonstrated previously that there are reduced ACE-2 levels in cardiovascular system in spontaneously hypertensive rats and in a rat model of myocardial infarction while overexpression of ACE-2 contributes to the attenuation of cardiovascular damage in these rats [10, 15-17]. An understanding of the regulation of these enzymes is clinically relevant in view of recent studies showing that ACE-2/ANG 1-7 expression is altered in pathological conditions such as diabetes, hypertension, and cardiovascular diseases [18-20].
However, recent studies indicated that ANG 1-7 has more active roles in RAAS. ANG 1-7 causes vasodilation, which antagonizes AT1-receptor mediated vasoconstriction. This effect seemed to be mediated by the bradykinin-NO (nitric oxide) pathway [13, 21]. Recently, Ishiyama et al., reported that olmesartan increased ACE-2 expression in the remodeling heart after myocardial infarction, which theoretically could contribute to the beneficial effects of ARB by facilitating increased cardiac ANG 1-7 formation [22]. It has also been reported that telmisartan induced a significant increases of ACE-2 gene and protein expressions in the myocardium of pressure-overloaded rats and also attenuates vascular hypertrophy in spontaneously hypertensive rats by the modulation of ACE-2 expression with a marked reversal of ERK1/2 and JNK phosphorylation signaling pathways [23-24]. Collectively, these results suggest that ANG 1-7 antagonized the pressor effect mediated by AT1-receptor stimulation, thereby resulting in a blood-pressure-lowering effect and an organ-protective effect, such as a reduction of cardiac hypertrophy and fibrosis and renal damage.
Despite many reports show the beneficial effect of ARBs in cardiovascular diseases [1-7, 11, 18-20, 22-24]. To date, few studies have examined the beneficial effects of telmisartan on this new axis of ACE-2/ANG 1-7/Mas receptor in cardiovascular diseases [23-25], but its effectiveness on this new axis of ACE-2/ANG 1-7/Mas receptor in a rat model of EAM is not yet known. The present study was designed to assess the cardioprotective effects of telmisartan on cardiac function using hemodynamic and echocardiographic parameters, neurohumoral factors such as serum ANG 1-7 and inflammation, oxidative stress, endoplasmic reticulum (ER) stress, myocardial apoptosis and signaling pathways in a rat model heart failure after EAM. Our findings reveal that administration of telmisartan attenuates myocardial remodeling in EAM rats by modulation of ACE-2/ANG 1-7/Mas receptor expression with a marked reversal of inflammatory events and stress signaling.
Materials and Methods
Chemicals
Unless otherwise stated, all reagents were of analytical grade and were purchased from Sigma (Tokyo, Japan). Telmisartan was generously provided by Boehringer Ingelheim GmbH (Ingelheim am Rhein, Germany).
Animals
Lewis rats (male, 8 weeks old) were purchased from Charles River Japan Inc. (Kanagawa, Japan). All experiments were performed in accordance with the guidelines of our institute [26]. Animals were kept in the departmental animal house under controlled conditions of 25±2 °C, relative humidity of 60±5% and a light-dark cycle of 12:12 h. They were fed with food pellets (Oriental Yeast Co., Ltd, Tokyo, Japan) and water ad libitum.
Immunization and treatment protocol
Lewis rats were injected in the footpads with antigen-adjuvant emulsion according to the procedure described previously. In brief, porcine cardiac myosin was dissolved in phosphate-buffered saline at 5 mg/ml and emulsified with an equal volume of complete Freund's adjuvant with 11 mg/ml Mycobacterium tuberculosis H37RA (Difco Lab., Detroit, MI, USA). EAM was induced in rats by immunization with 0.1 ml emulsion once by subcutaneous injection into their rear footpads (0.1 ml to each footpad). The morbidity of EAM was 100% in rats immunized by this procedure [2, 26]. After immunization, the Lewis rats were divided into two groups and received oral administration of telmisartan (10 mg/kg/day; Group-Tel-10) or vehicle (Group-EAM) for 21 days. Age-matched Lewis rats without immunization were used as normal controls (Group-N). The doses used in the experiments were determined on the basis of anti-inflammatory and antifibrotic properties of telmisartan as previously reported [26, 27]. We have reported that telmisartan (10 mg/kg/day) improved cardiac function and attenuated oxidative stress, cardiac remodeling (fibrosis and hypertrophy) and inflammatory mediators in rats with heart failure after EAM [26, 27].
Assessment of myocardial functions by hemodynamic and echocardiographic studies
To obtain hemodynamic data, rats were anesthetized with 2% halothane in oxygen during the surgical procedures. A catheter-tip transducer (Miller SPR 249; Miller Instruments, Houston, TX) was introduced into the left ventricle through the right carotid artery to determine peak LV pressure (LVP) and LV end-diastolic pressure (LVEDP), and the rates of intraventricular pressure rise and decline (±dP/dt) were recorded as described previously [26]. After instrumentation, the concentration of halothane was reduced to 0.5% to minimize the effects of the anesthetic on hemodynamic parameters. In addition, mean blood pressure (MBP) was measured in conscious rats by using the tail-cuff plethysmographic method [Softron BP-98A, Tokyo, Japan]. Echocardiographic studies were carried out with a 7.5-MHz transducer (Aloka Inc., Tokyo, Japan). The LV dimensions in diastole and systole and percent fractional shortening were estimated using the M-mode measurements.
Cardiac morphometric parameters
The body weight (BW) of rats was noted just before the surgical procedure. After the hemodynamic and echocardiographic analyses, the rats were sacrificed, and the myocardium was isolated and weighed to calculate the ratio of heart weight to body weight (HW/BW).
Measurement of ANG 1-7 by Enzyme-linked immunosorbent assay (ELISA)
Serum concentrations of ANG 1-7 was measured by using ELISA kits (Peninsula Laboratories, Belmont, CA) according to the manufacturer's instructions.
Immunohistochemical determination of myocardial angiotensin converting enzyme (ACE-2), Angiotensin (ANG 1-7) mas receptor
Formalin-fixed, paraffin-embedded cardiac tissue sections were used for immunohistochemical staining. After deparaffinization and hydration, the slides were washed in Tris-buffered saline (TBS; 10 mM/l Tris HCl, 0.85% NaCl, pH 7.5) containing 0.1% bovine serum albumin (BSA). Endogenous peroxidase activity was quenched by incubating the slides in methanol and 0.6% H2O2 in methanol. For antigen retrieval, the sections were pretreated with trypsin for 15 min at 37 °C. After overnight incubation with the primary antibody, i.e., rabbit polyclonal anti-ANG 1-7 mas receptor (diluted 1:100) (Alomone Labs Ltd, Jerusalem, Israel) antibody; goat polyclonal anti-ACE-2 (diluted 1:100) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4 °C, the slides were washed in TBS, horseradish peroxidase (HRP)-conjugated secondary antibody was then added, and the slides were further incubated at room temperature for 45 min. The slides were washed in TBS, incubated with diaminobenzidine tetrahydrochloride as the substrate, and counterstained with hematoxylin. A negative control without primary antibody was included in the experiment to verify the antibody specificity.
Immunofluorescence determination of glucose regulated protein-78 (GRP-78) and caspase-3
For immunofluorescence, tissues were fixed in 10% buffered formaldehyde solution and embedded in paraffin. Sections underwent microwave antigen retrieval, were blocked with 10% respective serums in phosphate-buffered saline, and were incubated with polyclonal goat anti- glucose regulated protein-78 (GRP-78) antibody (Santa Cruz Biotechnology); polyclonal rabbit anti-caspase-3 antibody (Cell Signaling Technology). Binding sites of the primary antibody were revealed with fluorescein isothiocyanate-conjugated secondary antibody (Sigma-Aldrich, St. Louis, MO, USA). Samples were visualized with a fluorescence microscope at 100X magnification (CIA-102; Olympus) [30].
