ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results and Discussions
Conclusion
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2011; 7(8):1171-1179. doi:10.7150/ijbs.7.1171 This issue Cite
Research Paper
Sphingobium Chlorophenolicum Dichlorohydroquinone Dioxygenase (PcpA) Is Alkaline Resistant and Thermally Stable
Wanpeng Sun1, Ramaswami Sammynaiken2, 3, Lifeng Chen1, Jason Maley2, Gabriele Schatte2, Yijiang Zhou4, Jian Yang1, ![]()
1. College of Pharmacy and Nutrition, University of Saskatchewan, 110 Science Place, Saskatoon, Saskatchewan S7N 5C9, Canada
2. Saskatchewan Structural Sciences Centre, University of Saskatchewan, 110 Science Place, Saskatoon, Saskatchewan S7N 5C9, Canada
3. Department of Biochemistry, University of Saskatchewan, 107 Wiggins Road, Saskatoon, Saskatchewan S7N 5E5, Canada
4. School of Medicine, Zhejiang University, Hangzhou, Zhejiang 310058, P.R. of China
Abstract
Dichlorohydroquinone dioxygenase (PcpA) is the ring-cleavage enzyme in the PCP biodegradation pathway in Sphingobium chlorophenolicum strain ATCC 39723. PcpA dehalogenates and oxidizes 2,6-dichlorohydroquinone to form 2-chloromaleylacetate, which is subsequently converted to succinyl coenzyme A and acetyl coenzyme A via 3-oxoadipate. Previous studies have shown that PcpA is highly substrate-specific and only uses 2,6-dichlorohydroquinone as its substrate. In the current study, we overexpressed and purified recombinant PcpA and showed that PcpA was highly alkaline resistant and thermally stable. PcpA exhibited two activity peaks at pH 7.0 and 10.0, respectively. The apparent kcat and Km were measured as 0.19 ± 0.01 s-1 and 0.24 ± 0.08 mM, respectively at pH 7.0, and 0.17 ± 0.01 s-1 and 0.77 ± 0.29 mM, respectively at pH 10.0. Electron paramagnetic resonance studies showed rapid oxidation of Fe(II) to Fe(III) in PcpA and the formation of a stable radical intermediate during the enzyme catalysis. The stable radical was predicted to be an epoxide type dichloro radical with the unpaired electron density localized on C3.
Keywords: Alkaline resistance, thermal stability, radical intermediate, EPR, biodegradation
Introduction
Pentachlorophenol (PCP) was first introduced into the environment as a wide-spectrum biocide in the 1920s. It was used primarily as an herbicide in farmlands and as a wood preservative in timber industry (1-3). However, severe diseases such as cancer, acute pancreatitis, immunodeficiency and neurological disorders are possible consequences after extensive exposure to PCP (4-7). PCP is currently listed as one of the major environmental pollutants in North America because of its widespread distribution, strong resistance to biodegradation, and high toxicity to both human and farm animals (4-10). PCP was banned from usage by the US Environmental Protection Agency in 1987 (11).
PCP is not a natural product and was expected to be highly resistant to biodegradation through its high and obstructive halogenations (12, 13). However, a number of soil and aquatic microorganisms, such as Sphingobium chlorophenolicum strain ATCC39723, have evolved biodegradation pathways over a very short period of time to degrade PCP in order to utilize its ring-cleavage products as their carbon source (14-17). These biodegradation pathways are still in the early stage of evolution and usually of very low metabolic efficiencies (18, 19).
The PCP biodegradation pathway in S. chlorophenolicum strain ATCC39723, which consists of six catalytic enzymes and one confirmed Lys-R type regulator, is the most characterized (15-27). Dichlorohydroquinone dioxygenase (PcpA), encoded by gene pcpA located on bases 14828-16527 in the 24 kb pEMAC fragment, contains 320 amino acid residues and requires Fe(II) for its activity (28). It has been shown that PcpA is PCP-inducible and its mRNA expression level increases after introducing PCP into the culture medium (28). PcpA dehalogenates and oxidizes 2,6-dichlorohydroquinone (2,6-DCHQ) into 2-chloromaleylacetate (2-CMA) (29, 30). It is highly substrate specific and only uses 2,6-DCHQ as its substrate (31). Because 2,6-DCHQ is a common metabolic intermediate of several chloroaromatic compounds, ring cleavage and dehalogenation catalyzed by PcpA are key steps involved in degradation of recalcitrant chlorophenols (31-33). Unlike the intradiol, extradiol and gentisate dioxygenases, PcpA catalyzes the cleavage of aromatic rings between a hydroxyl group and a chlorine group (29-31, 33). Sequence analysis suggests that PcpA belongs to the vicinal oxygen chelate (VOC) protein superfamily (33). Although its predicted tertiary structure differs significantly from that of the extradiol dioxygenases, PcpA may follow a catalytic mechanism similar to that of the extradiol dioxygenases (33).
