ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2011; 7(9):1401-1411. doi:10.7150/ijbs.7.1401 This issue Cite
Research Paper
Nitric Oxide is Involved in the Upregulation of IFN-γ and IL-10 mRNA Expression by CD8+ T Cells During the Blood Stages of P. chabaudi AS Infection in CBA/Ca Mice
M Legorreta-Herrera1 ![]() , S Rivas-Contreras1, JL Ventura-Gallegos2, A Zentella-Dehesa2,3
, S Rivas-Contreras1, JL Ventura-Gallegos2, A Zentella-Dehesa2,3
1. Laboratorio de Inmunología Molecular, Facultad de Estudios Superiores Zaragoza, Universidad Nacional Autónoma de México, Batalla 5 de mayo esq. Fuerte de Loreto, Iztapalapa 09230, México, D.F. México
2. Departamento de Medicina Genómica y Toxicología Ambiental, Instituto de Investigaciones Biomédicas, UNAM, México, D.F.
3. Departamento de Bioquímica, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán México, D.F.
Abstract
Nitric oxide (NO) is involved in the clearance of several types of bacteria, viruses and parasites. Although the roles of NO and CD8+ T cells in the immune response to malaria have been extensively studied, their actual contributions during the blood stages of malaria infection remain unclear.
In this work, we corroborate that serum NO levels are not associated with the in vivo elimination of the blood stages of Plasmodium chabaudi AS. In addition, we show that CD8+ T cells exhibit increased apoptosis and up regulate the expression of TNF-α mRNA on day 4 post-infection and IFN-γ and IL-10 mRNA on day 11 post-infection. Interestingly, only the levels of IFN-γ and IL-10 expression are affected when iNOS is inhibited with aminoguanidine (AG), suggesting that NO could be involved in the activation of CD8+ T cells during the blood stages of plasmodium infection.
Keywords: Nitric oxide, CD8+ T cells, apoptosis, Plasmodium.
1. Introduction
Malaria is still the cause of nearly 1 million deaths every year [1]. Much of the pathology of malaria is immune-mediated, which implies that the immune response needs to be carefully regulated. Some potential regulatory mechanisms include the following: apoptosis, the modification of cellular immune responses, the activities of self-regulating networks of effector molecules such as NO, IL-10 or IFN-γ [2-4], and CD8+ T cell activation. The optimal immune response to malaria infection is characterized by early, intense, pro-inflammatory cytokine-mediated effector mechanisms that kill or clear parasite-infected cells. This immediate pro-inflammatory response is just as rapidly suppressed by an anti-inflammatory response once parasite replication has been brought under control. The successful resolution of malaria infection may thus require a regulated progression from one type of immune response to another [5].
NO is a versatile component of the innate immune system, and iNOS-derived NO is involved in both the pathogenesis and control of several types of viral, bacterial and parasitic infections [6]. The increased production of endogenous NO during blood-stage malaria infection has been found to correlate with protection against Plasmodium falciparum [7]. Additionally, NO can be both cytotoxic and cytostatic to P. falciparum in vitro [8] and can inhibit the growth of P. falciparum in vitro [9]. Furthermore, NO is able to modulate the immune response via the regulation of apoptosis and the upregulation of cytokine mRNA expression [10-12]. Apoptosis modulates the development, maturation, activation and termination of the lymphocytic response [4, 13]. Apoptosis involves cytokines such as TNF-α and IFN-γ, and small molecules, such as NO and reactive oxygen intermediates [14], all of which are present at high concentrations in mice during acute infection with P. chabaudi AS [15].
However, controversy exists as to the role of nitric oxide (NO) in malaria because both protective and toxic effects of NO are frequently observed in parallel [16, 17].
CD8+ T cells are involved in both immune and regulatory responses and have been proposed to be essential for protective immunity against the liver stages of the Plasmodium life cycle [18, 19]. CD8+ T cells have also been demonstrated to confer protective immunity upon adoptive transfer [20]. In contrast, it has been reported that the presence or absence of CD8+ T cells does not change the disease outcome during the blood stages of malaria infection [21, 22]. Therefore, the role of CD8+ T cells in the establishment of protective immunity against blood-stage malaria remains a matter of controversy.
The roles of NO and CD8+ T cells in malaria have been extensively studied; however, their actual contributions to the immune response during the blood stages of malaria infection have remained controversial. Thus, an investigation of the impact of NO on CD8+ T cell apoptosis and on the expression of cytokines by CD8+ T cells is critical for defining the contributions of NO to the immune response during the blood stages of malaria infection.
In this work, we have manipulated NO levels in vivo to determine the effect on parasite elimination, upregulation of immune response-associated genes, apoptosis and cytokine mRNA levels produced by CD4+ T, CD8+ T and NK+ cells during blood-stage infection with P. chabaudi AS in CBA/Ca mice.
