ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2011; 7(8):1180-1187. doi:10.7150/ijbs.7.1180 This issue Cite
Research Paper
Early Exposure to Paraquat Sensitizes Dopaminergic Neurons to Subsequent Silencing of PINK1 Gene Expression in Mice
Hongxia Zhou1 ![]() , Cao Huang2, Jianbin Tong2, Xu-Gang Xia2
, Cao Huang2, Jianbin Tong2, Xu-Gang Xia2 ![]()
1. Department of Neurology, Anatomy & Cell Biology; Thomas Jefferson University, 1020 Locust Street, Philadelphia, PA 19107, USA
2. Department of Pathology, Anatomy & Cell Biology; Thomas Jefferson University, 1020 Locust Street, Philadelphia, PA 19107, USA
Abstract
Environmental exposure, genetic modification, and aging are considered risky for Parkinson's disease (PD). How these risk factors cooperate to induce progressive neurodegeneration in PD remains largely unknown. Paraquat is an herbicide commonly used for weed and grass control. Exposure to paraquat is associated with the increased incidence of PD. In contrast to familial PD, most sporadic PD cases do not have genetic mutation, but may suffer from partial dysfunction of neuron-protective genes as aging. Using conditional transgenic RNAi, we showed that temporal silencing of PINK1 expression in adult mice increased striatal dopamine, the phenotype that could not be induced by constitutive gene silencing. Moreover, early exposure to paraquat sensitized dopaminergic neurons to subsequent silencing of PINK1 gene expression, leading to a significant loss of dopaminergic neurons. Our findings suggest a novel pathogenesis of PD: exposure to environmental toxicants early in the life reduces the threshold of developing PD and partial dysfunction of neuron-protective genes later in the life initiates a process of progressive neurodegeneration to cross the reduced threshold of disease onset.
Keywords: Parkinson's disease, PINK1, pathogenesis, paraquat, RNAi, mice
Introduction
Parkinson disease (PD) results from progressive degeneration of dopaminergic neurons in the midbrain. PD is characterized by resting tremor, bradykinesia, and muscle rigidity (1). At present, no treatment can protect PD-susceptible neurons or prevent the disease from progression, although dopamine replenishment therapy temporally alleviates PD symptoms (2, 3). Limited knowledge of PD pathogenesis is a major roadblock in the development of effective therapy for this devastating disease. Recent advance in epidemiological studies suggests PD as a multifactorial disease. Aging, genetic and environmental factors are considered risky for PD, but how these risk factors interact to cause the disease remains largely unknown.
Occupational exposure to pesticides and cumulative exposure to the herbicide paraquat increase the risk of developing PD (4, 5). Repeated exposure to environmental toxicants is associated with the increased risk of PD, but no single environmental toxicant has been definitively linked to PD, suggesting that the other risk factors must be involved in the development of sporadic PD. Genetic factors influence the susceptibility to PD. The relatives of PD patients show an increased incidence of PD (6). While most PD cases are sporadic, about 10% of the cases have pathogenic mutations in the individual genes including alpha-synuclein, Leucine-Rich Repeat Kinase 2 (LRRK2), parkin, DJ-1, and PTEN-induced putative kinase 1 (PINK1) (7-13). Intriguingly, some PD patients carry mutation only in the one allele of the recessive genes parkin and PINK1 and some people carrying mutation in the dominant gene LRRK2 do not develop PD in their lives (14-16). The penetrance and expressivity of PD phenotypes appears to be determined not only by pathogenic mutation in the identified genes, but also by undefined factors.
Dysfunction of neuron-protective genes and exposure to toxicants may corporate to induce dopaminergic neurodegeneration in sporadic PD. Indeed, deletion of the neuron-protective genes such as parkin, DJ-1, and PINK1 sensitizes dopaminergic neurons to the toxicants MPTP and paraquat (17-24). Most sporadic PD cases do not have genetic mutation, but they may suffer from partial dysfunction of neuron-protective genes later in lives. For example, Parkin is S-nitrosylated in PD animal models and in PD patients (25, 26). S-nitrosylation inhibits Parkin ligase activity and induces accumulation of improperly folded and biologically inactive protein, causing a loss of the neuron-protective functions of Parkin. As aging, some neuron-protective genes may be partially inactivated due to epigenetic modification. Exposure to environmental toxicants and genetic failure may not occur concomitantly, but may occur sequentially. Perhaps, exposure to environmental toxicants early in the life causes accumulative effects and sensitizes dopaminergic neurons to the partial dysfunction of neuron-protective genes later in the life, causing sporadic PD. To obtain the partial dysfunction of a neuron-protective gene temporally in adult mice, we created conditional PINK1-RNAi transgenic mice, which were treated with paraquat before the PINK1 gene was partially silenced in the adulthood. We observed that early exposure to the environmental toxicant paraquat sensitized dopaminergic neurons to subsequent dysfunction of the PINK1 gene.
