ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2011; 7(9):1298-1310. doi:10.7150/ijbs.7.1298 This issue Cite
Research Paper
MIF Synergizes with Trypanosoma cruzi Antigens to Promote Efficient Dendritic Cell Maturation and IL-12 Production via p38 MAPK
Cesar A. Terrazas1, EriK Huitron1, Alicia Vazquez1, Imelda Juarez1, Griselda M. Camacho2, Elsa A. Calleja2, Miriam Rodriguez-Sosa1, ![]()
1. Unidad de Biomedicina, Facultad de Estudios Superiores-Iztacala, Universidad Nacional Autónoma de México (UNAM), C. P. 54090 Estado de México, Mexico.
2. Modulo de Instrumentación, Facultad de Estudios Superiores-Iztacala, Universidad Nacional Autónoma de México (UNAM), C. P. 54090 Estado de México, Mexico.
Abstract
Macrophage migration inhibitory factor (MIF) has been found to be involved in host resistance to several parasitic infections. To determine the mechanisms of the MIF-dependent responses to Trypanosoma cruzi, we investigated host resistance in MIF-/- mice (on the BALB/c background) during an intraperitoneal infection. We focused on the potential involvement of MIF in dendritic cell (DC) maturation and cytokine production. Following a challenge with 5 x 103 T. cruzi parasites, wild type (WT) mice developed a strong IL-12 response and adequate maturation of the draining mesenteric lymph node DCs and were resistant to infection. In contrast, similarly infected MIF-/- mice mounted a weak IL-12 response, displayed immature DCs in the early phases of infection and rapidly succumbed to T. cruzi infection. The lack of maturation and IL-12 production by the DCs in response to total T. cruzi antigen (TcAg) was confirmed by in vitro studies. These effects were reversed following treatment with recombinant MIF. Interestingly, TcAg-stimulated bone marrow-derived DCs from both WT and MIF-/- mice had increased ERK1/2 MAPK phosphorylation. In contrast, p38 phosphorylation was only upregulated in WT DCs. Reconstitution of MIF to MIF-/- DCs upregulated p38 phosphorylation. The MIF-p38 pathway affected MHC-II and CD86 expression as well as IL-12 production. These findings demonstrate that the MIF-induced early DC maturation and IL-12 production mediates resistance to T. cruzi infection, probably by activating the p38 pathway.
Keywords: Macrophage migration inhibitory factor, Trypanosoma cruzi, macrophages, dendritic cells, p38 MAPK, ERK1/2 MAPK.
Introduction
Chagas disease, which is caused by the protozoan parasite T. cruzi, is the most important parasitic infection in Latin America [1]. An estimated 10 million people carry the infection, while another 90 million live at risk [2, 3]. This parasitic disease is transmitted by the feces of bloodsucking insect vectors (Triatoma) [4]. T. cruzi has a fundamental role in inducing immunopathology and tissue damage in organs such as the heart, esophagus and colon by sequentially inducing inflammatory responses, cellular lesions and fibrosis [4-7]. Host resistance to experimental acute T. cruzi infection is dependent on both the innate and acquired immune responses mediated by macrophages (MOs), natural killer (NK) cells, CD4+ T cells, CD8+ T cells, and B cells [5, 8]. The innate immune response to T. cruzi involves the production of cytokines that have the capacity to regulate NK cell and MO activity, such as interferon (IFN)-γ, IFN-β, interleukin (IL)-12 and tumor necrosis factor (TNF)- α[5, 9-13], since genetically engineered mice lacking any of these cytokines fail to control parasitemia and rapidly succumb to T. cruzi infection [14, 15]. However, the mechanisms involved in the innate immune protection against T cruzi are not completely understood [13]. Part of the protective role of the innate cytokines may therefore be through the regulation of IL-12, which enhances TNF-α and IFN-γ production from NK cells [16]. Both TNF-α and IFN-γ are required for the induction of the nitric oxide-dependent microbicidal activity of MOs [17]. In contrast, the anti-inflammatory cytokines IL-4, IL-10 and transforming growth factor (TGF)-β mediate susceptibility to T. cruzi but are also required for preventing immune hyperactivity and organ immunopathology [7, 18, 19].
MIF is a pleiotropic cytokine that is produced by multiple different cell types, such as T cells, activated MOs, and dendritic cells (DCs) [20-23]. Upon release, MIF modulates the expression of several proinflammatory molecules, including cytokines (TNF-α, IFN-γ, IL-1β, IL-2, IL-6 and IL-8), nitric oxide, and COX2 [24-26]. Moreover, MIF is required for resistance to several pathogens that express TLR ligands. It is known that one of the main effects of MIF on the immune system is the upregulation of TLR4 expression [27], which contributes to the resistance of MIF-/- mice to LPS-induced sepsis [28].
It is known that MIF is critical for controlling acute T. cruzi infection [29], but the specific mechanism of MIF in the resistance to T. cruzi infection is not known.
MIF has the potential to modulate numerous intracellular signaling pathways, including several that are known to be involved in the transcription of inflammatory cytokines, such as IL-10, IL-13 and TGF-β, in MIF-induced macrophage activation [30]. For example, MIF deficiency impairs mitogen-activated protein kinase (MAPK) signaling induced by the proinflammatory cytokines IL-1 and TNF or by Ag-specific T cell activation [31-34], and MIF also activates the PI3K-Akt pathway [35, 36]. Whether MIF affects DC effector functions via its effects on these pathways has not yet been examined.
The aim of this study was to determine the effect of MIF-deficiency in DCs on protective immunity against T. cruzi infection. We examined the maturation of mesenteric lymph node-derived DCs from WT and MIF-/- mice after infection with T. cruzi in vivo or pulsing with TcAg in vitro. Our results indicate that the interaction of DCs with MIF during T. cruzi-induced maturation is required for the upregulation of MHC class II, costimulatory molecule expression and cytokine secretion of mature DCs. Moreover, we demonstrate a synergism between TcAg and MIF that favors DC maturation through p38 activation.
Materials and Methods
Mice. Eight-to-10-week-old male BALB/c mice were purchased from Harlan (México City, Mexico). MIF-/- mice were developed as described previously [37] and were backcrossed for more than ten generations to a BALB/c genetic background. The mice were maintained in a pathogen-free environment at our animal facility in accordance with institutional and national guidelines.
Parasite and peritoneal infections. The Mexican T. cruzi I Queretaro (Qro) (TBAR/MX/0000/Queretaro) strain was donated by Dr. Bertha Espinoza from I.I.B, UNAM. Mexico. Briefly, the T. cruzi Qro strain was isolated from the Triatoma barberi vector from the Queretaro State in central Mexico [38]. The strain was maintained by sequential murine passages. Blood trypomastigotes were obtained from previously infected BALB/c mice, counted, and adjusted with sterile phosphate-buffered saline (PBS) to contain 5 x 103 parasites in each 100 μl aliquot, and 100 μl was injected intraperitoneally (i.p.) into WT and MIF-/- male mice. Parasitemia was determined every seven days using a hemocytometer and blood diluted 1:50 in PBS with heparin.