In situ detection of superoxide production in hearts
To evaluate in situ superoxide production from hearts, unfixed frozen cross sections of the specimens were stained with dihydroethidium [DHE; Molecular Probes, Eugene, OR, USA] according to the previously validated method [28, 29]. Briefly, the unfixed frozen tissues were cut into 10-μm-thick sections and incubated with 10 μM DHE at 37 °C for 30 min in a light-protected humidified chamber. Fluorescence images were obtained using a fluorescence microscope equipped with a rhodamine filter. In the presence of superoxide, DHE is converted to the fluorescent molecule ethidium, which can then label nuclei by intercalating with DNA.
Western Immunoblotting
Myocardial tissue samples obtained from different groups were homogenized with lysis buffer. Protein concentrations in these homogenized samples were measured by the bicinchoninic acid method. For Western blots, proteins were separated by SDS-PAGE and identified with the following antibodies to quantify the myocardial levels of proteins: anti-Nox-4, anti-p67phox, anti-ACE-2, anti-cyclooxygenase-2 (Cox-2), anti-tumor necrosis factor-α (TNF-α), anti-glucose regulated protein-78 (GRP-78) and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) goat polyclonal antibodies; anti-p47phox, anti-p38 mitogen-activated protein kinase (p38 MAPK), anti-phospho-p38 MAPK, anti-c-Jun-N-terminal kinase (JNK), anti-phospho-JNK, anti-ERK, anti-phospho-ERK, anti-p38 MAPK-activated protein kinase-2, anti-phospho-p38 MAPK-activated mitogen kinase-2, anti-tumor necrosis factor receptor (TNFR)-associated factor (TRAF)-2, anti-NF-kB, anti-caspase-3, anti-caspase-7 and anti-ANG 1-7 mas receptor rabbit polyclonal antibody and anti-C/EBP homologous protein (CHOP), anti-iNOS, anti-Bcl-2 and anti-3-nitrotyrosine (3-NT) mouse polyclonal antibody (Santa Cruz Biotechnology). We used 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Bio-Rad, CA, USA), and electrophoretically transferred to nitrocellulose membranes. Membranes were blocked with 5% nonfat dry milk and 3% BSA (Sigma, St Louis, USA) in TBS-T (20 mM/l Tris, pH 7.6, 137 mM/l NaCl, and 0.05% Tween). After incubation with the primary antibody, the bound antibody was visualized with the respective HRP-coupled secondary antibody (Santa Cruz Biotechnology) and chemiluminescence developing agents (Amersham Biosciences, Buckinghamshire, UK). The level of GAPDH was estimated in every sample to check for equal loading of the sample. Films were scanned, and band densities were quantified with densitometric analysis using the Scion Image program (GT-X700, Epson, Tokyo, Japan).
Statistical analysis
All values are expressed as the means ± S.E.M. Statistical analysis of differences between the groups was performed by one-way ANOVA, followed by Tukey's or Bonferroni's method and the two tailed t-test when appropriate. P < 0.05 was considered as significant. For statistical analysis, GraphPad Prism 5 software (San Diego, CA, U.S.A) was used.
Results
Effects of telmisartan on myocardial functions
Although the heart rate was not different among the three groups of rats, central venous pressure (CVP) and LVEDP were significantly higher (5.98±0.6 vs 0.52±0.5 mm Hg (P < 0.01); 19.24±1.6 vs 4.12±5.6 mm Hg (P < 0.01), respectively) and ±dP/dt was significantly lower in vehicle-treated rats than in group N (2316±236 vs 7568±1046 mm Hg/s (P < 0.01); 2088±194 vs 7356±1560 mm Hg/s (P < 0.01), respectively), indicating systolic and diastolic dysfunction in vehicle-treated rats (Table 1). CVP and LVEDP were decreased more significantly in the telmisartan treated group compared with those in vehicle-treated group. Myocardial contractility parameters (rates of intraventricular pressure rise and decline) were also improved in EAM rats treated with telmisartan. In addition, MBP were significantly lowered in vehicle-treated rats than in group N rats and were marginally increased by treatment with telmisartan.
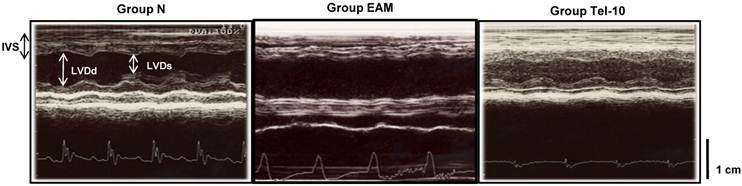
Echocardiographic studies showed increased LV dimensions in diastole and systole (7.8±0.8 vs 7.2±1.4 mm; 6.8±0.58 vs 3.8±0.36 mm (P < 0.01), respectively), and reduced fractional shortening (FS) (17.4±3.2 vs 41.5±2.5% (P < 0.01) and ejection fraction (EF) (38.0±10.5 vs 78.4±5.2% (P < 0.01) in group V rats, indicating impaired systolic function compared with group N rats (Table 1). Treatment with telmisartan more significantly decreased LV dimensions in systole and increased FS and EF when compared with those of vehicle-treated rats (Table 1 and Fig. 1).
Effects of telmisartan on morphometric parameters
HW and the ratio of HW/BW were significantly larger in group EAM than in group N rats (1.54±0.12 vs 1.20±0.05 g/kg (P < 0.01); 5.9±0.25 vs 3.5±0.14 g/kg (P < 0.01), respectively; Table 2). Telmisartan significantly reduced HW and the ratio of HW/BW when compared with those of vehicle-treated rats (Table 2).
Effects of telmisartan on serum ANG 1-7 levels
Serum ANG 1-7 concentrations were significantly decreased in vehicle-treated rats in comparison with those in group N rats (1.14±1.2 vs 0.73±0.8 ng/ml (P < 0.01)). Treatment with telmisartan further elevated the serum concentration of ANG 1-7 in comparison with those of vehicle-treated rats (Table 1).
Effects of telmisartan on inflammatory markers
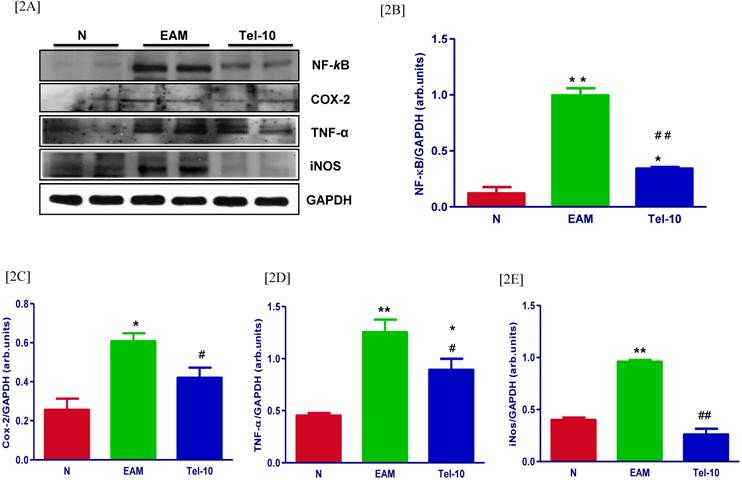
The myocardial protein levels of NF-kB, Cox-2, TNF-α and iNOS were significantly upregulated in group EAM rats (1.9, 2.4, 2.8 and 2.4-fold, respectively) compared with group N rats (Fig. 2A-2E). Telmisartan treatment significantly downregulated the protein levels of NF-kB, Cox-2, TNF-α and iNOS (2.9, 1.4, 1.4 and 3.6-fold, respectively) in the myocardium compared with those of vehicle-treated rats (Fig. 2A-2E).