This study demonstrates that the recombinant His6-tagged PcpA was alkaline resistant and thermally stable. PcpA catalyzed the ring-cleavage of 2,6-DCHQ through a relatively stable radical intermediate. This intermediate was determined to be an epoxide type dichloro radical from electron paramagnetic resonance (EPR) measurements and simulation of the EPR spectra.
Materials and Methods
PcpA and 2,6-DCHQ
The protocol used to overexpress and purify the enzymes involved in PCP biodegradation in S. chlorophenolicum strain ATCC39723 in our laboratory has been published previously (24, 25, 34). The purified PcpA had higher than 90% purity as examined on a 12% SDS-PAGE gel. PcpA was concentrated to 10 mg/mL using an Amicon® 10 kDa cutoff filter unit at 4 °C, aliquoted, and then stored at -80 °C in the presence of 5% glycerol. The substrate 2,6-DCHQ was purchased from 3B Scientific Corporation (Libertyville, Illinois, USA).
PcpA Assays
All experiments were carried out at least in triplicate. The steady-state enzyme kinetics of PcpA was monitored by UV-Visible absorption spectrometry using a Synergy™ HT microplate reader (Biotek Instruments, Winooski, Vermont, USA) and UV transparent 96 well plates. To study the effects of pH, the standard assay mixture (in 200 µL) contained 50 mM oxygen-saturated buffer/solution, 250 µM 2,6-DCHQ and 100 μM ascorbic acid. In a typical experiment, the reaction was started by the addition of 7 μg PcpA to the reaction mixture and monitored the change in absorbance at 305 nm (maximum absorption of 2,6-DCHQ) for 3 min at room temperature. With the exception of pH 5.0 where acetate was used as the buffer, all other pH values contained phosphate as the buffer/solution. The pH of each reaction mixture was measured to ensure that the addition of PcpA did not change the pH of the reaction system.
The steady-state enzyme kinetics of PcpA was examined in more detail for pH 7.0 and pH 10.0. The standard assay mixture (in 200 μL) contained 50 mM oxygen-saturated phosphate buffer, pH 7.0 or 10.0, 0.05-4.0 mM 2,6-DCHQ and 100 μM ascorbic acid. In a typical experiment, the reaction was started by the addition of 10 μg PcpA to the reaction mixture and monitored the change in absorbance at 305 nm for 20 min at room temperature. Both 50 mM phosphate buffer, pH 7.0 and the filtrate collected from concentrating the enzyme solution prior to storage were used as blanks, and neither solution showed activity towards 2,6-DCHQ. The apparent kcat and Km were determined by nonlinear regression of the initial reaction rate versus the 2,6-DCHQ concentration using the software GraphPad Prism (GraphPad Software, San Diego, California, USA).
Circular dichroism
Circular dichroism (CD) measurements were carried out on a PiStar-180 CD spectrometer (Applied Photophysics Ltd., Surrey, United Kingdom) using a 0.10 mm Quartz Suprasil cuvette (Hellma Canada, Concord, Ontario, Canada). The instrument was calibrated using (1S)-(+)-10-camphorsulfonic acid at 290.5 nm. Protein solutions (1 mg/mL PcpA, 50 mM phosphate buffer/solution, pH 7-14) were scanned from 260-185 nm in 0.5 nm steps at a scan rate of 10 nm/min and a bandwidth of 6 nm. The baseline was measured and subtracted using the corresponding pH phosphate buffer/solution or temperature. The secondary structural components of PcpA were deduced using the software CDNN version 2.1 (35).
Electron paramagnetic resonance
The EPR experiment was performed at low temperature (10K) to characterize the iron status and room temperature (~23 °C) to capture the reaction intermediate during PcpA catalysis with a fixed microwave frequency of 9.625 GHz on an X-band Bruker EMX EPR spectrometer (Bruker Canada, Milton, Ontario, Canada). All collected EPR spectra were corrected with the blank quartz EPR tube. The samples examined by the low temperature EPR included PcpA (1 mg/mL in 50 mM oxygen-saturated phosphate buffer pH 7.0), PcpA with ascorbic acid (concentration ranging from 25 to 250 µM), and the reaction mixture (50 mM oxygen-saturated phosphate buffer pH 7.0, 1 mg/mL PcpA, 250 µM DCHQ, and 100 µM ascorbic acid; incubated at room temperature for 2 min). For the room temperature EPR, the experimental samples were the reaction mixtures (50 mM oxygen-saturated phosphate buffer pH 7.0, 1 mg/mL PcpA, 250 µM DCHQ, and 0.025 - 2 mM ascorbic acid; incubated at room temperature for 2 min). Spin trap 5,5-dimethyl-pyrroline-N-oxide (DMPO) was used in the initial trials to enhance the radical signal during the catalysis. However, DMPO reduced the radical signal possibly by interacting with the radical intermediate. Since the radical signal was very stable, no spin trap was applied in the later experiments. A set of negative controls by replacing PcpA with phosphate buffer were examined and no EPR signals were observed. Another set of negative controls by replacing 2,6-DCHQ with phosphate buffer did not produce the radical signal either. No visible change was observed for the radical signal when ascorbic acid concentration was increased from 25 µM to 2 mM. Although EPR is often regarded as qualitative rather than quantitative, all samples were investigated at least in triplicate to ensure the observed EPR signals were real.