Preparation of T. cruzi total lysates. Trypomastigotes of T. cruzi that had been maintained by sequential culture in liver infusion tryptose medium were isolated, washed three times in PBS, and centrifuged at 1x104 rpm for 15 min. Protease inhibitors were added (0.1-2 μg/ml apotinin, 0.5-2 mM EDTA, 1-5 mM phenylmethyl fluoride, 1 μg/ml pepstatin, and 50 μg/ml TLCK (a-p-tosyl-L-Lysine chloromethyl ketone; all from Sigma-Aldrich, St. Louis, MO, USA), and the parasites were washed twice in sterile, ice-cold PBS by centrifugation at 200 g for 10 min at 4°C. The pellets were then frozen in liquid nitrogen and thawed at 37°C, and this process was repeated seven times. Parasite destruction was confirmed using a microscope. The parasite lysate was used as the total T. cruzi antigen (TcAg). The protein concentration was determined by the Lowry method [39], and the lysate was stored at -70ºC until use.
Cell preparations and culture conditions. Mesenteric cells and spleen cells were obtained from WT and MIF-/- mice under sterile conditions three, seven, ten and fifteen days after an i.p. T. cruzi infection. In brief, spleen or mesenteric lymph node tissues were minced and filtered to obtain cells, which were then washed and resuspended in Dulbecco's modified Eagle medium (DMEM; Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum (FCS), 2 mM L-glutamine, 0.25 U/ml penicillin and 100 μg/ml streptomycin (all from GIBCO, BRL, Grand Island, NY, USA). The cells from both tissues were resuspended at a concentration of 5 x 106 cells/ml and were analyzed by flow cytometry.
Bone marrow-derived murine DCs were differentiated from the bone marrow of uninfected WT and MIF-/- mice according to a version of a protocol previously described [40]. Briefly, bone marrow cells were flushed from the femurs and tibias of mice with DMEM supplemented 10% fetal calf serum (FCS), 2 mM L-glutamine, 0.25 U/ml penicillin and 100 μg/ml streptomycin (all from GIBCO). Red blood cells were lysed with 0.83% ammonium chloride (Sigma-Aldrich). After 72 h of incubation with 20 ng/ml recombinant murine granulocyte-macrophage colony-stimulating factor (rGM-CSF; Peprotech-Mexico, Mexico)-containing complete medium, non-adherent cells were discarded, and the remaining cells were fed with rGM-CSF-containing complete medium. On day 6 of the culture, the cells were fed with rGM-CSF-containing medium, and the non-adherent cells were used for experiments on day 6, at which point they stained positive (70-80%) for CD11c by flow cytometry. The cell suspensions were transferred to 24-well culture plates (Costar, Cambridge, MA, USA) at 5 x 105 cells/well and were stimulated with 50 μg/ml TcAg or with 1 μg/ml LPS (Sigma-Aldrich). For the DCs from MIF-/- mice, recombinant murine MIF (rMIF from R&D Systems, Minneapolis, MN, USA) was added (20 ng/ml) alone or together with TcAg or LPS. The plates were then incubated at 37ºC with 5% CO2 for 24 h. In some experiments, the DCs were incubated for two hours with a specific p38 inhibitor SB203580 at 10 µM (Invivogen, San Diego, CA, USA) before rMIF, TcAg or LPS treatment, and the inhibitor control samples were treated with a corresponding volume of DMSO. The supernatants were collected, centrifuged, aliquoted and frozen at -20ºC until use.
Cytokine measurements. The supernatants from the cell culture experiments described above were analyzed by ELISA to measure the production of MIF, IL-12, IFN-γ, IL-1β and TNF-α using kits as recommended by the manufacturer (Peprotech and R&D Systems). The optical density (OD) was measured after 5 minutes using an ELISA microplate reader (SpectraMax 250, Molecular Devices, USA) at 405 nm.
Flow cytometry. DCs were collected and stained for flow cytometric analysis. The DCs were washed in flow wash (Dulbecco´s PBS containing 1% FCS and 0.05% sodium azide) followed by incubation with allophycocyanin-conjugated anti-CD11c and fluorescein isothiocyanate or phycoerythrin (PE)-labeled anti-MHC-II, CD80, CD86, and OX40L Abs (all antibodies from BioLegend, San Diego, CA, USA) at 4°C for 30 min. After incubation, the cells were washed several times in buffer and were fixed in 1% paraformaldehyde (Sigma-Aldrich) and stored at 4°C in darkness before analysis using a FACS Calibur and Cell Quest software (Becton Dickinson, Franklin Lakes, NY, USA).
Detection of phosphorylation by flow cytometry. Bone marrow DCs that were obtained after 6 days of culture were washed and seeded without GM-CSF for 24 h to eliminate any remaining phosphorylation. Then 2 x 105 DCs were stimulated for 30 minutes with the different treatments. After stimulation, the DCs were fixed with 2% paraformaldehyde for 10 minutes at room temperature (RT). The cells were washed with staining buffer and permeabilized with ice-cold methanol for 10 minutes at 4ºC (all from Sigma-Aldrich). The cells were washed with staining buffer two times and were incubated 15 minutes with anti-CD16/32. A primary rabbit anti-mouse antibody against ERK or p38 (both from Biovision, San Diego, CA, USA) was added at 1:1000, and the cells were incubated for 20 minutes in the dark at RT. Finally, the cells were labeled with a secondary antibody, donkey anti-rabbit DyLight 488 (BioLegend), at 0.25 µg/106 cells for 20 minutes at RT in the dark, and flow cytometry was performed.
Statistical analysis. The data are expressed as the means +/- the standar error (SE). The statistical significance of the differences in the mean values was determined using Student's t test. The survival data are presented as Kaplan-Meier survival curves and were analyzed using the Log-rank test. P<0.05 was considered significant.
Results
Disease progression and parasitism in WT and MIF-/- mice over the course of T. cruzi infection
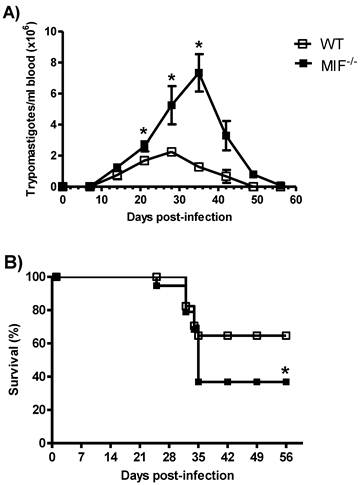
It has been shown that the genetic ablation of MIF rendered mice highly susceptible to intraperitoneal infection with T. cruzi [29]. WT and MIF-/- mice were infected by i.p. inoculation with 5 x 103 blood trypomastigotes of pathogenic strain (Queretaro) of T. cruzi. Both groups developed blood parasitemia on day 10 p.i., but on day 22 p.i., MIF-/- mice displayed significantly greater levels of parasitemia, which peaked at day 36 p.i. and could be detected until day 50 p.i. (Fig. 1A). In contrast, the maximum peak of parasitemia in WT mice was observed on day 27 p.i., and these mice controlled the infection by day 40. Interestingly, MIF-/- mice also began to succumb to infection as early as day 23 p.i., and only 37% of them survived until day 45. In contrast, 65% of WT mice survived the infection over the time-course examined (Fig. 1B).