Changes in hemodynamic and echocardiographic functional parameters after 3 weeks of treatment with telmisartan in rats with or without experimental autoimmune myocarditis
| Functional parameters | Group N (n=4) | Group EAM (n=6) | Group Tel-10 (n=6) |
|---|---|---|---|
| CVP (mmHg) | 0.60±0.3 | 5.12±0.6** | 2.14±1.2## |
| MBP (mmHg) | 94±8.0 | 80±10.0* | 84±7.0* |
| LVP (mmHg) | 112±4.0 | 74±7.0** | 84±4.8**# |
| LVEDP (mmHg) | 4.52±5.0 | 17.0±1.5** | 9.5±5.6**## |
| +dP/dt (mmHg/s) | 7456±954 | 2554±236** | 3704±335** |
| -dP/dt (mmHg/s) | 7244±1322 | 2300±194** | 3122±418** |
| HR (beats/min) | 355±8.3 | 301±19.5 | 296±19.8 |
| LVDd (mm) | 6.8±1.4 | 7.5±1.2 | 7.2±0.5 |
| LVDs (mm) | 3.5±0.5 | 7.2±0.8** | 5.5±0.70# |
| FS (%) | 38.5±2 | 19.5±3.8** | 30.7±5.4## |
| EF (%) | 72.4±5.5 | 39.8±8.2** | 62.56±6.8## |
Results are presented as the mean ± S.E.M. CVP, central venous pressure; MBP, mean blood pressure; LVP, left ventricular pressure; LVEDP, left ventricular end-diastolic pressure; {plus minus} dP/dt, rate of intra-ventricular pressure rise and decline; HR, heart rate; LVDd, left ventricular dimension in diastole; LVDs, left ventricular dimension in systole; FS, fractional shortening; EF, ejection fraction; group N, aged matched untreated rats; group EAM, rats with heart failure treated with vehicle; group Tel-10, rats with heart failure treated with telmisartan 10 mg/kg/day respectively; * P < 0.05 and ** P < 0.01 vs group N; # P < 0.05 and ## P < 0.01 vs group EAM.
Changes in morphometric and histopathological parameters and serum angiotensin 1-7 level after 3 weeks of treatment with telmisartan in rats with or without experimental autoimmune myocarditis
| Parameters | Group N (n=4) | Group EAM (n=6) | Group Tel-10 (n=6) |
|---|---|---|---|
| Body weight (g) | 325±5.2 | 238±3.8** | 250±4.2** |
| Heart weight (g) | 1.28±0.1 | 1.66±0.2** | 0.88±0.16## |
| HW / BW (g/kg) | 3.4±0.4 | 6.2±0.5** | 4.9±0.24## |
| Area of fibrosis (%) | 2.8±0.5 | 54.0±2.4** | 35.5±2.1## |
| Serum ANG 1-7 (ng/ml) | 1.04±1.5 | 0.68±0.7** | 0.96±1.3*# |
Results are presented as the mean ± S.E.M. HW/BW, ratio of heart weight to body weight; ANG 1-7, angiotensin 1-7; group N, aged matched untreated rats; group EAM, rats with heart failure treated with vehicle; group Tel-10, rats with heart failure treated with telmisartan 10 mg/kg/day, respectively; * P < 0.05 and ** P < 0.01 vs group N; # P < 0.05 and ## P < 0.01 vs group EAM.
Representative echocardiographic (M-mode) images showing the left ventricular dimensions. IVS: intra-ventricular septum; LVDd: left ventricular dimension in diastole; LVDs: left ventricular dimension in systole. Group N, age-matched untreated rats; group EAM, EAM rats administered with vehicle; group Tel-10, EAM rats treated with telmisartan (10 mg/kg/day) respectively. Scale bar indicates 1 cm.
Effects of telmisartan on myocardial protein expressions of inflammatory markers. [2A] Representative Western blots showing specific bands for NF-kB, Cox-2, TNF-α and iNOS, and GAPDH as an internal control. An equal amount of protein sample (30 μg) obtained from whole ventricular homogenate was applied in each lane. These bands are representative of five separate experiments. [2B-2E], Bar graph showing the densitometric analysis of the above Western blots. The mean density value of NF-kB, Cox-2, TNF-α and iNOS was expressed as a relative ratio to that of GAPDH. Each bar represents the mean ± S.E.M of 4 to 6 rats. Group N, age-matched untreated rats; group EAM, EAM rats administered with vehicle; group Tel-10, EAM rats treated with telmisartan (10 mg/kg/day) respectively. * P < 0.05 and **P<0.01 vs group N; # P < 0.05 and ## P < 0.01 vs group EAM.
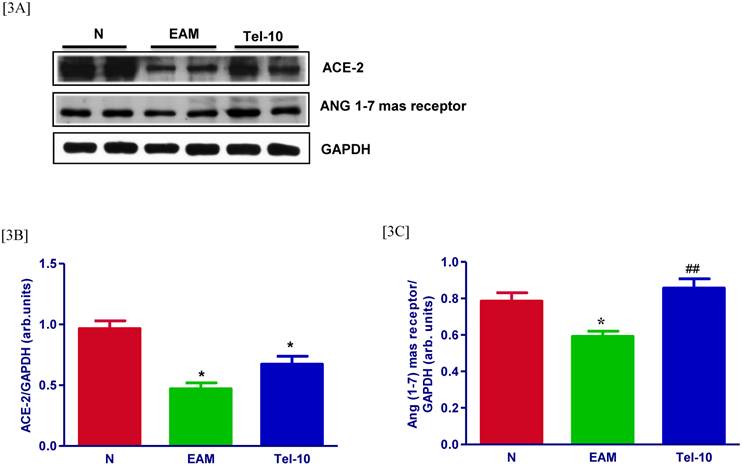
Effects of telmisartan on myocardial protein expressions of ACE-2 and ANG 1-7 mas receptor
The levels of ACE-2 and ANG 1-7 mas receptor in the myocardium were significantly downregulated in group EAM rats (0.7 and 0.5-fold, respectively) compared with those of group N rats (Fig. 3A-3C). After treatment with telmisartan, the levels of ACE-2 and ANG 1-7 mas receptor in the myocardium were upregulated (0.5 and 0.5-fold, respectively) compared with those of vehicle-treated rats (Fig. 3A-3C), but the effects were significant only in terms of Ang 1-7.
Effects of telmisartan on NADPH oxidase subunits
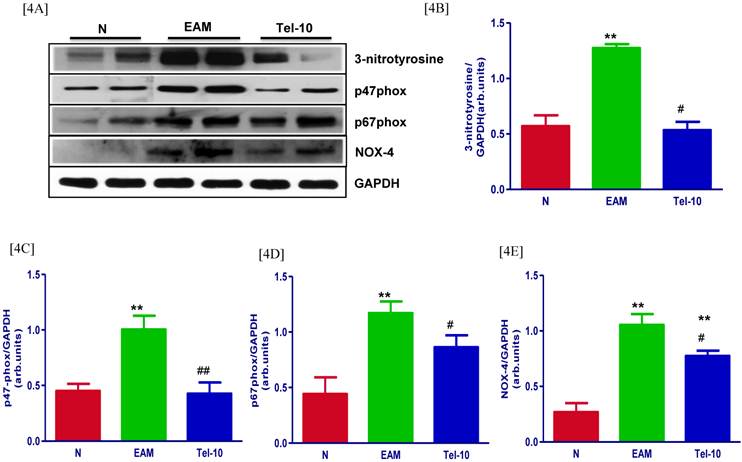
The myocardial protein levels of 3-NT, p47phox, p67phox and Nox-4 were significantly upregulated in group EAM rats (2.2, 2.2, 2.3 and 3.9-fold, respectively) compared with group N rats (Fig. 4A-4E). The myocardial protein levels of 3-NT, p47phox, p67phox and Nox-4 were significantly downregulated by telmisartan treatment (2.4, 2.3, 1.2 and 1.3-fold, respectively) compared with those in vehicle-treated rats (Fig. 4A-4E).