Results and Discussions
pH effect
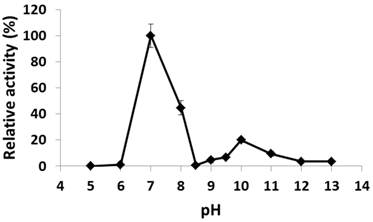
The recombinant His6-tagged PcpA was purified to > 90% purity as examined by SDS-PAGE. The pH effect on its catalytic activity was examined from pH 5.0 to 13.0. As shown in Figure 1, PcpA reached its maximal activity at pH 7.0, which is consistent with the observation that S. chlorophenolicum grows optimally around pH 7.0 (36). PcpA was sensitive to acidic pH and lost its activity at pH 5.0-6.0. On the basic pH side, a second activity peak was observed at pH 10.0 (~20-25% of the maximal activity). This observation was confirmed by repeating the experiment under finer grids around pH 10.0. The activity of PcpA dropped dramatically to < 1% of the maximal activity at pH 8.5, and then slowly rose to reach the second peak at pH 10.0. The activity of PcpA decreased again as the pH was higher than 10.0. However, PcpA maintained low but significant enzyme activity (~4% of maximal activity) at even pH 13.0. Because about 80% of PCP was used in combination with NaOH in wood preservation, most PCP-polluted sites may initially turn into alkaline soils (37). It is likely that a few microorganisms have evolved the ability to effectively degrade PCP under alkaline conditions, which is consistent with the observation that PCP biodegradation occurred more quickly under alkaline versus acidic conditions during soil remediation (37).
Effect of pH on PcpA enzymatic activity. The relative activity (%) for each pH was determined with respect to the activity at pH 7.0.
Alkaline resistance and thermal stability
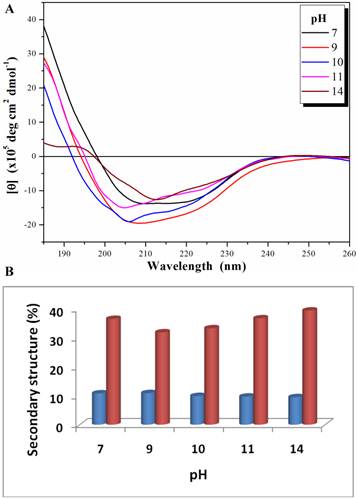
The biological function of an enzyme is closely related to its overall folding. To examine how PcpA kept its activity under high pH conditions, its secondary structure was determined at several pHs using CD spectroscopy. Both n→π* and π→π* transitions occurred in the far-UV spectral region, which corresponds to the electronic transitions within the amide backbone of proteins. The CD spectra for PcpA at selected pHs are shown in Figure 2A. It was determined that PcpA contains approximately 10% α-helix and 40% β-strand over the pH range tested (Figure 2B). Therefore, it is not unexpected for PcpA to be active even at high pHs because its secondary structure remains intact. Further pH tolerance studies showed that PcpA maintained its intact secondary structure and low but significant catalytic activity (~4% of maximal activity) even after four days of incubation in 50 mM phosphate solution, pH 13.0 (Supplementary Material: Figure S1).
Far-UV CD spectra (185-260 nm) for PcpA (1 mg/mL) at different pH (A), and the calculated percentage of the secondary structures with α-helix shown in blue and β-strand shown in coral (B). Each CD spectrum displayed is the average of 6 spectral measurements and smoothed with a 5-point Savitsky-Golay smoothing algorithm.