Materials and Methods
Transgenic mice and behavioral analysis
PINK1 RNAi transgenic mice have been characterized (27). Tamoxifen inducible Cre (CreERT) transgenic mice were purchased from Jackson Laboratories (28). Cre was driven by the ubiquitous hybrid promoter CAG and was expressed in all tissues (28). To minimize the leakage of Cre activity, male CreERT mice were crossed with female PINK1-RNAi mice to produce doubly transgenic offspring. Transgenic mice were identified by PCR analysis of tail DNA as described previously (27). As stated in the results, paraquat (Sigma) was administered to transgenic mice at the age of 14 days (0.3 mg/kg, subcutaneous injection; once) or at the age of 12 weeks (10mg/kg, twice a week, for 3 consecutive weeks). To induce a marked neurotoxicity, paraquat was repeatedly administered to selected mice at the postnatal ages and again in the adulthood. At the age of 16 weeks, selected mice were injected with 4-hydroxyl Tamoxifen (4-OHT; Sigma) to induce Cre activity and thus to activate PINK1 RNAi transgene. To achieve a full activation of PINK1 RNAi transgene, 4-OHT was administered by intraperitoneal injection for 5 consecutive days (50 mg/kg/day). The locomotor activity of transgenic mice within 30 minutes was measured by open field activity assay (Med Associates). Animal use followed NIH guidelines and the animal use protocol was approved by the Institutional Animal Care and Use Committees at the University.
Microdialysis and HPLC analysis
As described previously (29), the extracellular level of dopamine released into the striatum was measured by intra-brain microdialysis combined with HPLC. Anesthetized mice were implanted with a guide cannula the day before dialysis. On the following day, dialysis probe (BASi) was inserted through the guide cannula into the left striatum (coordinates: 0.5 mm anterior to bregma; 2 mm left from sagittal line; 1.5 mm ventral to the skull). Continuous perfusion was maintained with modified Ringer's solution (147 mm Na+, 4 mm K+, 1.3 mm Ca2+, 1 mm Mg2+, and 155.6 mm CI-). Dialysates were continuously collected and the dialysates of every 20-minute were pooled as one sample for HPLC analysis. Basal dopamine release was established within two hours when comparable levels of dopamine released were obtained in three consecutive samplings (20 minutes each). The DAT inhibitor nomifensine was applied to dialysis solution (200 µM) for 40 minutes (two samplings) and enhanced dopamine release was then measured. Dopamine concentration in dialysates was instantly measured by HPLC. The content of striatal dopamine was measured by HPLC as described previously (27). Briefly, the striatum was dissected from mouse brain and was homogenized in 0.1 M perchloric acid. Dopamine in cleared lysates was measured by HPLC. Tissue pellets were dissolved in 1M NaOH and protein concentration was determined. The contents of striatal dopamine were adjusted with protein concentration.
Analysis of gene expression by PCR
PCR was done as described previously (30). In brief, total RNA was isolated from mouse tissues and the total RNA of 1µg was reversely transcribed to cDNA with oligo-dT primer (RT kit, Invitrogen). The resulting cDNA was used PCR amplification. The following primers were used to amplify specific mouse genes: primers 5'-GCTTGCCAATCCCTTCTATG-3' (forward) and 5'-CTCTCGCTGGAGCAGTGAC-3' (reverse) for PINK1; primers 5'-CGGTATAATGGTGGAGTTG-3' (forward) and 5'-ACCCTTAAGTTCAGCGTTACT-3' (reverse) for L17; primers 5'-CAGTGTGGACTTCTACCGTC-3' (forward) and 5'-AACTGGAGAAGGCAATCAGC-3' (reverse) for DAT; And primers 5'- CTATCCTCATGGACCACAAC-3' (forward) and 5'-GCTCAGTCACTCTCAGATTC-3' (reverse) for VMAT2. Cycling conditions were 2 min at 94°C followed by 26 or 30 cycles of 15 seconds at 94°C, 30 seconds at 60°C, and 20 seconds at 72°C. PCR products were resolved on 2% agarose gels.