Effects of telmisartan on myocardial protein expressions of ACE-2 and ANG 1-7 mas receptor. [3A], Representative Western blots showing specific bands for ACE-2, ANG 1-7 mas receptor, and GAPDH as an internal control. An equal amount of protein sample (30 μg) obtained from whole ventricular homogenate was applied in each lane. These bands are representative of three separate experiments. [3B-3C], Bar graph showing the densitometric analysis of the above Western blots. The mean density value of ACE-2 and ANG 1-7 mas receptor is expressed as the relative ratio to that of GAPDH. Each bar represents the mean ± S.E.M of 4 to 6 rats. Group N, age-matched untreated rats; group EAM, EAM rats administered with vehicle; group Tel-10, EAM rats treated with telmisartan (10 mg/kg/day) respectively. * P < 0.01 vs group N; ## P < 0.01 vs group EAM.
Effects of telmisartan on myocardial protein expressions of 3-NT, p47phox, p67phox and Nox-4. [4A] Representative Western blots showing specific bands for 3-NT, p47phox, p67phox and Nox-4, and GAPDH as an internal control. An equal amount of protein sample (30 μg) obtained from whole ventricular homogenate was applied in each lane. These bands are representative of five separate experiments. [4B-4E], Bar graph showing the densitometric analysis of the above Western blots. The mean density value of 3-NT, p47phox, p67phox and Nox-4 was expressed as a relative ratio to that of GAPDH. Each bar represents the mean ± S.E.M of 4 to 6 rats. Group N, age-matched untreated rats; group EAM, EAM rats administered with vehicle; group Tel-10, EAM rats treated with telmisartan (10 mg/kg/day) respectively. **P<0.01 vs group N; # P < 0.05 and ## P < 0.01 vs group EAM.
Effects of telmisartan on myocardial content of ACE-2 and ANG 1-7 mas receptor
Myocardial immunoreactivity for ACE-2 and ANG 1-7 mas receptor was apparently weaker in vehicle-treated rats (Fig. 5A-5B). Telmisartan treatment significantly increased the myocardial content of ACE-2 and ANG 1-7 mas receptor compared with those of vehicle-treated rats (Fig. 5A-5B).
Effects of telmisartan on superoxide production in hearts
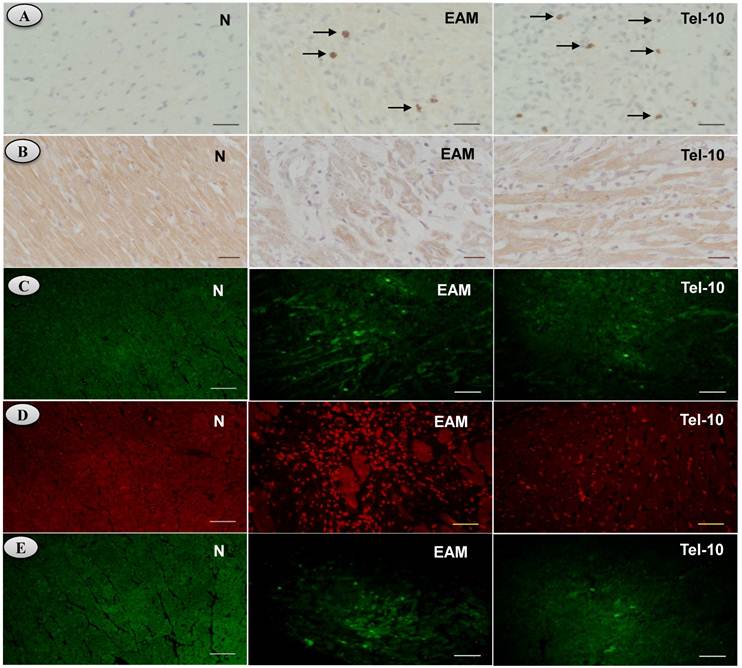
We used the fluorescent probe DHE, which has been used to detect intracellular superoxide formation [28, 29]. Fig. 5D shows the intracellular red fluorescence due to the intercalation of ethidium into DNA in the heart of vehicle-treated rats after 21days of myosin immunization compared to the normal rats. Myosin-induced enhancement of ethidium fluorescence was inhibited in telmisartan treated rats compared with that of vehicle-treated rats (Fig. 5D), indicating an overall reduced oxidative stress.
Effects of telmisartan on myocardial protein expression of ER stress markers
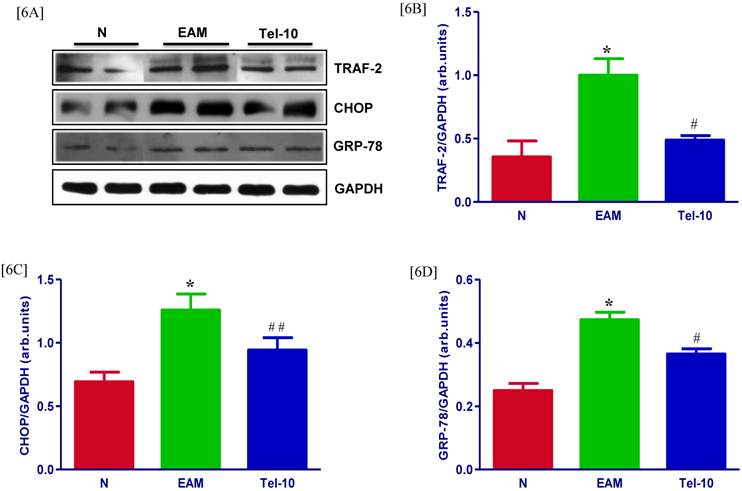
The protein levels of TRAF-2, CHOP and GRP-78 in the myocardium were significantly upregulated in group EAM rats (2.8, 1.3 and 1.6-fold, respectively) compared with group N rats (Fig. 6A-6D). The protein levels of TRAF-2, CHOP and GRP-78 were downregulated in telmisartan treated rats (2.0, 1.3 and 1.4-fold, respectively) compared with those of vehicle-treated rats (Fig. 6A-6D). In addition, we have measured the myocardial content of GRP-78 by Immunofluorescence, and the myocardial immune reactivity for GRP-78 was significantly increased in the vehicle-treated rats compared with those of group N rats (Fig. 5E) and these increased immuno reactivity was significantly decreased by telmisartan treated rats than those in group EAM rats (Fig. 5E).
Effects of telmisartan on myocardial content (ACE-2 and ANG 1-7), apoptosis, superoxide production and ER stress in rats with EAM induced by autoimmune myocarditis. [5A-5B] Immunohistochemistry of angiotensin converting enzyme (ACE-2), angiotensin (ANG) 1-7 mas receptor [counterstained with hematoxylin; ×400]. [5C], Immunofluorescence determination of caspase-3 positive cells. [5D] In situ superoxide production (bright area) using DHE-staining in the rat myocardium (x200). [5E] Immunofluorescence determination of glucose regulated protein-78 (GRP-78) (x100). Group N, age-matched untreated rats; group EAM, EAM rats administered with vehicle; group Tel-10, EAM rats treated with telmisartan (10 mg/kg/day) respectively. Scale bar indicates 20 μm.