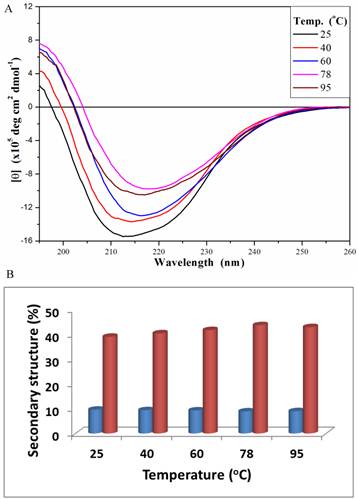
The secondary structure of PcpA was also investigated under different temperatures at the representative pH of 7.0 (Figure 3A). At 25 oC, the peak minimum from the CD spectrum was around 212 nm; and this peak minimum red-shifted to around 216 nm with an increase in temperature. The secondary structure components of PcpA were found to contain approximately 10% α-helix and 40% β-sheet over the temperature range tested (Figure 3B). Although the signal for the CD spectrum was lower at 95 oC compared to 25 oC, it did indicate that there was a significant amount of PcpA remaining folded in the solution at high temperature. As an Fe(II)-dependent dioxygenase, it is difficult to compare the activity of PcpA under different temperatures because the iron status in PcpA and the oxygen concentration in the reaction mixture are closely correlated with temperature. Therefore, we just measured the activity of PcpA at room temperature after exposing at 95 oC for 10 min. PcpA was found to retain about 25% of its maximal activity. The loss of activity for PcpA is likely due to the oxidation of Fe(II) to Fe(III). It should be noted that when a protein becomes completely unfolded in solution, the π-bond delocalization will be lost along the amide backbone, resulting in a CD signal of zero in the far-UV range. Unfortunately, the current setup on the CD spectrometer limits measurements above 100 oC.
Far-UV CD spectra (195-260 nm) for PcpA (1mg/mL, 50 mM phosphate buffer, pH 7.0) at different temperature (A), and the calculated percentage of the secondary structures with α-helix shown in blue and β-strand shown in coral (B). Each CD spectrum displayed is the average of 6 spectral measurements and smoothed with a 5-point Savitsky-Golay smoothing algorithm.
In spite of its low catalytic activity, PcpA has to tolerate environment challenges, such as extreme pH and high temperature, as a newly evolved enzyme. Over the progress of molecular evolution, an enzyme usually evolves a way of lowering the activation energy and recognizing different substrate states (38). Since mutations could occur at random in a protein sequence during evolution, an enzyme may lose other characteristics such as its environmental tolerance (39). PcpA might be a good example of enzymes with broad adaptability and compromised efficiency in the early stage of evolution. Recently, Dai and Copley performed random mutation and efficiency selection to improve the PCP-degrading enzyme activities (26). It would be interesting to see whether these mutations affect the pH and temperature tolerances for PcpA. In addition, a structural model of PcpA indicated the presence of three histidine and one glutamate residues at the catalytic active site (33), suggesting the ionization state of these residues may also play an important role in determining PcpA enzyme activity.
Kinetic parameters at pH 7.0 and 10.0
As a Fe(II)-dependent dioxygenase, the kinetic measurement of PcpA is not trivial. In the previous studies, an aerobic reconstitution of purified PcpA apoenzyme with Fe(II) was applied immediately before enzyme activity assay (30, 33). We questioned the validity of this method as S. chlorophenolicum is an aerobic bacterium and PcpA should present as a mixture of both Fe(II) and Fe(III) forms. It is impractical to have PcpA present only in the Fe(II) form in S. chlorophenolicum under natural environmental conditions. Our EPR studies discussed below also showed that the Fe(III) form was present in relatively small quantities and the reducing agent ascorbic acid could only reduce less than 20 percent of Fe(III) into Fe(II). Therefore, the reconstituted PcpA is likely not biologically relevant, so we eliminated this step in the current study. Since NAD(P)H is required for several reactions in the PCP biodegradation pathway and may exist in a reasonably high concentration in S. chlorophenolicum, we added 100 µM ascorbic acid to the PcpA reaction system to mimic the presence of NAD(P)H. In addition, ascorbic acid stabilized substrate 2,6-DCHQ as it could be oxidized to a very stable 2,6-dichlorosemiquinone radical in less than 30 sec under aqueous conditions (revealed by EPR; Supplementary Material: Figure S2).
Because PcpA is thermally stable, the filtrate of the PcpA sample from an Amicon® 10 kDa cutoff filter unit was used as the blank control instead of boiled PcpA. The enzyme activity of PcpA was assayed by monitoring the loss of 2,6-DCHQ in the reaction system. The apparent kcat and Km were measured at 0.19 ± 0.01 s-1 and 0.24 ± 0.08 mM, respectively at pH 7.0, and 0.17 ± 0.01 s-1 and 0.77 ± 0.29 mM, respectively at pH 10.0. The almost identical kcat values suggested that PcpA would exhibit the same enzymatic efficiency between pH 7.0 and 10.0 in the presence of excessive substrate. The three-fold increase in Km at pH 10.0 was likely due to decreased binding affinity for 2,6-DCHQ, which, in turn, might be caused by changes of the ionization state of the active site polar amino acids. At pH 10.0, there would be more 2,6-DCHQ present in the deprotonated and negatively charged form as compared to pH 7.0. The negative charge might hinder the binding of 2,6-DCHQ to the substrate binding pocket of PcpA because the predicted active site glutamate residue would be negatively charged and the histidine residues would be uncharged at pH 10 (33). The apparent kcat at pH 7.0 was only about one tenth of that for the reconstituted PcpA (kcat = 2.1 s-1) (30); whereas the apparent Km at pH 7.0 was much higher than the previously reported Km for the reconstituted enzyme (Km = 13.5 µM) (30). This suggested that Fe(II) was critical not only for the ring-cleavage reaction but also for the 2,6-DCHQ binding. Depending on the intracellular influence to the presence of the Fe(II) form of PcpA, the apparent kinetic parameters of PcpA in the S. chlorophenolicum cells may vary from the current results. However, it is unlikely for PcpA to reach the catalytic efficiency level of the reconstituted enzyme, because the biodegradation of PCP involves several oxidative reactions and PcpA can not present only in the Fe(II) form within the S. chlorophenolicum cells. Regardless of the reaction systems, it is still unclear how the small portion of Fe(III) form resulted in about ten-fold decrease in kcat and more than fifteen-fold increase in Km for PcpA. An important question needed to be addressed in future studies is the binding affinity of 2,6-DCHQ to the Fe(III) form of PcpA.