MIF is required to survive to Trypanosoma cruzi infection. WT and MIF-/- mice were infected i.p. with 3x103 trypomastigotes. The number of blood trypomastigotes per mouse was determined by counting in a Neubauer chamber (A). The survival curve of infected MIF-/- and WT mice is also shown (B). The data are expressed as the means with standard error (SE) and are representative of three independent experiments. Six animals were used in each group. *P< 0.002 using Student´s t test in (A) and *P< 0.006 using a Log-rank test (B).
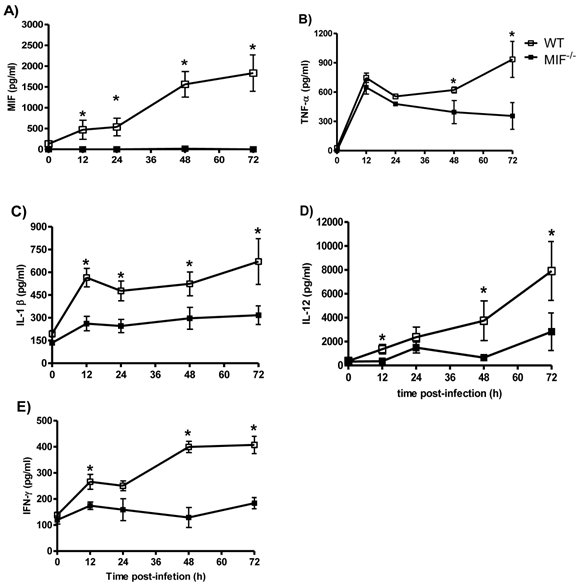
Proinflammatory cytokines are impaired in MIF-/- mice during early T. cruzi infection
It has been shown that the increased susceptibility and mortality of MIF-/- mice correlates with an altered production of IL-12, IFN-γ, TNF-α and IL-1β [29]. We sought to investigate how early the decreased production of these cytokines could be detected. We compared the MIF, IL-12, IFN-γ, TNF-α and IL-1β levels in the sera of T. cruzi-infected WT and MIF-/- mice as early as 12 hours post-infection until 72 hours post-infection. After infection with T. cruzi, WT mice displayed rapid production of TNF-α, IL-1β, IL-12 and IFN-γ (Fig. 2A to E), and the levels were increased at 72 hours after infection. Importantly, MIF secretion was detectable at least 12 h after infection. In contrast, MIF-/- mice displayed a significant impairment in the production of all of the cytokines listed in the early infection (Fig. 3A and E).
Dynamics of DC activation in response to T. cruzi infection
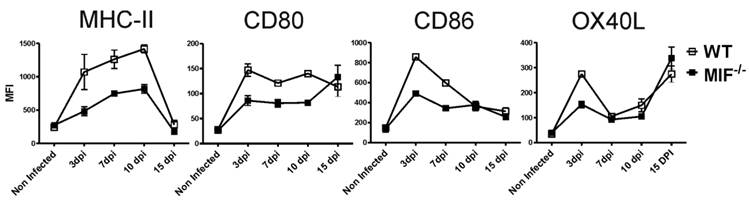
To gain insight into the mechanisms involved in the MIF-dependent resistance to T. cruzi infection, we evaluated whether a lack of MIF would affect in vivo DC maturation during T. cruzi infection. We measured the kinetics of DC maturation marker expression in the mesenteric lymph nodes from day 3 to 15 after infection. As shown in figure 3, there was a striking reduction in MHC-II expression on DCs at day 3 post-infection in MIF-/- mice relative to WT animals. Additionally, lower expression levels of costimulatory molecules, such as CD80, CD86 and OX40L, were also observed in MIF-/- mice, particularly on day 3 after i.p. infection. These findings demonstrate that the absence of endogenous MIF affects the expression of molecules related to antigen presentation in DCs.
T. cruzi antigen-driven stimulation of MHC class II and costimulatory molecule expression by DCs is dependent on autocrine MIF
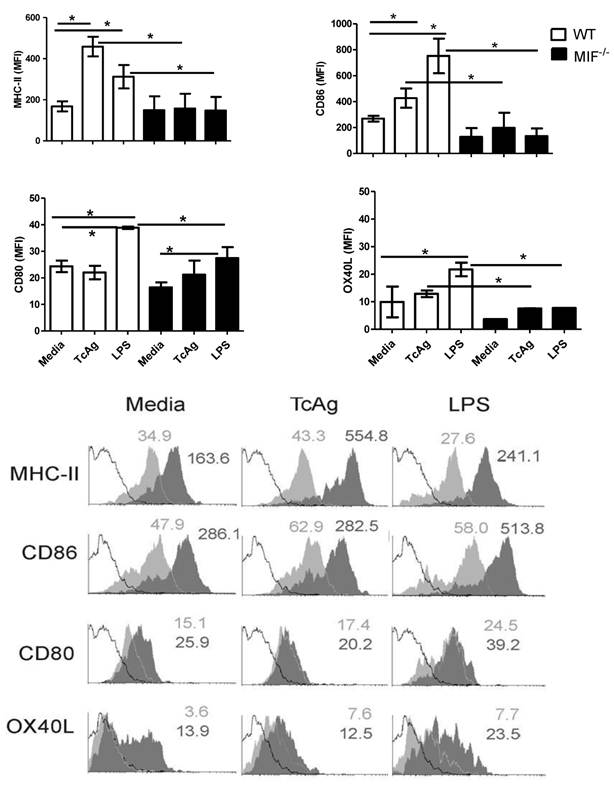
To compare the capability of TcAg to induce the maturation of DCs in MIF-/- versus WT mice, we pulsed bone marrow-derived DCs with TcAg (100 μg/ml) for 24 h, and the expression of MHC-II and costimulatory molecules by CD11c+ DC was evaluated by comparing the expression levels with those induced by the microbe-derived stimulator LPS, which was chosen for its known ability to mature and activate DCs [12]. TcAg treatment induced the maturation of WT DC, resulting in an upregulation of several maturation markers (MHC-II, CD86, CD80, and OX40L) (Fig. 4A). Interestingly, the expression of these molecules was lower on MIF-/- DCs following exposure to either LPS or TcAg as compared to WT DCs (Figure 4B). These findings demonstrate that the absence of endogenous MIF affects the expression of molecules related to antigen presentation in DCs.
Proinflammatory cytokines are diminished in the absence of MIF. The levels of MIF (A), TNF-α (B), IL-1β (C), IL-12 (D), and IFN-γ (E) in the sera from MIF-/- and WT mice infected with 3x103 blood trypomastigotes of T. cruzi. For the assessment of systemic cytokine production, the mice were bled at the indicated time points, and the levels of cytokines were determined by ELISA as described above. The values presented are the mean ± SE of triplicate samples with 6 animals per time point per group. * P< 0.05 using Student's t-test.