Histology and stereological cell counting
TH immunostaining and stereological cell counting were done as described previously (27, 31). Mouse brain was cut on a Cryostat into serial sections of 30 µm. Every fourth section through the SNpc was immunostained for TH (Pel-Freez; 1: 1000). TH positive neurons in the SNpc were estimated by stereological cell counting.
Statistical analysis
Statistical significance among group means was assessed by ANOVA followed by Tukey's post hoc test. In all analyses, the null hypothesis was rejected at the level of 0.05.
Results
Exposure to the herbicide paraquat increases the risk of PD (5), and thus we chose paraquat as an example of environmental toxicants. Previous studies showed that repeated exposure to paraquat causes severe damage to dopaminergic system (32). To assess whether early exposure to environmental toxicants sensitizes dopaminergic neurons to later dysfunction of neuron-protective genes, we first determined an optimal dosage of paraquat that is sufficient to induce a moderate loss of dopaminergic neurons in the mouse strains established in our lab (Figure 1). We treated male C57BL6 mice with paraquat by subcutaneous injection and observed a moderate, but significant, loss of dopaminergic neurons in the SNpc and a marked depletion of striatal dopamine (Figure 1). Some mice were injected of paraquat during postnatal development and again in adulthood and a greater loss of dopaminergic neurons was induced by repeated exposure to paraquat (Figure 1). Paraquat caused accumulative damage to dopaminergic neurons in mice. We further examined whether dopaminergic neurotransmission would be compensated in wildtype mice after the toxicant is removed. As expected, lost dopaminergic neurons were never restored (Figure 2a). In contrast, synaptic release of dopamine was significantly increased in the mice after paraquat was withdrawn (Figure 2b). Synaptic release of dopamine is virtually related to dopaminergic function and thus, the findings suggest that the dopaminergic function may be compensated after the risk factor is removed.
Recessive mutation of the PINK1 gene is linked to familial PD (10). We chose PINK1 as an example of neuron-protective genes to examine PD pathogenesis. We have created conditional transgenic mice that express PINK1 RNAi upon Cre-mediated activation of the transgene and display a significant knockdown of PINK1 gene expression by the RNAi (27). Constitutive depletion of PINK1 by gene knockout or by transgenic RNAi does not induce dopaminergic neuron death in the mutant mice (27, 33). To avoid any potential compensation for gene loss during mouse development, we used inducible Cre to activate PINK1 RNAi transgene in adult mice and thus to silence PINK1 gene expression in the adulthood (Figure 3a). Similar to constitutive expression of PINK1 RNAi (27), temporal expression of PINK1 RNAi silenced the expression of PINK1 and did not affect the expression of the dopamine transporter (DAT) and the vesicular monoamine transporter 2 (VMAT2) genes in transgenic mice (Figure 3a). Consistent with constitutive gene silencing (27), temporal silencing of PINK1 gene expression failed to induce dopaminergic neuron death in transgenic mice at advanced ages (Figure 3c). Unexpectedly, temporal silencing of PINK1 gene expression in adult mice increased striatal dopamine and enhanced locomotor activity (Figure 3b and 3d). The effect of PINK1 gene silencing on striatal dopamine was diminished as aging (Figure 3). Temporal silencing of PINK1 gene expression in adult mice disturbed dopamine homeostasis, uncovering the phenotypes that were obscured possibly by developmental compensation.
To test whether early exposure to environmental toxicants sensitizes dopaminergic neurons to partial dysfunction of neuron-protective genes, we first administered paraquat to PINK1-RNAi transgenic mice and then silenced the expression of the PINK1 gene by inducing Cre activity (Figure 3a). Surprisingly, a greater depletion of striatal dopamine and dopaminergic neurons was induced by a combination of early exposure to paraquat and subsequent silencing of PINK1 gene expression, compared to either of the individual treatments (Figure 4). The results suggest that environmental exposure and genetic defects may sequentially occur and synergistically induce a progressive degeneration of dopaminergic neurons in PD.