EPR characterization
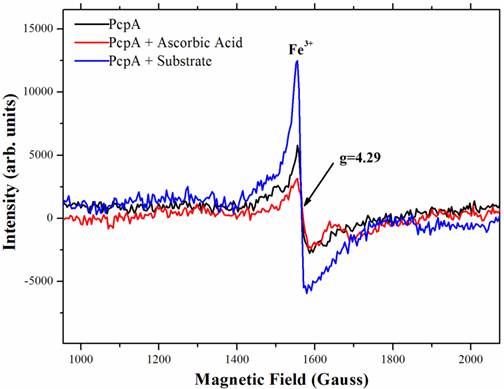
To characterize the function of iron during catalysis, low-temperature (10 K) EPR spectra were collected for several PcpA samples. Figure 4 showed the high spin signal of Fe(III) in PcpA at g = 4.29. This is the high spin form of Fe(III) in a rhombic environment. Previous studies on the Fe(II)-dependent extradiol protocatechuate 4,5-dioxygeanse showed an axial type Fe(III) ion signal at g = 6.0 (40), which is rare for a non-heme iron. We did not observe this axial type Fe(III) signal in PcpA. Upon adding 100 µM ascorbic acid to the PcpA sample, the high spin Fe(III) signal at g = 4.29 was reduced, suggesting that a portion of the Fe(III) ion was reduced to Fe(II) ion. However, this reduction was less than 20% of the original Fe(III) in PcpA even at 250 µM ascorbic acid. The high spin Fe(III) signal at g = 4.29 increased dramatically for the reaction sample containing PcpA, ascorbic acid and 2,6-DCHQ, indicating that the Fe(II) ion was oxidized to Fe(III) ion during the enzymatic catalysis. Therefore, the ring-cleavage reaction catalyzed by PcpA is likely through a Fe(II)-superoxide intermediate. Assuming the g = 4.29 signal for the reaction sample represented all iron, the Fe(III) form would be no more than 15% in the purified PcpA sample. From the EPR results, it is evident that the Fe(III) form was formed by oxidation of Fe(II) ion rather than incorporation of the Fe(III) ion during the enzyme overexpression. The structural model of PcpA showed that the Fe(II) was coordinated by amino acids His11, His227, Glu276 and Tyr266; and mutations of these amino acids into alanine residues almost depleted the whole enzyme activity (33).
Radical intermediate
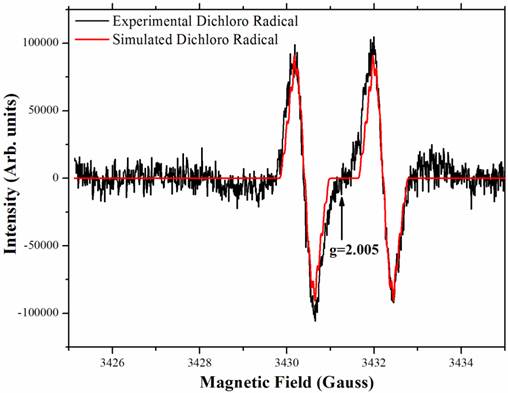
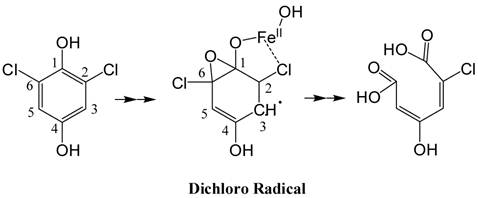
The PcpA-catalyzed ring cleavage reaction of 2,6-DCHQ was studied by EPR at room temperature. As shown in Figure 5, a stabilized radical intermediate was discovered. Two lines were measured with g = 2.005 and hyperfine splitting of 1.8 Gauss. Further examination suggested that these lines are wider than expected for the simple hyperfine of a single hydrogen atom. The linewidth, lineshape and structural features that are barely discernable above noise suggest a complex unresolved spectrum. EPR spectral simulation revealed that the radical was a dichloro radical, and the potential structure was determined to be an epoxide type radical with the unpaired electron density on C3 (Figure A/Scheme 1). The spin 1/2 of the hydrogen atom on C5 gave rise to the doublet and spin 3/2 of the Cl atoms gave rise to the nearly resolved superhyperfine structure. This superhyperfine was unresolved due to the Cl nuclear g value of 0.547 compared to 5.58 for hydrogen. The two Cl atoms stabilized the radical structure, which has been predicted to be stable in previous EPR experiments (41). Hydrolysis of this dichloro radical cleaves the aromatic ring to generate the catalytic product 2-CMA. To further examine whether ascorbic acid can affect the stability of the dichloro radical, different concentrations (25 µM-2 mM) of ascorbic acid were examined in the experiment and we did not observe any change for the radical.