Effects of telmisartan on myocardial protein expressions of TRAF-2, CHOP and GRP-78. [6A], Representative Western blots showing specific bands for TRAF-2, CHOP, GRP-78, and GAPDH as an internal control. An equal amount of protein sample (30 μg) obtained from whole ventricular homogenate was applied in each lane. These bands are representative of four separate experiments. [6B-6D] Bar graph showing the densitometric analysis of the above Western blots. The mean density value of TRAF-2, CHOP and GRP-78 is expressed as the relative ratio to that of GAPDH. Each bar represents the mean ± S.E.M of 4 to 6 rats. Group N, age-matched untreated rats; group EAM, EAM rats administered with vehicle; group Tel-10, EAM rats treated with telmisartan (10 mg/kg/day) respectively; * P < 0.01 vs group N; # P < 0.05 and ## P < 0.01 vs group EAM.
Effects of telmisartan on cardiac apoptosis
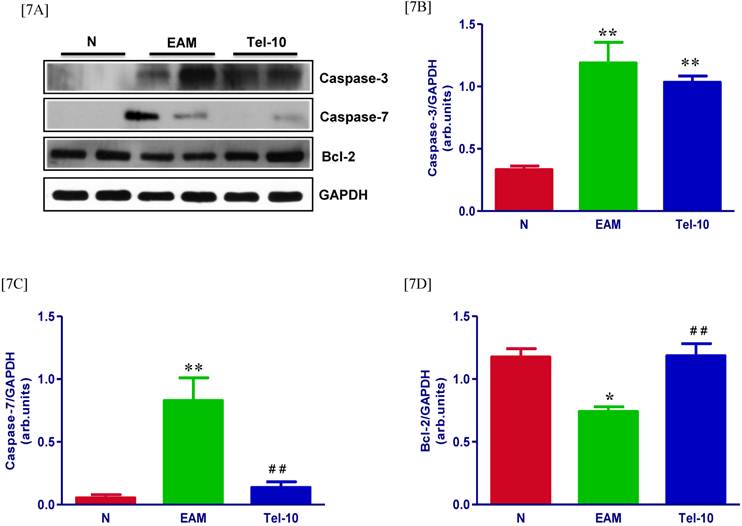
The protein levels of caspase-3 and 7 in the myocardium were significantly upregulated in group EAM rats (4.2 and 5-fold, respectively) compared with group N rats (Fig. 7A-7C). Though, these levels were attenuated in telmisartan treated rats (0.5 and 4.5-fold, respectively) compared with those of vehicle-treated rats (Fig. 7A-7C), but the effect was significant only in-terms of caspase-7. In addition we observed decreased caspase-3 positive cells in the telmisartan treated group compared with those of vehicle-treated rats (Fig. 5C). In contrast, myocardial protein level apoptotic regulator molecule bcl-2 was significantly decreased in vehicle-treated rats than group N rats (Fig. 7D) and these levels were significantly increased in the telmisartan treated rats.
Effects of telmisartan on myocardial protein expressions of signaling pathways
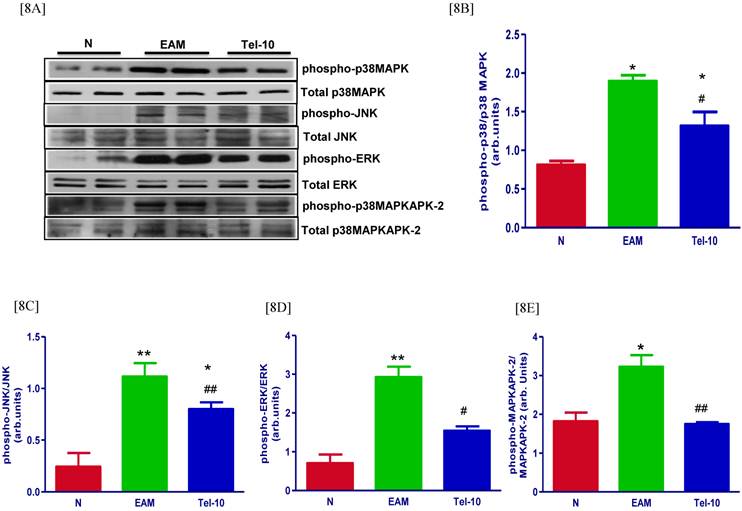
Myocardial protein expression of phospho-p38 MAPK, phospho-JNK, phospho-ERK and phospho-p38 MAPKAPK-2 was markedly increased in group EAM rats (1.6, 4.6, 4.1 and 1.8-fold, respectively) compared with that in group N rats (Fig. 8A-8E) and these protein levels were significantly decreased by telmisartan treatment (1.4, 1.4, 1.8 and 1.8-fold, respectively) compared with those of vehicle-treated rats (Fig. 8A-8E).
Effects of telmisartan on myocardial apoptosis and its marker molecules. [7A-7C] Representative Western blots showing specific bands for caspase-3, caspase-7 and bcl-2, and GAPDH as an internal control. An equal amount of protein sample (30 μg) obtained from whole ventricular homogenate was applied in each lane. These bands are representative of four separate experiments. [7A-7C], Bar graph showing the densitometric analysis of the above Western blots. The mean density value of caspase-3, caspase-7 and bcl-2 is expressed as the relative ratio to that of GAPDH. Each bar represents the mean ± S.E.M of 4 to 6 rats. Group N, age-matched untreated rats; group EAM, EAM rats administered with vehicle; group Tel-10, EAM rats treated with telmisartan (10 mg/kg/day), respectively; * P < 0.05 and **P<0.01 vs group N; ## P < 0.01 vs group EAM.
Discussion
From previous studies, using angiotensin type 1 receptor (AT1R) blockers were found to be effective in improving cardiac function and in preventing myocardial remodeling in various experimental heart failure models. However, whether AT1R blockers are better than ACE inhibitors on heart failure remains unknown, and it may differ according to the pathogenesis of heart failure. The results of present study demonstrate that the treatment with oral ARB (telmisartan) improved both systolic (+dP/dt, % EF and % FS) and diastolic (−dP/dt and LVEDP) functions, increased in neurohormonal parameter such as serum ANG 1-7, and reduced the severity of acute EAM in rats, and that the cardioprotection of ARB occur via the modulation of ACE2/ANG 1-7/Mas receptor expression with marked suppression of inflammation, oxidative stress, endoplasmic reticulum (ER) stress, myocardial apoptosis and signaling pathways.
Over the last decade, evidence accumulated that ANG 1-7 has cardiovascular protective effects [31] and counteracts detrimental effects of ANG II under pathophysiological conditions [32]. These effects may relate to the heptapeptide's ability of vasorelaxation post myocardial infarction and its blood pressure lowing effects under hypertensive conditions. It has been reported that loss of ACE-2 accelerates maladaptive LV remodeling in response to myocardial infarction through increased production of inflammatory cytokines such as interferon-gamma, interleukin-6, and the chemokine, monocyte chemoattractant protein-1, as well as increased phosphorylation of ERK1/2 and JNK1/2 signaling pathways and treatment with irbesartan reduced nicotinamide-adenine dinucleotide phosphate (NADPH) oxidase activity, infarct size, matrix matelloproteinase activation, and myocardial inflammation, ultimately resulting in improved post-myocardial infarction ventricular function [33]. Further, ANG-(1-7) or captopril treatment reduced the expression of several genes of inflammation involved in the NF-kappaB signaling pathway, as a result it reduces cardiac ischemia-induced dysfunction in diabetic hypertensive rats [34]. Interestingly, we could observe increased myocardial protein levels of inflammatory cytokines, such as TNF-α, Cox-2, iNOS, and NF-kB in rats with EAM, and these changes in protein levels were significantly decreased by telmisartan treated group (Fig. 2A-2E). Further, we have observed an increased expression of ACE-2 and ANG 1-7 mas receptor in the rats treated with ARBs compared with those of vehicle-treated rats (Fig. 3 and 5A-5B). These results suggest that reduced levels of ACE-2/ANG1-7 facilitates adverse myocardial remodeling in EAM rats by potentiating the ANG-II effects by means of the AT1 receptors, and treatment with telmisartan prevents myocardial remodeling through augmentation of ACE-2/ANG1-7, as a result it decreases the inflammatory events in EAM rats.