Surface expression of MHC-II, CD80, CD86 and OX40L on mesenteric DCs in T. cruzi-infected WT and MIF-/- mice. MIF-/- or WT mice were infected i.p. with 5 x 103 blood trypomastigotes. DCs from the mesenteric lymph nodes were harvested at 0, 3, 7, 10 and 15 days post-infection, and 1 x 107 cells/ml were processed by flow cytometric analysis. CD11c+ cells were analyzed for the expression of MHC-II, CD80, CD86 and OX40L. Open squares correspond to WT DCs, and solid black squares correspond to MIF-/- DCs. The flow cytometry profiles are representative of three experiments. *P <0.05 with respect to WT using Student's t-test.
MIF-/- bone marrow-derived DCs fail to mature after TcAg stimulation in vitro. (A) Immature DCs were incubated with TcAg (50 μg/ml) or LPS (1 μg/ml) for 24 h, and activation was determined by measuring the expression of MHC-II, CD80, CD86 and OX40L. B) One of three representative histograms from bone marrow-derived DCs is shown. The open white histogram represents the isotype control, the closed black histogram represents the bone marrow-derived DCs from WT mice, and the closed gray histogram represents DCs from MIF-/- mice.
T. cruzi antigen-pulsed MIF-/- DCs have impaired antigen-specific cytokine production
The cytokine profile generated by APCs during the early interaction with pathogen-derived molecules is critical for the polarization of cellular responses. We investigated this profile in the culture supernatants of bone marrow DCs following a 24 h pulse with TcAg. WT and MIF-/- DC upregulated the secretion of the proinflammatory cytokines IL-12 and TNF-α in response to TcAg stimulation, but the levels of both cytokines were significantly higher in the WT DCs than in the MIF-/- DCs (Figure 5A and B).
MIF is important for DC IL-12 responses to TcAg. Bone marrow-derived DCs from WT or MIF-/- mice were incubated with TcAg (50 μg/ml) or LPS (1 μg/ml) for 24 h, and activation was determined by measuring the level of IL-12 (p40) (A) or TNF-α released into supernatants by ELISA. The data are the average of three mice in each group. The black bars indicate MIF-/- mice. An asterisk indicates statistical significance (*P < 0.05) using Student's t-test.
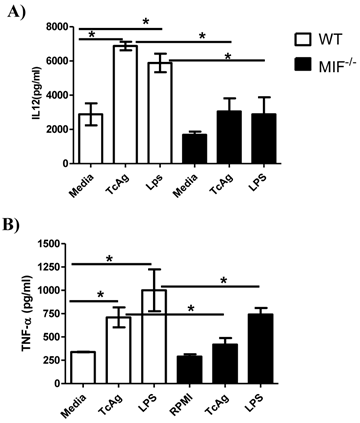
We also determined the levels of the regulatory cytokine IL-10 and found that IL-10 production was impaired in MIF-/- DCs (data not shown). These results suggest that TcAg promotes a cytokine profile that is dominated by the presence of the proinflammatory cytokines IL-12 and TNF-α in WT DCs, whereas the same stimulus induces a poor proinflammatory cytokine profile characterized by a lower level of production of IL-12 and TNF-α in MIF-/- DCs.
T. cruzi antigen enhances ERK activation independent of MIF, but p38 phosphorylation depends on TcAg-MIF synergism in bone marrow-derived DCs
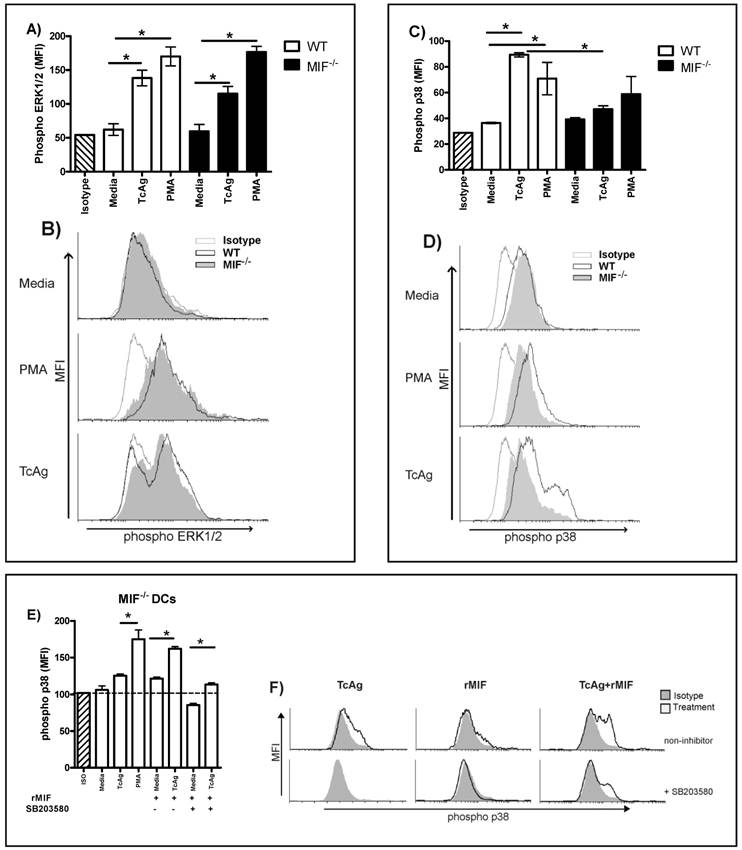
LPS, TcAg and other TLR ligands activate MAPK and the subsequent cytokine production [12, 41, 42]. We analyzed the activation of reported key regulators involved in cytokine production and DC differentiation [43, 44], such as extracellular signal-regulated kinase (ERK)-1/2 MAPK, which has been implicated in blocking IL-12 in DCs through the stabilization of c-fos, and the p38 MAPK pathway, which has been implicated in IL-12 production [45, 46].
DCs were stimulated with phorbol-12-myristatc-13 acetate (PMA) as a positive control or TcAg for 30 min. TcAg and PMA increased the phosphorylation of the ERK1/2 MAPK pathway in WT and MIF-/- DCs to a similar level (Fig. 6A, B). As shown in Fig. 6C and D, TcAg induced p38 phosphorylation on WT DC; but unlike PMA, TcAg had no effect on p38 phosphorylation in MIF-/- DC. Thus, the early recognition of TcAg induces ERK1/2 phosphorylation in DCs, but synergism between MIF and TcAg apparently induces DC activation via the p38 pathway.
To confirm whether the activation of p38 is associated with MIF secretion, we pulsed MIF-/- DCs with TcAg or PMA and restored MIF with exogenous rMIF. We found that TcAg or rMIF leads to a slight upregulation of p38 in MIF-/- DCs, but when both rMIF and TcAg were added to MIF-/- DC cultures, a strong upregulation of p38 was observed (Fig. 6E, F). As shown in Fig. 6E and F, the inhibitor of the p38 pathway, SB203580, significantly reduced the signal activation during the TcAg plus rMIF treatment (P < 0.001). In contrast, TcAg plus rMIF without the inhibitor did not reduce the p38 signal activation (Fig. 6 E and F). Thus, our results indicate that MIF and TcAg synergize to induce DC activation via the p38 pathway.