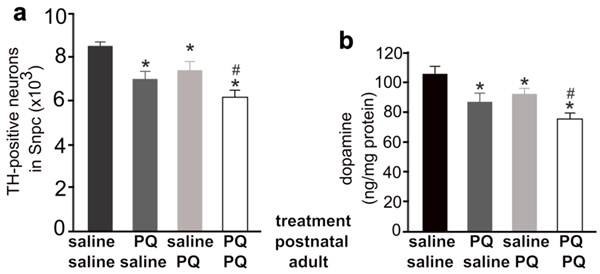
Different combination of paraquat and saline was administered to the mice at the postnatal and the adult ages. a, Stereological cell counting revealed a significant loss of TH-positive neurons in the SNpc of paraquat (PQ)-treated mice. PQ or saline was administered to male mice at the age of 14 days (0.3mg/kg, once) and further at the age of 12 weeks (10mg/kg, twice a week, for 3 consecutive weeks). One week after last PQ injection, the mice were killed for analysis. b, HPLC analysis revealed the contents of dopamine in the striatum of PQ-treated and -untreated mice. Data are means ± SEM (n = 7-8). * p < 0.05 compared to the control mice only injected of saline; # p < 0.05 compared to the mice injected of paraquat at the age of 14 days.
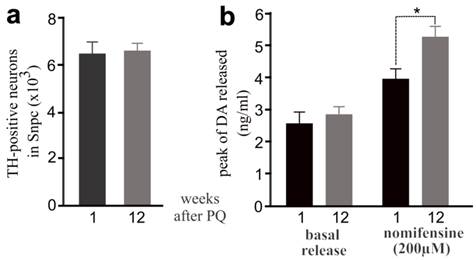
Elevation of dopamine release was observed in mice after PQ treatment. a, Stereological cell counting revealed no progressive loss of TH-positive neurons in PQ-treated mice after paraquat withdrawal. b, Intra-striatum microdialysis revealed extracellular levels of dopamine released from dopaminergic terminals. Male mice were injected with PQ at the age of 14 days (0.3mg/kg, once) and further at the age of 12 weeks (10mg/kg, twice a week, for 3 consecutive weeks). Microdialysis was performed in the mice at 1 or 12 weeks after last injection of PQ. By the end of microdialysis, the mice were terminated for stereological cell counting. Data are means ± SEM (n = 5). * p < 0.05.
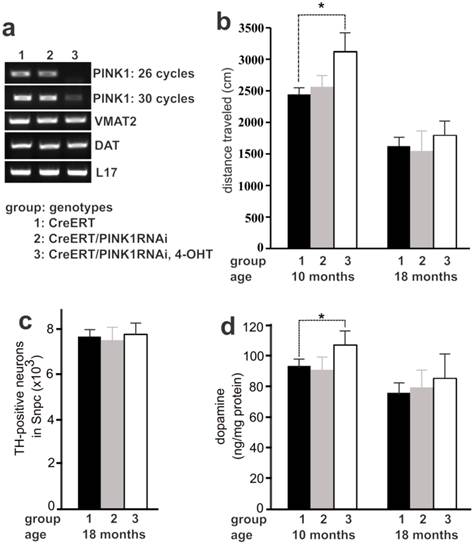
Temporal silencing of PINK1 expression in adult mice disturbed dopaminergic function. a, PCR analysis revealed a significant knockdown of PINK1 expression by RNAi. In CreERT and PINK1-RNAi double transgenic mice, Cre activity was induced by injection of 4-hydroxyl Tamoxifen (4-OHT). Gene expression in the midbrain was analyzed by PCR one month after 4-OHT induction. b, Locomotor activity in 30 minutes was measured by open field activity assay. Mice at the age of 16 weeks were treated with 4-OHT to induce gene silencing and were tested for locomotor activity at 10 months and again at 18 months of age. Data are means ± SEM (n = 6). * p < 0.05. c, Stereological cell counting revealed the number of TH-positive neurons in the SNpc. Data are means ± SEM (n = 5). d, HPLC measured the contents of striatal dopamine. Mice of individual group were terminated at the age of 10 or 18 months for cell counting. Data are means ± SEM (n = 5). * p < 0.05.