Myocardial expressions of phospho-p38 MAPK, phospho-JNK, p38 MAPK, JNK, phospho-ERK, ERK, phospho-p38MAPKAPK-2 and p38-MAPKAPK-2. [8A] Representative western blots showing specific bands for phospho-p38 MAPK, phospho-JNK, p38 MAPK, JNK, phospho-ERK, ERK, phospho-p38MAPKAPK-2 and p38-MAPKAPK-2 as an internal control. An equal amount of protein sample (30 μg) obtained from whole ventricular homogenate was applied in each lane. These bands are representative of eight separate experiments. [8B-8E], Bar graph showing the densitometric analysis of the above Western blots. The mean density value of phospho-p38 MAPK, phospho-JNK, phospho-ERK and phospho-p38MAPKAPK-2 was expressed as a ratio relative to that of p38 MAPK, JNK, ERK and p38-MAPKAPK-2. Each bar represents the mean ± S.E.M of 4 to 6 rats. Group N, age-matched untreated rats; group EAM, EAM rats administered with vehicle; group Tel-10, EAM rats treated with telmisartan (10 mg/kg/day) respectively; * P < 0.01 and **P<0.01 vs group N; # P < 0.05 and ## P < 0.01 vs group EAM.
Several studies have described that oxidative stress, apoptosis is involved in the pathogenesis and development of cardiac diseases including heart failure, atherosclerosis and hypertension [35-37]. Many studies have shown that blockade of ANG-II significantly reduces the levels of proinflammatory mediators and oxidative stress products in various models of inflammation [38-41]. Recent study reported that, local RAS was found to be highly activated as manifested by the increased ACE expression and ANG-II level, and decreased ACE-2 expression and ANG 1-7 level in the heart [42]. As the key peptide of the RAS, ANG-II exerts a variety of effects via its type 1 receptor, including vasoconstriction, sodium retention, cell proliferation and apoptosis, proinflammation and oxidative stress [43-44]. Overproduction of reactive oxygen species (ROS) causes oxidative stress, and has been implicated in the pathophysiology of cardiovascular disease. Contrary to ANG-II, ANG 1-7 has vasodilation, anti-inflammation and anti-proliferation effects. Benter et al., reported that treatment with ANG 1-7 constitutes a potential therapeutic strategy to alleviate NOX-mediated oxidative stress and to reduce renal dysfunction in diabetic hypertensive rats [45]. In addition, irbesartan, an AT1 receptor blocker, diminished maladaptive LV remodeling in response to myocardial infarction through reduced NADPH oxidase activity and upregulation of ACE-2 levels [30]. In the present study, we confirmed the utility of ACE-2/ANG 1-7 to alleviate ROS activation in the myocardial remodeling in EAM rats by several independent indices such as immunohistological and western immunoblotting detection of ACE-2, ANG 1-7 mas receptor expression, superoxide production measured by DHE staining, apoptosis and increased expression of NADPH oxidase subunits (Figs 3, 5 and 7), respectively. The expression of NADPH oxidase subunits were significantly downregulated by telmisartan treatment in comparison with that of vehicle-treated rats (Fig 4). Our present data may link the involvement of ACE-2/ANG 1-7 axis in the attenuation of myocardial remodeling process via, at least in part, a pathway of reduced ROS activation. On the basis of our results, we consider that telmisartan protects the heart from myocarditis partly through the suppression of apoptosis and oxidative stress.
Multiple signal transduction pathways mediated through G-protein-coupled receptors have been implicated in cardiac dysfunction and hypertrophy under the pathophysiological circumstances of heart failure [46]. The endoplasmic reticulum (ER) is a highly dynamic organelle that participates in the folding of secretory and membrane proteins. Several signaling pathways are initiated to cope with ER stress, which are designated as the unfolded protein response (UPR) [47]. Studies reported that, induction of GRP78 has been widely used as a marker for ER stress and the onset of UPR [48]. Furthermore, GRP78 serves as a master modulator for the UPR network by binding to the ER stress sensors such as protein kinase R (PKR)-like ER kinase, inositol requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6) and inhibiting their activation [48]. Oxidative stress, hypoxia, and enhanced protein synthesis in failing hearts could all potentially enhance ER stress. Emerging data has indicated that excessive and/or prolonged ER stress leads to the initiation of the apoptotic processes promoted by transcriptional induction of CHOP or by the activation of c-Jun-N-terminal kinase (JNK) and/or caspase-12-dependent pathway [49]. On the other hand, it has been reported that ANG-II upregulated ER chaperones and induced apoptosis in cultured adult rat cardiac myocytes [50]. In contrast, ANG-(1-7) reduces the agonist-mediated increase in protein synthesis and mitogen-activated protein kinases (MAPK) signaling in cultured myocytes and because plasma ANG-(1-7) levels are increased after treatment with AT1 receptor blockers [51-52], suggest that ANG-(1-7) may participate in the improvement in cardiac function. Furthermore, Tallant and Clark showed that ANG-(1-7) treatment blocked the ANG II-stimulated phosphorylation and activation of ERK1 and ERK2 [12]. Furthermore, Koka et al demonstrated that treatment with AT1 receptor blocker or blockade of ERK1/2 or MAPK by either specific inhibitor was able to abolish ANG II-induced ACE2 downregulation in human kidney tubular cells which suggest that ANG II-induced ACE2 downregulation has been shown to be associated with activation of ERK1/2 and p38 MAP kinase pathways [53]. Moreover, it has been reported that telmisartan induced a significant increases of ACE-2 gene and protein expressions in the myocardium of pressure-overloaded rats and also attenuates vascular hypertrophy in spontaneously hypertensive rats by the modulation of ACE-2 expression with a marked reversal of ERK1/2 and JNK phosphorylation signaling pathways [23-24]. In the present study, telmisartan treatment significantly downregulated the myocardial protein expressions of MAPK signaling pathways and ER stress markers including TRAF-2, CHOP and GRP-78 compared with those of vehicle-treated rats (Figs. 5E, 6 and 8). Taken together, our results suggest that telmisartan treatments regulate oxidative stress, ER stress and signaling pathways in myosin-induced heart failure through modulating ACE-2/ANG1-7/Mas receptor axis. However, attenuation of myocardial remodeling events through ACE-2/ANG1-7/Mas receptor axis in EAM rats remains to be determined.
Our study raises the possibility that, in addition to ANG-II antagonism, the activation of the ACE-2-ANG-(1-7)-Mas receptor axis might become an alternative approach to treat cardiovascular disease. In conclusion, our results indicate that telmisartan reduced the severity of EAM significantly and the mechanism of disease amelioration by this inhibitory agent can be at least partly explained by the suppression of inflammatory markers, oxidative & ER stress, myocardial apoptosis and signaling pathways, which resulted in the improvement of myocardial function.