TcAg enhances phospho-ERK independent of MIF, but phospho-38 is related to MIF modulation. Bone marrow-derived DCs from WT and MIF-/- mice were pulsed for 30 min with TcAg or PMA as described. ERK (A) and p38 (C) phosphorylation was determined by flow cytometry. One representative histogram for ERK (B) and p38 (D) phosphorylation of three is shown. (E) MIF-/- DCs pulsed with TcAg, rMIF, TcAg+rMIF or PMA as control with or without the p38 inhibitor. (F) Representative histogram for MIF-/- DCs pulsed with rMIF or TcAg+rMIF with or without the inhibitor. An asterisk indicates statistical significance (*p < 0.05 using Student's t-test).
The p38 pathway is involved in the T. cruzi-MIF modulatory response
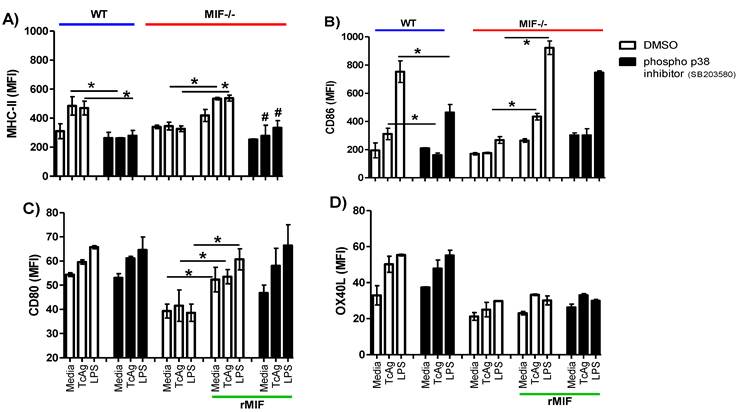
To determine whether the activation of p38 is associated with costimulatory molecule expression, we pre-treated WT-DCs with the inhibitor SB203580 or the vehicle (as described in the materials and methods section) followed by incubation with TcAg or LPS for 30 min. Next, we measured DC maturation marker expression, specifically that of MHC-II, CD86, CD80 and OX40L. As expected, WT DCs stimulated with TcAg or LPS displayed enhanced expression of costimulatory molecules (Fig. 7A-D). The inhibition of phospho- p38 only impacted MHC-II and CD86 expression during TcAg stimulation (P < 0.001), and no differences in CD80 and OX40L expression were found. Further, the inhibitor did not affect costimulatory molecule expression upon LPS treatment (Figure 7C-D). As previously demonstrated, MIF-/- DCs had impaired costimulatory molecule expression in response to both TcAg and LPS stimulation (Fig. 7A-D). When we added exogenous rMIF, a recovery of the expression of MHC-II, CD86 and CD80 was evident, but this recovery was not observed for OX40L. Again, p38 inhibition only impacted MHC-II and CD86 expression (Fig. 7 A, B). Thus, our results confirm that the p38 pathway is involved in a specific T. cruzi-MIF modulatory response in DCs.
p38 phosphorylation is MIF-dependent. DCs (2 x 105 cells/ml) were pretreated for 2 h with the p38 inhibitor SB203580 (10 μM) or the vehicle (DMSO), then were stimulated with TcAg (50 μg/ml) or LPS (1 μg/ml) for 18 h, p38 phosphorylation was determined by flow cytometry. A) MHC-II, B) CD86, C) CD80 and D) OX40L expression is shown. One representative experiment of two is shown. An asterisk indicates statistical significance (*P < 0.05 using Student's t-test).
Discussion
Antigen presenting cells, such as DCs, are responsible for initiating the primary immune response and for the development of immunity and/or tolerance [47-49]. They are endowed with a unique capacity to “sense” pathogens or pathogen-derived molecules through receptors with specificity for pathogen-associated molecular patterns (PAMPs) [50]. T. cruzi can be recognized by DC through different transmembrane proteins called Toll-like receptors (TLRs) by recognizing conserved microbial structures. For example T. cruzi-derived Tc52 and glycosylphosphatidylinositol (GPI) can bind TLR2 [51-53], glycoinositolphospholipids (GIPLs), which are a subset of GPI anchors from the epimastigote state of T. cruzi, can bind to TLR4 [54] and CpGs bind to TLR9 to activate DCs [48]. Indeed, the lack of adaptor proteins to TLRs, such as MyD88, leads to an impairment of MAPK phosphorylation and in consequence, impaired IL-12 and TNF-α production [55]. These deficiencies have an important role in mediating susceptibility, meaning that MyD88-/- DCs are more susceptible to parasite infection [56, 57].
It is well documented that several T. cruzi molecules have stimulatory effects [52, 53, 58]. It has been also demonstrated that some of these molecules trigger the phosphorylation of MAPK and IκB in macrophages and DCs [52, 55]. Recently, it has been demonstrated that T. cruzi or uncharacterized T. cruzi molecules target DC maturation and/or functionality [59-63] and are associated with high levels of IL-10 production by DCs [64]. Inhibition of ERK1/2 MAPK down-regulated IL-10 production and restored the stimulatory capacity of DCs, showing the importance of this pathway in DC modulation [12] .
Recently, the receptors that mediate the cellular responses supported by MIF were identified [65-67]. MIF binds to CD74, which is the cell surface form of the class II invariant chain (li) [65]. However, depending on the cellular context and the stimulation status, MIF triggers several signaling pathways. For example, in B lymphocytes, tumor cells and MO, MIF leads to the activation of ERK1/2 MAPK and PI3K/AKT signaling, resulting in cell proliferation, enhanced survival, and gene expression of the proinflammatory chemokine CXCL8 [36, 65, 68-70]. MIF induces matrix metallopeptidase (MMP)-9 expression in murine MO mainly via the ERK1/2 MAPK pathway [71]. MMP-2 production in rheumatoid synovial fibroblasts requires the activation of protein kinase C and the JNK and SRC signaling pathways by MIF [72]. During septic shock, MIF mediates the phosphorylation of JNK [73]. MIF also regulates macrophage chemotactic responses via MAPK and Rho GTPase [31]. In lung adenocarcinoma cells, MIF promotes JNK-dependent AP-1 transactivation and the subsequent transcriptional regulation of CXCL8 [74].
MIF also has inhibitory properties. For example, the release of MIF inhibits signaling through JNK in ischemic cardiomocytes, triggering the phosphorylation of the pro-apoptotic protein BAD with an ensuing increase in cell death [75]. When the MIF target cells are pre-stimulated by stress, MIF inhibits or attenuates the JNK MAPK pathway. The activation of the JNK pathway in fibroblasts stimulated by TNF or UV irradiation-induced stress is blocked by higher concentrations of MIF [76].