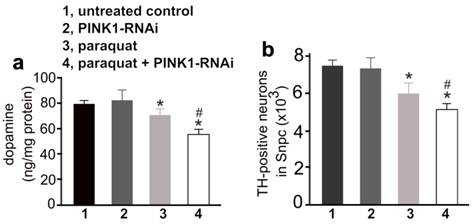
Early exposure to paraquat sensitized dopaminergic neurons to subsequent silencing of PINK1 gene expression. a, HPLC measured the contents of striatal dopamine in mice. The CreERT single (1, untreated control; 3, paraquat) and the CreERT/PINK1-RNAi double transgenic mice (group 2 and 4) were injected with paraquat or saline at the age of 14 days and further at the age of 12 weeks (10mg/kg, twice a week, for 3 consecutive weeks). One week later, the mice were injected with 4-OHT to induce Cre activity and thus to activate PINK1-RNAi. At the age of 20 months, the mice were terminated for analysis. b, Stereological cell counting revealed the number of TH-positive neurons in the SNpc. Data are means ± SEM (n = 10). * p < 0.05 compared to untreated control; # p < 0.05 compared to group 3 (paraquat).
Discussion
Exposure to environmental toxicants, genetic modification of critical genes, and physiological aging are suggested contributing to PD pathogenesis. We showed that repeated exposure to paraquat caused accumulative damage to dopaminergic neurons in mice. By transgenic RNAi, we silenced PINK1 expression selectively in adult mice and observed a marked change to dopamine homeostasis that could not be induced by constitutive silencing of PINK1 expression. Early exposure to paraquat sensitized dopaminergic neurons to subsequent silencing of PINK1 expression in aged mice. Our findings suggest a potential pathogenesis for PD: exposure to environmental toxicants early in the life reduces the threshold of developing PD; partial dysfunction of neuron-protective genes later in the life continuously compromises dopaminergic function and eventually causes the system to cross the reduced threshold of developing PD.
Paraquat is an herbicide commonly used for weed and grass control. Exposure to paraquat is associated with the increased risk of PD (5). In the most studies, paraquat is administered by intraperitoneal injection and few studies (32) show a statistically significant loss of striatal dopamine content, although a trend of dopamine loss is always observed. We first tested intraperitoneal injection of paraquat and observed a severe loss of treated mice (data not shown). Compared to intraperitoneal injection of MPTP, subcutaneous injection of MPTP induces a more severe loss of dopaminergic neurons with less variation in phenotypic expression. We then examined the subcutaneous injection of paraquat and observed a significant loss of striatal dopamine content. Our findings in mice are in great consistency with previous findings in rats that are subcutaneously injected with paraquat (34). Intraperitoneal injection may lead to an abrupt absorption of neurotoxicant such as paraquat and cause a remarkable fluctuation of blood and tissue paraquat, leading to varied phenotypes. By contrast, subcutaneous injection of paraquat or MPTP may cause less fluctuation of the chemicals in tissues, producing a relatively constant effect. Consistent with previous findings (32, 35-39), our results confirmed that exposure to paraquat caused substantial and accumulative damage to dopaminergic neurons in mice. The detrimental effects of environmental toxicants may accumulate, but may not cause a progressive loss of dopaminergic neurons unless recurrent exposure to toxicants happens frequently. In fact, the reduction in synaptic release of dopamine was partly compensated after paraquat was removed though the lost neurons were never restored. The finding suggests that dopaminergic dysfunction stopped progression after the environmental toxicant was eliminated. Exposure to environmental toxicants reduces the threshold of PD onset, and continuous presence of risk factors is required to induce progressive degeneration of dopaminergic neurons and to cross the reduced threshold of disease onset. Such a lasting insult is likely from an endogenous source in sporadic PD.
As aging, the functions of some neuron-protective genes may be compromised (25, 26). While pathogenic mutation of the two PINK1 alleles definitely causes PD (10), mutation of the one PINK1 allele also increases PD susceptibility (40, 41), suggesting that PINK1 has dose-dependent effects on dopaminergic neurons. Unlike familial PD, sporadic PD may not have inheritable mutation in neuron-protective genes, but may suffer from partial dysfunction of the beneficial genes later in the life (25, 26). In contrast to previous studies (42-44), our study applied transgenic RNAi technology to partially silence the expression of the neuron-protective gene PINK1 selectively in adult mice. Compared to constitutive gene knockout, temporal silencing of PINK1 expression in adult mice increased the content of striatal dopamine, possibly because it avoided developmental compensation for the gene loss. Interestingly, simultaneous deletion of parkin, DJ-1, and PINK1 also increases striatal dopamine in the triple knockout mice at advanced ages (45). By contrast, deleting any of the three genes (parkin, DJ-1, and PINK1) alone fails to induce the phenotype in the lifetime of single-gene knockout mice (33, 46-48). Intriguingly, striatal dopamine was decreased to the control level in our PINK1-RNAi mice at advanced ages. Alteration to dopamine neurotransmission may be an early event in the process of dopaminergic neurodegeneration.