Acknowledgements
This research was supported by a Yujin Memorial Grant, Ministry of Education, Culture, Sports, Science and Technology, Japan, and by a grant from the promotion and Mutual Aid Corporation for Private Schools, Japan. We also express our sincere gratitude to Dr. Masaki Nagata (Division of Oral and Maxillofacial Surgery, Niigata University Graduate School of Medical and Dental Sciences, Niigata, Japan) for carrying out the RT-PCR analysis in this study.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Kawai C. From myocarditis to cardiomyopathy: mechanisms of inflammation and cell death. Circulation. 1999;99:1091-1100
2. Kodama M, Matsumoto Y, Fujiwara M, Masani F, Izumi T, Shibata A. A novel experimental model of giant cell myocarditis induced in rats by immunization with cardiac myosin fraction. Clin Immunol Immunopathol. 1990;57:250-262
3. Kodama M, Zhang S, Hanawa H, Shibata A. Immunohistochemical characterization of infiltrating mononuclear cells in the rat heart with experimental autoimmune giant cell myocarditis. Clin Exp Immunol. 1992;90:330-335
4. Fuse K, Kodama M, Ito M, Okura Y, Kato K, Hanawa H, Aoki S, Aizawa Y. Polarity of helper T cell subsets represents disease nature and clinical course of experimental autoimmune myocarditis in rats. Clin Exp Immunol. 2003;134:403-408
5. Binah O. Pharmacologic modulation of the immune interaction between cytotoxic lymphocytes and ventricular myocytes. J Cardiovasc Pharmacol. 2001;38:298-316
6. Ferrario CM, Strawn WB. Role of the renin-angiotensin-aldosterone system and proinflammatory mediators in cardiovascular disease. Am J Cardiol. 2006;98:121-128
7. Schmieder RE, Hilgers KF, Schlaich MP, Schmidt BM. Renin-angiotensin system and cardiovascular risk. Lancet. 2007;369:1208-1219
8. Santos RA, Ferreira AJ, Simoes ESAC. Recent advances in the angiotensin-converting enzyme2-angiotensin (1-7)-mas axis. Exp Physiol. 2008;93:519- 527
9. Krum H, Gilbert RE. Novel therapies blocking the renin-angiotensin-aldosterone system in the management of hypertension and related disorders. J Hypertens. 2007;25:25-35
10. Igase M, Strawn WB, Gallagher PE, Geary RL, Ferrario CM. Angiotensin II AT1 receptors regulate ACE2 and angiotensin-(1-7) expression in the aorta of spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2005;289:H1013-1019
11. Kassiri Z, Zhong J, Guo D, Basu R, Wang X, Liu PP, Scholey JW, Penninger JM, Oudit GY. Loss of angiotensin-converting enzyme 2 accelerates maladaptive left ventricular remodeling in response to myocardial infarction. Circ Heart Fail. 2009;2:446-455
12. Tallant EA, Clark MA. Molecular mechanisms of inhibition of vascular growth by angiotensin-(1-7). Hypertension. 2007;42:574-579
13. Ferrario CM, Chappell MC, Tallant EA, Brosnihan KB, Diz DI. Counterregulatory actions of angiotensin-(1-7). Hypertension. 1997;30:535-541
14. Roks AJ, van Geel PP, Pinto YM, Buikema H, Henning RH, de Zeeuw D, van Gilst WH. Angiotensin-(1-7) is a modulator of the human renin-angiotensin system. Hypertension. 1999;4:296-301
15. Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, Oliveira-dos-Santos AJ, da Costa J, Zhang L, Pei Y, Scholey J, Ferrario CM, Manoukian AS, Chappell MC, Backx PH, Yagil Y, Penninger JM. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417(6891):822-828
16. Zhong JC, Huang DY, Yang YM, Li YF, Liu GF, Song XH, Du K. Upregulation of angiotensin-converting enzyme 2 by all-trans retinoic acid in spontaneously hypertensive rats. Hypertension. 2004;44(6):907-912
17. Zhao YX, Yin HQ, Yu QT, Qiao Y, Dai HY, Zhang MX, Zhang L, Liu YF, Wang LC, Liu de S, Deng BP, Zhang YH, Pan CM, Song HD, Qu X, Jiang H, Liu CX, Lu XT, Liu B, Gao F, Dong B. ACE2 overexpression ameliorates left ventricular remodeling and dysfunction in a rat model of myocardial infarction. Hum Gene Ther. 2010;21:1545-1554
18. Tallant EA, Ferrario CM, Gallagher PE. Cardioprotective role for angiotensin-(1-7) and angiotensin converting enzyme 2 in the heart. Future Cardiol. 2006;2:335-342
19. Trask AJ, Ferrario CM. Angiotensin-(1-7): pharmacology and new perspectives in cardiovascular treatments. Cardiovasc Drug Rev. 2007;25:162-174
20. Grobe JL, Mecca AP, Lingis M, Shenoy V, Bolton TA, Machado JM, Speth RC, Raizada MK, Katovich MJ. Prevention of angiotensin II-induced cardiac remodeling by angiotensin-(1-7). Am J Physiol Heart Circ Physiol. 2007;292:H736-742
21. Brosnihihan KB, Li P, Ferrario CM. Angiotensin-(1-7) dilates canine coronary arteries through kinins and nitric oxide. Hypertension. 1996;27:523-528
22. Ishiyama Y, Gallagher PE, Averill DB, Tallant EA, Brosnihan KB, Ferrario CM. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension. 2004;43:970-976
23. Wang LJ, Ma H, Liao XX, He JG, Zhang WW, Tian F, Cai YM, Gu HB, Hao YH, Hu XS, Zou HM, Zhou QL. An experimental study of expression of angiotension converting enzyme 2 in myocardium and effect of telmisartan treatment in pressure-overloaded rats. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2008;20:218-222
24. Zhong JC, Ye JY, Jin HY, Yu X, Yu HM, Zhu DL, Gao PJ, Huang DY, Shuster M, Loibner H, Guo JM, Yu XY, Xiao BX, Gong ZH, Penninger JM, Oudit GY. Telmisartan attenuates aortic hypertrophy in hypertensive rats by the modulation of ACE2 and profilin-1 expression. Regul Pept. 2011;166:90-97
25. Lakshmanan AP, Watanabe K, Thandavarayan RA, Sari FR, Harima M, Giridharan VV, Soetikno V, Kodama M, Aizawa Y. Telmisartan attenuates oxidative stress and renal fibrosis in streptozotocin induced diabetic mice with the alteration of angiotensin-(1-7) mas receptor expression associated with its PPAR-γ agonist action. Free Radic Res. 2011;45:575-584
26. Sukumaran V, Watanabe K, Veeraveedu PT, Thandavarayan RA, Gurusamy N, Ma M, Yamaguchi K, Suzuki K, Kodama M, Aizawa Y. Telmisartan, an angiotensin-II receptor blocker ameliorates cardiac remodeling in rats with dilated cardiomyopathy. Hypertens Res. 2010;33:695-702
27. Sukumaran V, Watanabe K, Veeraveedu PT, Ma M, Gurusamy N, Rajavel V, Suzuki K, Yamaguchi K, Kodama M, Aizawa Y. Telmisartan ameliorates experimental autoimmune myocarditis associated with inhibition of inflammation and oxidative stress. Eur J Pharmacol. 2011;652(1-3):126-35
28. Miller Jr FJ, Gutterman DD, Rios CD, Heistad DD, Davidson BL. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ Res. 1998;82:1298-1305
29. Takaya T, Kawashima S, Shinohara M, Yamashita T, Toh R, Sasaki N, Inoue N, Hirata K, Yokoyama M. Angiotensin II type 1 receptor blocker telmisartan suppresses superoxide production and reduces atherosclerotic lesion formation in apolipoprotein E-deficient mice. Atherosclerosis. 2006;186:402-410
30. Thandavarayan RA, Watanabe K, Ma M, Gurusamy N, Veeraveedu PT, Konishi T, Zhang S, Muslin AJ, Kodama M, Aizawa Y. Dominant-negative p38alpha mitogen-activated protein kinase prevents cardiac apoptosis and remodeling after streptozotocin-induced diabetes mellitus. Am J Physiol Heart Circ Physiol. 2009;297(3):H911-9