Characterization of the iron status during PcpA catalysis by low temperature EPR. The experiment was undertaken at 10K with a fixed microwave frequency of 9.625 GHz on an X-bank Bruker EMX EPR spectrometer. A clear rhombic Fe(III) signal with g = 4.29 was observed for the prepared PcpA sample (shown in black). When ascorbic acid (25 - 250 µM) was added to the sample, the EPR signal was slightly reduced (shown in red). However, the Fe(III) signal increased dramatically after the enzymatic reaction was initiated (shown in blue). The reaction mixture contained 50 mM oxygen-saturated phosphate buffer, pH 7.0, 1 mg/mL PcpA, 250 µM 2,6-DCHQ and 100 µM ascorbic acid.
The stable radical intermediate revealed by room temperature EPR during 2,6-DCHQ ring cleavage. The reaction mixture (volume: 0.5 mL) contained 50 mM oxygen-saturated phosphate buffer, pH 7.0, 1 mg/mL PcpA, 250 µM 2,6-DCHQ and ascorbic acid with different concentrations (25 µM - 2 mM). The reaction mixture was incubated at room temperature for 2 min before being injected into a quartz tube to collect the EPR signal. Simulation based on the predicted epoxide-type dichloro radical (Figure A/Scheme 1) perfectly matched the observed signal.
(Scheme 1): Predicted structure of the stable radical intermediate during PcpA catalysis. The EPR simulation indicated that the radical intermediate is likely an epoxide type dichloro radical with the unpaired electron density on C3.
Conclusion
The S. chlorophenolicum PCP biodegradation pathway is still in the early stage of molecular evolution and of very low metabolic efficiency (19). PcpA, the Fe(II)-dependent ring-cleavage enzyme in the pathway, catalyzes the dehalogenation and oxidation of 2,6-DCHQ to generate 2-CMA. In this study, we showed that recombinant PcpA exhibited two activity peaks at pH 7.0 and 10.0, respectively. The apparent kcat and Km were measured as 0.19 ± 0.01 s-1 and 0.24 ± 0.08 mM, respectively at pH 7.0, and 0.17 ± 0.01 s-1 and 0.77 ± 0.29 mM, respectively at pH 10.0. PcpA was also shown to be alkaline resistant and thermally stable. It maintained its secondary structure as well as low but significant activity even after four days of incubation in 50 mM phosphate solution, pH 13.0. Because about 80% of PCP was used in combination with NaOH in wood preservation (37), it is likely that S. chlorophenolicum may have not only quickly assembled a pathway to degrade PCP but also evolved its enzymes to tolerate the alkaline conditions in spite of that its optimal growth condition is at pH 7.0 (36). Rapid oxidation of Fe(II) to Fe(III) in PcpA and the formation of a stable epoxide-type dichloro radical intermediate were observed during the enzyme catalysis using electron paramagnetic resonance. Further investigations have been initiated on studying the detailed catalytic mechanism of PcpA, especially the role of the Fe(II) ion, understanding why PcpA lost its catalytic activity under acidic pHs, and establishing a standard protocol to measure the PcpA activity at different temperatures.
Supplementary Material
Fig.S1 - CD spectrum of PcpA (1 mg/mL) after 4 days incubation in 50 mM phosphate solution, pH 13.0. Fig.S2 - The stable dichlorosemiquinone radical formed from rapid oxidation of 2,6-dichlorohydroquinone (less than 30 seconds) was revealed by room temperature EPR.