Given that environmental toxicants cause accumulative damage to dopaminergic neurons, exposure to toxicants any time in the life will reduce the threshold of developing PD and will increase the incidence of PD. Immature brain is vulnerable to neurotoxins (32), and exposure to environmental toxicants early in the life may play an important role in PD pathogenesis. Previous studies have shown that complete and constitutive deletion of neuron-protective genes such as parkin, DJ-1, and PINK1 sensitizes dopaminergic neurons to neurotoxins (17-24). Previous studies all test the effects of environmental toxicants on dopaminergic system after a neuroprotective gene is permanently inactivated in mice. By striking contrast, we used a reverse order of gene-environment interaction to examine how PD risky factors corporate to induce the disease. We assessed the effects of early paraquat treatment on the phenotypic expression caused by partial and temporal depletion of the PINK1 gene, testing a new theory on PD pathogenesis. Conditional transgenic RNAi provides an unprecedented tool for temporal, partial gene depletion in animal models. Temporal silencing of PINK1 expression by transgenic RNAi induced a significant loss of dopaminergic neurons in aged mice, which received paraquat treatment early in their lives. Endogenous and exogenous toxins may execute toxicity to dopaminergic neurons via varying mechanisms (49-53). Since RNAi may cause nonspecific toxicity or side-effects, it could not be ruled out whether the outcome in PINK1-RNAi transgenic mice is a combined effect of environmental toxicant, gene silencing, and RNAi toxicity. Regardless of mechanisms, sequential insults from multiple hits may cause accumulative damage to dopaminergic neurons and any lasting insults may initiate a progressive degeneration of dopaminergic neurons, inducing PD onset. The novelty of this hypothetic PD pathogenesis warrants further clinical and epidemiological studies.
Acknowledgements
This work is supported by grants from the National Institute of Neurological Disorders and Stroke and National Institute of Environmental Health Sciences (NS064042 and ES016760 to X.G.X). The content is the responsibility of the authors and does not necessarily represent the official view of the funding institutes.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Schulz JB. Mechanisms of neurodegeneration in idiopathic Parkinson's disease. Parkinsonism Relat Disord. 2007;13(Suppl 3):S306-308
2. Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson's disease. Science. 2003;302(5646):819-822
3. Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson's disease. Neuron. 2010;66(5):646-661
4. Baldi I, Lebailly P, Mohammed-Brahim B, Letenneur L, Dartigues JF, Brochard P. Neurodegenerative diseases and exposure to pesticides in the elderly. Am J Epidemiol. 2003;157(5):409-414
5. Liou HH, Tsai MC, Chen CJ, Jeng JS, Chang YC, Chen SY, Chen RC. Environmental risk factors and Parkinson's disease: a case-control study in Taiwan. Neurology. 1997;48(6):1583-1588
6. Foltynie T, Sawcer S, Brayne C, Barker RA. The genetic basis of Parkinson's disease. J Neurol Neurosurg Psychiatry. 2002;73(4):363-370
7. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG. et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276(5321):2045-2047
8. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605-608
9. Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G. et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299(5604):256-259
10. Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S. et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304(5674):1158-1160
11. Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P. et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601-607
12. Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS. et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44(4):595-600
13. Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK. Parkinson's disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci. 2006;26(1):41-50