Direct evidence for role the of MIF in the activation on DCs came from a study on the role of MIF in Toxoplasma gondii infection in which MIF-deficient DCs had impaired IL-12 and TNF-α production in response to T. gondii antigen [22]. Here, we show that DCs from an in vivo T. cruzi infection or pulsed in vitro with TcAg had impaired IL-12 production costimulatory molecule expression. These data demonstrate that the action of MIF is the result of its direct impact on DCs and that the production of IL-12 favors the anti-parasite effector mechanism during T. cruzi infection.
To elucidate the intracellular events involved in the MIF synergism with T. cruzi antigens in DCs, we examined ERK1/2 and p38 phosphorylation. We found that ERK1/2 phosphorylation was enhanced to a similar degree in both WT and MIF-/- DCs pulsed with TcAg. This upregulation of ERK1/2 has been associated with the overproduction of IL-10 by DCs exposed to live parasites combined with LPS, which synergize to induce a regulatory phenotype that is associated with impaired costimulatory molecule expression by DCs [12]. Here, we observed a similar level of ERK1/2 phosphorylation in both WT and MIF-/- DCs stimulated with TcAg, indicating that ERK1/2 activation is not dependent on MIF. Moreover, we found high levels of IL-12 secretion and MHC-II and CD86 upregulation after TcAg exposure in WT, but not MIF-/- DCs, despite the high phosphorylation of ERK1/2 in MIF-/- DCs, confirming that the ERK1/2 pathway is not involved in the impaired DC maturation and perhaps is implicated in the regulatory DC phenotype as was demonstrated by Pocini et al. [64].
The fact that despite the phosphorylation of ERK1/2, we detected high expression of IL-12, MHC-II and CD86 in WT DCs may be explained by the use of total antigen (TcAg); some of molecules inside the parasite can be released and activate TLRs, such as TLR2, TLR4 or TLR9 in DCs, and favor the balance toward inflammatory MAPK mediators, such as p38. This conjecture is supported by the observation that macrophages exposed to GPI from T. cruzi induces p38 phosphorylation, but live T. cruzi, in which these molecules could be hidden from immune recognition, does not [12, 55].
Consistent with this hypothesis, we found enhanced p38 phosphorylation in WT DCs, but not in MIF-/- DCs, which correlates with the lack of IL-12 production and the impaired maturation observed for MIF-/- DCs. This supports the hypothesis that the intracellular events mediated by MIF that favor the effective maturation and IL-12 production in DCs pulsed with TcAg are carried out through the p38 signaling pathway.
This hypothesis was tested using exogenous MIF. We found that rMIF slightly upregulated p38 phosphorylation in MIF-/- DCs. Similarly, TcAg only slightly increased p38 phosphorylation. However, using TcAg and rMIF together as stimuli, p38 was highly phosphorylated, indicating a synergism between TcAg and MIF leading to efficient DC maturation. Moreover, the addition of rMIF to MIF-/- DCs restored IL-12 and TNF-α production as well as the expression of maturation molecules after LPS or TcAg treatment. Due to the lack of p38 phosphorylation and the fact that rMIF restored DC maturation in MIF-/- DCs in response to TcAg, we hypothesized that the impaired maturation occurs through the p38 pathway.
Finally, we demonstrated that the restored cytokine production and costimulatory molecule expression were p38 dependent. Using a selective p38 inhibitor, we found that only IL-12, MHC-II and CD86 were downregulated. All these findings demonstrate a key role for MIF in the modulation of p38 pathway and suggest that this regulatory effect involves parasite-DC interactions. Thus, MAPKs appear to be influenced by MIF, wherein MIF favors the balance toward inflammatory cytokine production through p38 phosphorylation, in response to this protozoon.
Acknowledgements
We thank Biol. Ana Federica Chavez for technical assistance. This work was supported by PAPIIT-UNAM Grant IN213009-3 and in part by PAPCA 2010-2011 Grant 13. C.T.A. and A.V.M. are fellows of the National Council of Science and Technology (CONACYT).
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Miles M.A, Llewellyn M.S, Lewis M.D. et al. The molecular epidemiology and phylogeography of Trypanosoma cruzi and parallel research on Leishmania: looking back and to the future. Parasitology. 2009;136(12):1509-28
2. Coura J.R, Dias J.C. Epidemiology, control and surveillance of Chagas disease: 100 years after its discovery. Mem Inst Oswaldo Cruz. 2009;104(Suppl 1):31-40
3. Ocana-Mayorga S, Llewellyn M.S, Costales J.A. et al. Sex, subdivision, and domestic dispersal of Trypanosoma cruzi lineage I in southern Ecuador. PLoS Negl Trop Dis. 2010;4(12):e915
4. Parker E.R, Sethi A. Chagas disease: coming to a place near you. Dermatol Clin. 2011;29(1):53-62
5. Junqueira C, Caetano B, Bartholomeu D.C. et al. The endless race between Trypanosoma cruzi and host immunity: lessons for and beyond Chagas disease. Expert Rev Mol Med. 2010;15(12):e29
6. Rodrigues C.M, Valadares H.M, Francisco A.F. et al. Coinfection with different Trypanosoma cruzi strains interferes with the host immune response to infection. PLoS Negl Trop Dis. 2010;4(10):e846
7. Maya J.D, Orellana M, Ferreira J. et al. Chagas disease: Present status of pathogenic mechanisms and chemotherapy. Biol Res. 2010;43(3):323-31
8. Parodi C, Padilla A.M, Basombrio M.A. Protective immunity against Trypanosoma cruzi. Mem Inst Oswaldo Cruz. 2009;104(Suppl 1):288-94
9. Sathler-Avelar R, Vitelli-Avelar D.M, Teixeira-Carvalho A. et al. Innate immunity and regulatory T-cells in human Chagas disease: what must be understood?. Mem Inst Oswaldo Cruz. 2009;104(Suppl 1):246-51
10. Dosreis G.A. Evasion of immune responses by Trypanosoma cruzi, the etiological agent of Chagas disease. Braz J Med Biol Res. 2011;44(2):84-90
11. Gutierrez F.R, Guedes P.M, Gazzinelli R.T. et al. The role of parasite persistence in pathogenesis of Chagas heart disease. Parasite Immunol. 2009;31(11):673-85
12. Poncini C.V, Gimenez G, Pontillo C.A. et al. Central role of extracellular signal-regulated kinase and Toll-like receptor 4 in IL-10 production in regulatory dendritic cells induced by Trypanosoma cruzi. Mol Immunol. 2010;47(11-12):1981-8