Acknowledgements
This work was supported by a Discovery Grant from the Natural Sciences and Engineering Research Council (NSERC) of Canada, a New Investigator Establishment Grant and a New Investigator Equipment Grant from the Saskatchewan Health Research Foundation (SHRF), and a New Opportunities Fund from the Canada Foundation for Innovation (CFI). The CD and EPR measurements were completed at the Saskatchewan Structural Sciences Centre, which is supported by the University of Saskatchewan.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Frank R, Braun HE, Ripley BD, Clegg BS. Contamination of rural ponds with pesticides, 1971-85, Ontario, Canada. Bull Environ Contam Toxicol. 1990;44:401-9
2. Ingram LL, Tarlton K. Effect of physical properties of pentachlorophenol and creosote components on vaporization from treated wood: Review of prior data. Forest Prod J. 2005;55:86-9
3. Thompson TS, Treble RG, Mariasegaram M. Determination of atmospheric contamination by pentachlorophenol using pine needles located near treated utility pole storage sites. Bull Environ Contam Toxicol. 1997;59:548-55
4. Sai K, Kang KS, Hirose A, Hasegawa R, Trosko JE, Inoue T. Inhibition of apoptosis by pentachlorophenol in v-myc-transfected rat liver epithelial cells: relation to down-regulation of gap junctional intercellular communication. Cancer Lett. 2001;173:163-74
5. Khurana V, Barkin JS. Pancreatitis induced by environmental toxins. Pancreas. 2001;22:102-5
6. Daniel V, Huber W, Bauer K, Suesal C, Mytilineos J, Melk A, Conradt C, Opelz G. Association of elevated blood levels of pentachlorophenol (PCP) with cellular and humoral immunodeficiencies. Arch Environ Health. 2001;56:77-83
7. Peper M, Ertl M, Gerhard I. Long-term exposure to wood-preserving chemicals containing pentachlorophenol and lindane is related to neurobehavioral performance in women. Am J Ind Med. 1999;35:632-41
8. D'Angelo EM, Reddy KR. Aerobic and anaerobic transformations of pentachlorophenol in wetland soils. Soil Sci Soc Am J. 2000;64:933-43
9. Tisch M, Faulde MK, Maier H. Genotoxic effects of pentachlorophenol, lindane, transfluthrin, cyfluthrin, and natural pyrethrum on human mucosal cells of the inferior and middle nasal conchae. Am J Rhinol. 2005;19:141-51
10. Villela IV, de Oliveira IM, da Silva J, Henriques JA. DNA damage and repair in haemolymph cells of golden mussel (Limnoperna fortunei) exposed to environmental contaminants. Mutat Res. 2006;605:78-86
11. McAllister KA, Lee H, Trevors JT. Microbial degradation of pentachlorophenol. Biodegradation. 1996;7:1-40
12. Dimmock JR. Problem solving learning: applications in medicinal chemistry. Am J Pharm Educ. 2000;64:1
13. Valenti P, Recanatini M, Da Re P, Cima L, Giusti P. Halogenated dimefline-type derivatives. Archiv der Pharmazie. 2000;316:421-26
14. Warner JR, Behlen LS, Copley SD. A trade-off between catalytic power and substrate inhibition in TCHQ dehalogenase. Biochemistry. 2008;47:3258-65
15. Xun L, Orser CS. Purification of a Flavobacterium pentachlorophenol-induced periplasmic protein (PcpA) and nucleotide sequence of the corresponding gene (pcpA). J Bacteriol. 1991;173:2920-6
16. Xun L, Orser CS. Purification and properties of pentachlorophenol hydroxylase, a flavoprotein from Flavobacterium sp. strain ATCC 39723. J Bacteriol. 1991;173:4447-53
17. Xun L, Topp E, Orser CS. Purification and characterization of a tetrachloro-p-hydroquinone reductive dehalogenase from a Flavobacterium sp. J Bacteriol. 1992;174:8003-7
18. McCarthy DL, Claude AA, Copley SD. In vivo levels of chlorinated hydroquinones in a pentachlorophenol-degrading bacterium. Appl Environ Microbiol. 1997;63:1883-8
19. Dai M, Rogers JB, Warner JR, Copley SD. A previously unrecognized step in pentachlorophenol degradation in Sphingobium chlorophenolicum is catalyzed by tetrachlorobenzoquinone reductase (PcpD). J Bacteriol. 2003;185:302-10
20. Copley SD. Evolution of a metabolic pathway for degradation of a toxic xenobiotic: the patchwork approach. Trends Biochem Sci. 2000;25:261-5
21. Miyauchi K, Adachi Y, Nagata Y, Takagi M. Cloning and sequencing of a novel meta-cleavage dioxygenase gene whose product is involved in degradation of γ-hexachlorocyclohexane in Sphingomonas paucimobilis. J Bacteriol. 1999;181:6712-9