14. Clark LN, Afridi S, Karlins E, Wang Y, Mejia-Santana H, Harris J, Louis ED, Cote LJ, Andrews H, Fahn S, Waters C, Ford B, Frucht S, Ottman R, Marder K. Case-control study of the parkin gene in early-onset Parkinson disease. Arch Neurol. 2006;63(4):548-552
15. Bonifati V, Rohe CF, Breedveld GJ, Fabrizio E, De Mari M, Tassorelli C, Tavella A, Marconi R, Nicholl DJ, Chien HF, Fincati E, Abbruzzese G, Marini P, De Gaetano A, Horstink MW. et al. Early-onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology. 2005;65(1):87-95
16. Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 2007;6(7):652-662
17. Gegg ME, Cooper JM, Schapira AH, Taanman JW. Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS ONE. 2009;4(3):e4756
18. Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW, Duchen MR, Abramov AY. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell. 2009;33(5):627-638
19. Haque ME, Thomas KJ, D'Souza C, Callaghan S, Kitada T, Slack RS, Fraser P, Cookson MR, Tandon A, Park DS. Cytoplasmic Pink1 activity protects neurons from dopaminergic neurotoxin MPTP. Proc Natl Acad Sci U S A. 2008;105(5):1716-1721
20. Sallinen V, Kolehmainen J, Priyadarshini M, Toleikyte G, Chen YC, Panula P. Dopaminergic cell damage and vulnerability to MPTP in Pink1 knockdown zebrafish. Neurobiol Dis. 2010;40(1):93-101
21. Gonzalez-Polo R, Niso-Santano M, Moran JM, Ortiz-Ortiz MA, Bravo-San Pedro JM, Soler G, Fuentes JM. Silencing DJ-1 reveals its contribution in paraquat-induced autophagy. J Neurochem. 2009;109(3):889-898
22. Yang W, Chen L, Ding Y, Zhuang X, Kang UJ. Paraquat induces dopaminergic dysfunction and proteasome impairment in DJ-1-deficient mice. Hum Mol Genet. 2007;16(23):2900-2910
23. Dinis-Oliveira RJ, Remiao F, Carmo H, Duarte JA, Navarro AS, Bastos ML, Carvalho F. Paraquat exposure as an etiological factor of Parkinson's disease. Neurotoxicology. 2006;27(6):1110-1122
24. Ved R, Saha S, Westlund B, Perier C, Burnam L, Sluder A, Hoener M, Rodrigues CM, Alfonso A, Steer C, Liu L, Przedborski S, Wolozin B. Similar patterns of mitochondrial vulnerability and rescue induced by genetic modification of alpha-synuclein, parkin, and DJ-1 in Caenorhabditis elegans. J Biol Chem. 2005;280(52):42655-42668
25. Yao D, Gu Z, Nakamura T, Shi ZQ, Ma Y, Gaston B, Palmer LA, Rockenstein EM, Zhang Z, Masliah E, Uehara T, Lipton SA. Nitrosative stress linked to sporadic Parkinson's disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci U S A. 2004;101(29):10810-10814
26. Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM. S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science. 2004;304(5675):1328-1331
27. Zhou H, Falkenburger BH, Schulz JB, Tieu K, Xu Z, Xia XG. Silencing of the Pink1 gene expression by conditional RNAi does not induce dopaminergic neuron death in mice. Int J Biol Sci. 2007;3(4):242-250
28. Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244(2):305-318
29. Xia XG, Schmidt N, Teismann P, Ferger B, Schulz JB. Dopamine mediates striatal malonate toxicity via dopamine transporter-dependent generation of reactive oxygen species and D2 but not D1 receptor activation. J Neurochem. 2001;79(1):63-70
30. Hongxia Zhou BHF, Jörg B Schulz, Kim Tieu, Zuoshang Xu, Xu Gang Xia. Silencing of the Pink1 Gene Expression by Conditional RNAi Does Not Induce Dopaminergic Neuron Death in Mice. Int J Biol Sci. 2007;3(4):242-250
31. Tian T, Huang C, Tong J, Yang M, Zhou H, Xia XG. TDP-43 Potentiates Alpha-synuclein Toxicity to Dopaminergic Neurons in Transgenic Mice. Int J Biol Sci. 2011;7(2):234-243
32. Cory-Slechta DA, Thiruchelvam M, Barlow BK, Richfield EK. Developmental pesticide models of the Parkinson disease phenotype. Environ Health Perspect. 2005;113(9):1263-1270
33. Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, Bonsi P, Zhang C, Pothos EN, Shen J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci U S A. 2007;104(27):11441-11446