31. Langeveld B, van Gilst WH, Tio RA, Zijlstra F, Roks AJ. Angiotensin-(1-7) attenuates neointimal formation after stent implantation in the rat. Hypertension. 2005;45:138-141
32. Iwata M, Cowling RT, Gurantz D, Moore C, Zhang S, Yuan JX, Greenberg BH. Angiotensin-(1-7) binds to specific receptors on cardiac fibroblasts to initiate antifibrotic and antitrophic effects. Am J Physiol Heart Circ Physiol. 2005;289:H2356-2363
33. Kassiri Z, Zhong J, Guo D, Basu R, Wang X, Liu PP, Scholey JW, Penninger JM, Oudit GY. Loss of angiotensin-converting enzyme 2 accelerates maladaptive left ventricular remodeling in response to myocardial infarction. Circ Heart Fail. 2009;2:446-455
34. Al-Maghrebi M, Benter IF, Diz DI. Endogenous angiotensin-(1-7) reduces cardiac ischemia-induced dysfunction in diabetic hypertensive rats. Pharmacol Res. 2009;59:263-268
35. Tsutsui H, Kinugawa S, Matsushita S. Oxidative stress and mitochondrial DNA damage. Circ J. 2008;72(suppl A):31- 37
36. Dhalla NS, Temsah RM, Netti Cadan T. Role of oxidative stress in cardiovascular diseases. J Hypertens. 2000;18:655- 673
37. Kannan K, Jain SK. Oxidative stress and apoptosis. Pathophysiology. 2000;7:153- 163
38. Kjeldsen SE, Julius S. Hypertension mega-trials with cardiovascular end points: effect of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers. Am Heart J. 2004;148:747-754
39. Neri Serneri GG, Boddi M, Modesti PA, Coppo M, Cecioni I, Toscano T, Papa ML, Bandinelli M, Lisi GF, Chiavarelli M. Cardiac angiotensin II participates in coronary microvessel inflammation of unstable angina and strengthens the immunomediated component. Circ Res. 2004;94:1630-1637
40. Puvi N, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxydase activity upstream mediators. Circ Res. 2002;91:406-413
41. Willemsen JM, Westerink JW, Dallinga-Thie GM, Van Zonneveld AJ, Gaillard CA, Rabelink TJ, de Koning EJ. Angiotensin-II type 1 receptor blockade improves hyperglycemia-induced endothelial dysfunction and reduces proinflammatory cytokine release from leukocytes. J Cardiovasc Pharmacol. 2007;49:6-12
42. Liu CX, Hu Q, Wang Y, Zhang W, Ma ZY, Feng JB, Wang R, Wang XP, Dong B, Gao F, Zhang MX, Zhang Y. Angiotensin-Converting Enzyme 2 Overexpression Remarkably Ameliorated Glomerular Injury in a Rat Model of Diabetic Nephropathy: A Comparison with ACE Inhibition. Mol Med. 2010 In press
43. Oudit GY, Kassiri Z, Patel MP, Chappell M, Butany J, Backx PH, Tsushima RG, Scholey JW, Khokha R, Penninger JM. Angiotensin II-mediated oxidative stress and inflammation mediate the age-dependent cardiomyopathy in ACE2 null mice. Cardiovasc Res. 2007;75:29-39
44. Zhang GX, Ohmori K, Nagai Y, Fujisawa Y, Nishiyama A, Kimura S. Role of AT1 receptor in isoproterenol-induced cardiac hypertrophy and oxidative stress in mice. J Mol Cell Cardiol. 2007;42:804-811
45. Benter IF, Yousif MH, Dhaunsi GS, Kaur J, Chappell MC, Diz DI. Angiotensin-(1-7) prevents activation of NADPH oxidase and renal vascular dysfunction in diabetic hypertensive rats. Am J Nephrol. 2008;28:25-33
46. Molkentin JD, Dorn II GW. Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu Rev Physiol. 2001;63:391-396
47. Bernales S, Papa FR, Walter P. Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol. 2006;22:487-508
48. Schro¨der M, Kaufman RJ. The mammalian unfolded protein response. Annual Review of Biochemistry. 2005;74:739-789
49. Oyadomari S, Araki E, Mori M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic β-cells. Apoptosis. 2002;7:335-345
50. Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, Yutani C, Ozawa K, Ogawa S, Tomoike H, Hori M, Kitakaze M. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705-712
51. Campbell DJ, Kladis A, Duncan AM. Nephrectomy, converting enzyme inhibition, and angiotensin peptides. Hypertension. 1993;22:513-522
52. Kohara K, Brosnihan KB, Chappell MC, Khosla MC, Ferrario CM. Angiotensin-(1-7). A member of circulating angiotensin peptides. Hypertension. 1991;17:131-138
53. Koka V, Huang XR, Chung AC, Wang W, Truong LD, Lan HY. Angiotensin II up-regulates angiotensin I-converting enzyme (ACE), but down-regulates ACE2 via the AT1-ERK/p38 MAP kinase pathway. Am J Pathol. 2008;172:1174-1183
Author Biography
Mr Vijayakumar Sukumaran obtained master degree in Pharmacy (Pharmaceutical chemistry) in 2002 from K. M. College of pharmacy (Dr. MGR. Medical University, Tamil Nadu, India) and worked as a lecturer at JSS University (India) as well as at INTI International University (Malaysia). Currently, as a doctoral researcher [Department of Clinical Pharmacology, Niigata University of Pharmacy and Applied Life Sciences (NUPALS, Japan)], he is engaged in full time research activities. So far, he has been working in two different labs, and in all those labs he has contributed significantly to initiate and carrying out novel projects. To credit, he has published 26 research articles in well reputed journals and also one book chapter in Advances in medicine and Biology [In press, 2011]. In addition, he has communicated 3 papers in good impact journals and presented 9 papers in National and International Conferences related to his research work. In his Master Degree project, he initiated a study to screen the 2, 3-disubstituted quinazolin-4(3H)-ones as analgesic and anti-inflammatory agents in Prof. Meena's lab, Dr. MGR. Medical University, India. His research interest's spreads cover the fields of cardiovascular disease, diabetic complications, nephropathy, toxicity studies, DSS-induced colitis and metabolic syndrome.
Prof. Kenichi Watanabe obtained his MD in 1974 followed by a Medical PhD from Niigata Medical University in 1985 and a Pharmaceutical PhD from Shizuoka Prefectural University in 1995. Currently, he is head of the Department of Clinical Pharmacology, NUPALS, Niigata City, Japan. Prior to NUPALS, Prof. Kenichi Watanabe had held faculty appointments at Niigata University Graduate School of Medical and Dental Sciences, Niigata, Japan. His areas of expertise include heart failure, diabetes, hypertension, inflammation, cardiac imaging, cell signaling and metabolic syndrome. He has over 100 publications in his credit.
![]() Corresponding author: Professor Kenichi Watanabe, MD, Ph.D., Department of Clinical Pharmacology, Faculty of Pharmaceutical Sciences, Niigata University of Pharmacy and Applied Life Sciences, 265-1 Higashizima, Niigata city, 956-8603, Japan. Tel.: +81 250 255267; fax: +81 250 25 5021. E-mail address: watanabeac.jp (Kenichi Watanabe); svkumar1979com (Vijayakumar Sukumaran)
Corresponding author: Professor Kenichi Watanabe, MD, Ph.D., Department of Clinical Pharmacology, Faculty of Pharmaceutical Sciences, Niigata University of Pharmacy and Applied Life Sciences, 265-1 Higashizima, Niigata city, 956-8603, Japan. Tel.: +81 250 255267; fax: +81 250 25 5021. E-mail address: watanabeac.jp (Kenichi Watanabe); svkumar1979com (Vijayakumar Sukumaran)
Received 2011-2-24
Accepted 2011-8-23
Published 2011-9-8