13. Kuehn C.C, Oliveira L.G, Santos C.D. et al. Prior and concomitant dehydroepiandrosterone treatment affects immunologic response of cultured macrophages infected with Trypanosoma cruzi in vitro?. Vet Parasitol. 2011;177(3-4):242-6
14. Marinho C.R, Nunez-Apaza L.N, Martins-Santos R. et al. IFN-gamma, but not nitric oxide or specific IgG, is essential for the in vivo control of low-virulence Sylvio X10/4 Trypanosoma cruzi parasites. Scand J Immunol. 2007;66(2-3):297-308
15. Michailowsky V, Silva N.M, Rocha C.D. et al. Pivotal role of interleukin-12 and interferon-gamma axis in controlling tissue parasitism and inflammation in the heart and central nervous system during Trypanosoma cruzi infection. Am J Pathol. 2001;159(5):1723-33
16. Silva J.S, Machado F.S, Martins G.A. The role of nitric oxide in the pathogenesis of Chagas disease. Front Biosc. 2003;8:s314-25
17. Gutierrez F.R, Mineo T.W, Pavanelli W.R. et al. The effects of nitric oxide on the immune system during Trypanosoma cruzi infection. Mem Inst Oswaldo Cruz. 2009;104(Suppl 1):236-45
18. Laucella S.A, Rottenberg M.E, de Titto E.H. Role of cytokines in resistance and pathology in Trypanosoma cruzi infection. Rev Argent Microbiol. 1996;28(2):99-109
19. Abrahamsohn I.A, Coffman R.L. Trypanosoma cruzi: IL-10 TNF, IFN-gamma, and IL-12 regulate innate and acquired immunity to infection. Exp Parasitol. 1996;84(2):231-44
20. Awandare G.A, Ouma C, Keller C.C. et al. A macrophage migration inhibitory factor promoter polymorphism is associated with high-density parasitemia in children with malaria. Genes Immun. 2006;7(7):568-75
21. Amaral F.A, Fagundes C.T, Guabiraba R. et al. The role of macrophage migration inhibitory factor in the cascade of events leading to reperfusion-induced inflammatory injury and lethality. Am J Pathol. 2007;171(6):1887-93
22. Terrazas C.A, Juarez I, Terrazas L.I. et al. Toxoplasma gondii: impaired maturation and pro-inflammatory response of dendritic cells in MIF-deficient mice favors susceptibility to infection. Exp Parasitol. 2010;126(3):348-58
23. Popa C, van Lieshout A.W, Roelofs M.F. et al. MIF production by dendritic cells is differentially regulated by Toll-like receptors and increased during rheumatoid arthritis. Cytokine. 2006;36(1-2):51-6
24. Cooke G, Armstrong M.E, Donnelly S.C. Macrophage migration inhibitory factor (MIF), enzymatic activity and the inflammatory response. Biofactors. 2009;35(2):165-8
25. Xin D, Rendon B.E, Zhao M. et al. The MIF homologue D-dopachrome tautomerase promotes COX-2 expression through beta-catenin-dependent and -independent mechanisms. Mol Cancer Res. 2010;8(12):1601-9
26. Balachandran S, Gadekar P.K, Parkale S. et al. Synthesis and biological activity of novel MIF antagonists. Bioorg Med Chem Lett. 2011;21(5):1508-11
27. Roger T, David J, Glauser M.P. et al. MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature. 2001;414(6866):920-4
28. Calandra T, Echtenacher B, Roy D.L. et al. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat Med. 2000;6(2):164-70
29. Reyes J.L, Terrazas L.I, Espinoza B. et al. Macrophage migration inhibitory factor contributes to host defense against acute Trypanosoma cruzi infection. Infect Immun. 2006;74(6):3170-9
30. Juttner S, Bernhagen J, Metz C.N. et al. Migration inhibitory factor induces killing of Leishmania major by macrophages: dependence on reactive nitrogen intermediates and endogenous TNF-alpha. J Immunol. 1998;161(5):2383-90
31. Fan H, Hall P, Santos L.L. et al. Macrophage migration inhibitory factor and CD74 regulate macrophage chemotactic responses via MAPK and Rho GTPase. J Immunol. 2011;186(8):4915-24
32. Toh M.L, Aeberli D, Lacey D. et al. Regulation of IL-1 and TNF receptor expression and function by endogenous macrophage migration inhibitory factor. J Immunol. 2006;177(7):4818-25
33. Aeberli D, Yang Y, Mansell A. et al. Endogenous macrophage migration inhibitory factor modulates glucocorticoid sensitivity in macrophages via effects on MAP kinase phosphatase-1 and p38 MAP kinase. FEBS Lett. 2006;580(3):974-81
34. Santos L.L, Dacumos A, Yamana J. et al. Reduced arthritis in MIF deficient mice is associated with reduced T cell activation: down-regulation of ERK MAP kinase phosphorylation. Clin Exp Immunol. 2008;152(2):372-80
35. Gore Y, Starlets D, Maharshak N. et al. Macrophage migration inhibitory factor induces B cell survival by activation of a CD74-CD44 receptor complex. J Biol Chem. 2008;283(5):2784-92
36. Lue H, Thiele M, Franz J. et al. Macrophage migration inhibitory factor (MIF) promotes cell survival by activation of the Akt pathway and role for CSN5/JAB1 in the control of autocrine MIF activity. Oncogene. 2007;26(35):5046-59
37. Bozza M, Satoskar A.R, Lin G. et al. Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J Exp Med. 1999;189(2):341-6
38. Espinoza B, Rico T, Sosa S. et al. Mexican Trypanosoma cruzi T. cruzi I strains with different degrees of virulence induce diverse humoral and cellular immune responses in a murine experimental infection model. J Biomed Biotechnol. 2010:890672
39. Lowry O.H, Rosebrough N.J, Farr A.L. et al. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193(1):265-75
40. Inaba K, Inaba M, Romani N. et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176(6):1693-702
41. Kotlyarov A, Neininger A, Schubert C. et al. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat Cell Biol. 1999;1(2):94-7
42. Dong C, Davis R.J, Flavell R.A. MAP kinases in the immune response. Annu Rev Immunol. 2002;20:55-72
43. Qian C, Jiang X, An H. et al. TLR agonists promote ERK-mediated preferential IL-10 production of regulatory dendritic cells (diffDCs), leading to NK-cell activation. Blood. 2006;108(7):2307-15
44. Park S.J, Nakagawa T, Kitamura H. et al. IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J Immunol. 2004;173(6):3844-54
45. Agrawal S, Agrawal A, Doughty B. et al. Cutting edge: different Toll-like receptor agonists instruct dendritic cells to induce distinct Th responses via differential modulation of extracellular signal-regulated kinase-mitogen-activated protein kinase and c-Fos. J Immunol. 2003;171(10):4984-9
46. Jackson A.M, Mulcahy L.A, Porte J. et al. Role of mitogen-activated protein kinase and PI3K pathways in the regulation of IL-12-family cytokines in dendritic cells and the generation of T H-responses. Eur Cytokine Netw. 2010;21(4):319-28