34. Shimizu K, Matsubara K, Ohtaki K, Fujimaru S, Saito O, Shiono H. Paraquat induces long-lasting dopamine overflow through the excitotoxic pathway in the striatum of freely moving rats. Brain Res. 2003;976(2):243-252
35. Brooks AI, Chadwick CA, Gelbard HA, Cory-Slechta DA, Federoff HJ. Paraquat elicited neurobehavioral syndrome caused by dopaminergic neuron loss. Brain Res. 1999;823(1-2):1-10
36. Thiruchelvam M, Richfield EK, Baggs RB, Tank AW, Cory-Slechta DA. The nigrostriatal dopaminergic system as a preferential target of repeated exposures to combined paraquat and maneb: implications for Parkinson's disease. J Neurosci. 2000;20(24):9207-9214
37. Thiruchelvam M, Richfield EK, Goodman BM, Baggs RB, Cory-Slechta DA. Developmental exposure to the pesticides paraquat and maneb and the Parkinson's disease phenotype. Neurotoxicology. 2002;23(4-5):621-633
38. Thiruchelvam M, Brockel BJ, Richfield EK, Baggs RB, Cory-Slechta DA. Potentiated and preferential effects of combined paraquat and maneb on nigrostriatal dopamine systems: environmental risk factors for Parkinson's disease?. Brain Res. 2000;873(2):225-234
39. Cory-Slechta DA, Thiruchelvam M, Richfield EK, Barlow BK, Brooks AI. Developmental pesticide exposures and the Parkinson's disease phenotype. Birth Defects Res A Clin Mol Teratol. 2005;73(3):136-139
40. Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat Rev Neurosci. 2006;7(3):207-219
41. Abou-Sleiman PM, Muqit MM, McDonald NQ, Yang YX, Gandhi S, Healy DG, Harvey K, Harvey RJ, Deas E, Bhatia K, Quinn N, Lees A, Latchman DS, Wood NW. A heterozygous effect for PINK1 mutations in Parkinson's disease?. Ann Neurol. 2006;60(4):414-419
42. Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441(7097):1162-1166
43. Kim RH, Smith PD, Aleyasin H, Hayley S, Mount MP, Pownall S, Wakeham A, You-Ten AJ, Kalia SK, Horne P, Westaway D, Lozano AM, Anisman H, Park DS, Mak TW. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc Natl Acad Sci U S A. 2005;102(14):5215-5220
44. Song DD, Shults CW, Sisk A, Rockenstein E, Masliah E. Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp Neurol. 2004;186(2):158-172
45. Kitada T, Tong Y, Gautier CA, Shen J. Absence of nigral degeneration in aged parkin/DJ-1/PINK1 triple knockout mice. J Neurochem. 2009;111(3):696-702
46. Von Coelln R, Thomas B, Savitt JM, Lim KL, Sasaki M, Hess EJ, Dawson VL, Dawson TM. Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc Natl Acad Sci U S A. 2004;101(29):10744-10749
47. Chen L, Cagniard B, Mathews T, Jones S, Koh HC, Ding Y, Carvey PM, Ling Z, Kang UJ, Zhuang X. Age-dependent motor deficits and dopaminergic dysfunction in DJ-1 null mice. J Biol Chem. 2005;280(22):21418-21426
48. Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, Tong Y, Martella G, Tscherter A, Martins A, Bernardi G, Roth BL, Pothos EN, Calabresi P, Shen J. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron. 2005;45(4):489-496
49. Chu CT. A pivotal role for PINK1 and autophagy in mitochondrial quality control: implications for Parkinson disease. Hum Mol Genet. 2010;19(R1):R28-37
50. Dagda RK, Cherra SJ3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem. 2009;284(20):13843-13855
51. Cui M, Aras R, Christian WV, Rappold PM, Hatwar M, Panza J, Jackson-Lewis V, Javitch JA, Ballatori N, Przedborski S, Tieu K. The organic cation transporter-3 is a pivotal modulator of neurodegeneration in the nigrostriatal dopaminergic pathway. Proc Natl Acad Sci U S A. 2009;106(19):8043-8048
52. Cui M, Tang X, Christian WV, Yoon Y, Tieu K. Perturbations in mitochondrial dynamics induced by human mutant PINK1 can be rescued by the mitochondrial division inhibitor mdivi-1. J Biol Chem. 2010;285(15):11740-11752
53. Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrane J, Moore DJ, Dawson VL, Grailhe R, Dawson TM. et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107(1):378-383
Author contact
![]() Corresponding author: H.Z. (Hongxia.zhouedu) or X.G.X (xugang.xiaedu)
Corresponding author: H.Z. (Hongxia.zhouedu) or X.G.X (xugang.xiaedu)
Received 2011-9-27
Accepted 2011-10-20
Published 2011-10-26