22. Wang H, Tiirola MA, Puhakka JA, Kulomaa MS. Production and characterization of the recombinant Sphingomonas chlorophenolica pentachlorophenol 4-monooxygenase. Biochem Biophys Res Commun. 2001;289:161-6
23. Xun L, Topp E, Orser CS. Diverse substrate range of a Flavobacterium pentachlorophenol hydroxylase and reaction stoichiometries. J Bacteriol. 1992;174:2898-902
24. Chen L, Maloney K, Krol E, Zhu B, Yang J. Cloning, overexpression, purification, and characterization of the maleylacetate reductase from Sphingobium chlorophenolicum strain ATCC 53874. Curr Microbiol. 2009;58:599-603
25. Chen L, Yang J. Biochemical characterization of the tetrachlorobenzoquinone reductase involved in the biodegradation of pentachlorophenol. Int J Mol Sci. 2008;9:198-212
26. Dai M, Copley SD. Genome shuffling improves degradation of the anthropogenic pesticide pentachlorophenol by Sphingobium chlorophenolicum ATCC 39723. Appl Environ Microbiol. 2004;70:2391-7
27. Huang Y, Xun R, Chen G, Xun L. Maintenance role of a glutathionyl-hydroquinone lyase (PcpF) in pentachlorophenol degradation by Sphingobium chlorophenolicum ATCC 39723. J Bacteriol. 2008;190:7595-600
28. Cai M, Xun L. Organization and regulation of pentachlorophenol-degrading genes in Sphingobium chlorophenolicum ATCC 39723. J Bacteriol. 2002;184:4672-80
29. Lee JY, Xun L. Purification and characterization of 2,6-dichloro-p-hydroquinone chlorohydrolase from Flavobacterium sp. Strain ATCC 39723. J Bacteriol. 1997;179:1521-4
30. Xun L, Bohuslavek J, Cai M. Characterization of 2,6-dichloro-p-hydroquinone 1,2-dioxygenase (PcpA) of Sphingomonas chlorophenolica ATCC 39723. Biochem Biophys Res Commun. 1999;266:322-5
31. Chanama S, Crawford RL. Mutational analysis of pcpA and its role in pentachlorophenol degradation by Sphingomonas (Flavobacterium) chlorophenolica ATCC 39723. Appl Environ Microbiol. 1997;63:4833-8
32. Milliken CE, Meier GP, Watts JEM, Sowers KR, May HD. Microbial anaerobic demethylation and dechlorination of chlorinated hydroquinone metabolites synthesized by basidiomycete fungi. Appl Environ Microbiol. 2004;70:385-92
33. Machonkin TE, Holland PL, Smith KN, Liberman JS, Dinescu A, Cundari TR, Rocks SS. Determination of the active site of Sphingobium chlorophenolicum 2,6-dichlorohydroquinone dioxygenase (PcpA). J Biol Inorg Chem. 2010;15:291-301
34. Su Y, Chen L, Bandy B, Yang J. The catalytic product of pentachlorophenol 4-monooxygenase is tetrachlorohydroquinone rather than tetrachlorobenzoquinone. Open Microbiol J. 2008;2:100-6
35. Böhm G, Muhr R, Jaenicke R. Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 1992;5:191-5
36. Crawford RL. Biodegradation of pentachlorophenol. US: US Patent No 4713340. 1987
37. Bellin CA, O'Connor GA, Jin YT. Sorption and degradation of pentachlorophenol in sludge-amended soils. J Environ Qual. 1990;19:603-8
38. Albery WJ, Knowles JR. Evolution of enzyme function and the development of catalytic efficiency. Biochemistry. 1976;15:5631-40
39. Fujiwara S, Lee SG, Haruki M, Kanaya S, Takagi M, Imanaka T. Unusual enzyme characteristics of aspartyl-tRNA synthetase from hyperthermophilic archaeon Pyrococcus sp. KOD1. FEBS Lett. 1996;394:66-70
40. Arciero DM, Lipscomb JD, Huynh BH, Kent TA, Münck E. EPR and Mössbauer studies of protocatechuate 4,5-dioxygeanse. Characterization of a new Fe2+ environment. J Biol Chem. 1983;258:14981-91
41. Song Y, Buettner GR, Parkin S, Wagner BA, Robertson LW, Lehmler HJ. Chlorination increases the persistence of semiquinone free radicals derived from polychlorinated biphenyl hydroquinones and quinones. J Org Chem. 2008;73:8296-304
Author contact
![]() Corresponding author: Jian Yang, College of Pharmacy and Nutrition, University of Saskatchewan, 110 Science Place, Saskatoon, Saskatchewan S7N 5C9, Canada. Tel: 306-966-6361; Fax: 306-966-6377; E-mail: jian.yangca
Corresponding author: Jian Yang, College of Pharmacy and Nutrition, University of Saskatchewan, 110 Science Place, Saskatoon, Saskatchewan S7N 5C9, Canada. Tel: 306-966-6361; Fax: 306-966-6377; E-mail: jian.yangca
Received 2011-6-30
Accepted 2011-10-17
Published 2011-10-25