47. Steinman R.M, Hawiger D, Liu K. et al. Dendritic cell function in vivo during the steady state: a role in peripheral tolerance. Ann N Y Acad Sci. 2003;987:15-25
48. Terrazas C.A, Terrazas L.I, Gomez-Garcia L. Modulation of dendritic cell responses by parasites: a common strategy to survive. J Biomed Biotechnol. 2010:357106
49. Lanzavecchia A, Sallusto F. Dynamics of T lymphocyte responses: intermediates, effectors, and memory cells. Science. 2000;290(5489):92-7
50. Manickasingham S.P, Edwards A.D, Schulz O. et al. The ability of murine dendritic cell subsets to direct T helper cell differentiation is dependent on microbial signals. Eur J Immunol. 2003;33(1):101-7
51. Almeida I.C, Gazzinelli R.T. Proinflammatory activity of glycosylphosphatidylinositol anchors derived from Trypanosoma cruzi: structural and functional analyses. J Leukoc Biol. 2001;70(4):467-77
52. Ouaissi A, Guilvard E, Delneste Y. et al. The Trypanosoma cruzi Tc52-released protein induces human dendritic cell maturation, signals via Toll-like receptor 2, and confers protection against lethal infection. J Immunol. 2002;168(12):6366-74
53. Campos M.A, Almeida I.C, Takeuchi O. et al. Activation of Toll-like receptor-2 by glycosylphosphatidylinositol anchors from a protozoan parasite. J Immunol. 2001;167(1):416-23
54. Oliveira A.C, Peixoto J.R, de Arruda L.B. et al. Expression of functional TLR4 confers proinflammatory responsiveness to Trypanosoma cruzi glycoinositolphospholipids and higher resistance to infection with T. cruzi. J Immunol. 2004;173(9):5688-96
55. Ropert C, Almeida I.C, Closel M. et al. Requirement of mitogen-activated protein kinases and I kappa B phosphorylation for induction of proinflammatory cytokines synthesis by macrophages indicates functional similarity of receptors triggered by glycosylphosphatidylinositol anchors from parasitic protozoa and bacterial lipopolysaccharide. J Immunol. 2001;166(5):3423-31
56. Bafica A, Santiago H.C, Goldszmid R. et al. Cutting edge: TLR9 and TLR2 signaling together account for MyD88-dependent control of parasitemia in Trypanosoma cruzi infection. J Immunol. 2006;177(6):3515-9
57. Ropert C, Gazzinelli R.T. Regulatory role of Toll-like receptor 2 during infection with Trypanosoma cruzi. J Endotoxin Res. 2004;10(6):425-30
58. Belaunzaran M.L, Wainszelbaum M.J, Lammel E.M. et al. Phospholipase A1 from Trypanosoma cruzi infective stages generates lipid messengers that activate host cell protein kinase c. Parasitology. 2007;134(Pt 4):491-502
59. Alba Soto C.D, Mirkin G.A, Solana M.E. et al. Trypanosoma cruzi infection modulates in vivo expression of major histocompatibility complex class II molecules on antigen-presenting cells and T-cell stimulatory activity of dendritic cells in a strain-dependent manner. Infect Immun. 2003;71(3):1194-9
60. Van Overtvelt L, Vanderheyde N, Verhasselt V. et al. Trypanosoma cruzi infects human dendritic cells and prevents their maturation: inhibition of cytokines HLA-DR, and costimulatory molecules. Infect Immun. 1999;67(8):4033-40
61. Brodskyn C, Patricio J, Oliveira R. et al. Glycoinositolphospholipids from Trypanosoma cruzi interfere with macrophages and dendritic cell responses. Infect Immun. 2002;70(7):3736-43
62. Chaussabel D, Pajak B, Vercruysse V. et al. Alteration of migration and maturation of dendritic cells and T-cell depletion in the course of experimental Trypanosoma cruzi infection. Lab Invest. 2003;83(9):1373-82
63. Vray B, Camby I, Vercruysse V. et al. Up-regulation of galectin-3 and its ligands by Trypanosoma cruzi infection with modulation of adhesion and migration of murine dendritic cells. Glycobiology. 2004;14(7):647-57
64. Poncini C.V, Alba Soto C.D, Batalla E. et al. Trypanosoma cruzi induces regulatory dendritic cells in vitro. Infect Immun. 2008;76(6):2633-41
65. Leng L, Metz C.N, Fang Y. et al. MIF signal transduction initiated by binding to CD74. J Exp Med. 2003;197(11):1467-76
66. Bernhagen J, Krohn R, Lue H. et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13(5):587-96
67. Ortolano S, Hwang I.Y, Han S.B. et al. Roles for phosphoinositide 3-kinases, Bruton's tyrosine kinase, and Jun kinases in B lymphocyte chemotaxis and homing. Eur J Immunol. 2006;36(5):1285-95
68. Binsky I, Haran M, Starlets D. et al. IL-8 secreted in a macrophage migration-inhibitory factor- and CD74-dependent manner regulates B cell chronic lymphocytic leukemia survival. Proc Natl Acad Sci U S A. 2007;104(33):13408-13
69. Shi X, Leng L, Wang T. et al. CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity. 2006;25(4):595-606
70. Mitchell R.A, Metz C.N, Peng T. et al. Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J Biol Chem. 1999;274(25):18100-6
71. Yu X, Lin S.G, Huang X.R. et al. Macrophage migration inhibitory factor induces MMP-9 expression in macrophages via the MEK-ERK MAP kinase pathway. J Interferon Cytokine Res. 2007;27(2):103-9
72. Pakozdi A, Amin M.A, Haas C.S. et al. Macrophage migration inhibitory factor: a mediator of matrix metalloproteinase-2 production in rheumatoid arthritis. Arthritis Res Ther. 2006;8(4):R132
73. Lin X, Sakuragi T, Metz C.N. et al. Macrophage migration inhibitory factor within the alveolar spaces induces changes in the heart during late experimental sepsis. Shock. 2005;24(6):556-63
74. Coleman A.M, Rendon B.E, Zhao M. et al. Cooperative regulation of non-small cell lung carcinoma angiogenic potential by macrophage migration inhibitory factor and its homolog, D-dopachrome tautomerase. J Immunol. 2008;181(4):2330-7
75. Qi D, Hu X, Wu X. et al. Cardiac macrophage migration inhibitory factor inhibits JNK pathway activation and injury during ischemia/reperfusion. J Clin Invest. 2009;119(12):3807-16
76. Kleemann R, Hausser A, Geiger G. et al. Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature. 2000;408(6809):211-6
Author contact
![]() Corresponding author: Miriam Rodriguez-Sosa, Unidad de Biomedicina, FES-Iztacala, UNAM. Av. de los Barrios # 1, Los Reyes Iztacala, Tlalnepantla, 54090. Edo. de México, Mexico. Phone: (+5255) 5623-1333 Fax: (+5255) 5623 1138. E-mail: rodriguezmiztacala.unam.mx
Corresponding author: Miriam Rodriguez-Sosa, Unidad de Biomedicina, FES-Iztacala, UNAM. Av. de los Barrios # 1, Los Reyes Iztacala, Tlalnepantla, 54090. Edo. de México, Mexico. Phone: (+5255) 5623-1333 Fax: (+5255) 5623 1138. E-mail: rodriguezmiztacala.unam.mx
Received 2011-9-1
Accepted 2011-10-1
Published 2011-10-25