2. Materials and Methods
2.1 Mice and parasites
The CBA/Ca mice were kindly donated by Dr. W. Jarra (National Institute for Medical Research, London). The mice were bred, fed and maintained in a specific pathogen-free environment at the FES Zaragoza-Universidad Nacional Autónoma de México animal house facilities in accordance with the institutional and national official guideline NOM-062-ZOO-1999 for the use and care of laboratory animals.
Plasmodium chabaudi AS was kindly donated by Dr. W. Jarra (National Institute for Medical Research, London).
2.2 Infection and treatment
For infection, batches of sex- and age-matched (6-8 weeks) CBA/Ca mice were inoculated intravenously (i.v.) with 5 x 104 P. chabaudi AS-parasitised erythrocytes (PE). Each batch of infected mice was subdivided into three groups of six to eight animals. The first group was treated with aminoguanidine (AG), a selective inhibitor of iNOS, twice daily (300 mg/kg dissolved in 100 µL sterile PBS) [23]. The second group was treated with lipopolysaccharide (LPS, 1 mg/kg), and a third group was intraperitoneally (i.p.) injected with PBS daily for three days. In the first two experiments, mice were treated at day 4; in the next two experiments, they were treated at day 11; and in the last two experiments, mice were treated at day 21 post-infection. For additional controls, a parallel batch of non-infected mice was divided into three groups. One group was treated with AG, another with LPS and the third with PBS at the same doses described above. Three hours after the last injection, the mice were sacrificed under ether anaesthesia.
2.3 Blood sampling
Parasitaemia was evaluated daily by examination of Giemsa-stained thin blood smears. Microscopic enumeration of parasitaemia was performed under oil using a Zeiss Standard 20 microscope (Cark Zeiss LTD, Welwyn Garden City). Parasitaemias of 0.5% and above were determined by counting the number of parasitised erythrocytes present in a total of 200 red blood cells. Lower levels of parasitaemia were assessed by counting the number of parasitised erythrocytes present in 50 fields. The course of infection in each group is shown as the geometric mean of the percentage of parasitaemia.
2.4 Nitric oxide quantification
Nitrate concentration in the serum was evaluated according to the procedure described by Jacobs et al. [15]. Fifteen microlitres of serum from each mouse were incubated for 15 min at room temperature with 5 µL of nitrate reductase (5 U/mL; Boehringer Mannheim, Laval, Quebec, Canada) and 15 µL of NADPH (1.25 mg/mL; Boehringer). After incubation, 100 µL of Griess reagent (1% sulphanilamide, 0.1% N-(1-naphthyl)ethylenediamine dihydrochloride and 1% orthophosphoric acid (Sigma Chemical Co. St. Louis, MO, USA) and 100 µL of trichloroacetic acid (10% aqueous solution) were added, and this mixture was incubated for 10 min at room temperature. Next, the protein precipitates were removed by centrifugation at 13,000 rpm for 5 min, and 100 µL of each supernatant was transferred to a 96-well flat-bottom plate. The nitrate concentration was evaluated in an ELISA reader (Stat Fax Plate Translator USA at 540 nm) using a standard curve made from sodium nitrate (Sigma) diluted in pooled serum from uninfected control CBA/Ca mice that were treated in the same way as the samples.
2.5 Apoptosis detection
Externalisation of phosphatidylserine was detected by annexin-V-FLUOS (Roche Diagnostics, Basel, Switzerland), as described previously [24]. Briefly, 1 x 106 spleen cells were suspended in 200 µL reaction buffer (10 mM HEPES/NaOH, pH 7.4; 140 mM NaCl and 5 mM CaCl2) and 2 µL of annexin-V-FLUOS. After incubation for 15 min in the dark at room temperature, the cells were rinsed with reaction buffer and suspended in 400 µL of reaction buffer containing 10 µg/mL of propidium iodide. Fluorescence was analysed using a FACSCalibur flow cytometer and CellQuestTM software (Becton Dickinson, San Jose, CA, USA). For each sample, 40,000 events were acquired and analysed. The dead cells were excluded by gating for PI negative cells, and the percentage of annexin V positive cells (early apoptosis) was determined after correction for non-specific/background fluorescence.
2.6 Cell phenotype characterisation
The suspensions of 1 x 106 spleen cells were washed in PBS and stained with pre-calibrated dilutions of PE-conjugated anti-mouse -CD3, -CD4, -CD8 or -B220 monoclonal antibodies (BD Pharmingen, San Diego, CA, USA). The cells were incubated for 30 min at 4 °C, washed twice with fluorescence activated cell sorter (FACS) buffer (0.1% bovine serum albumin and 0.1% sodium azide/PBS) and suspended in 100 µL of FACS buffer. The cells were then stained with annexin V, as described in the previous section.
2.7 Cell phenotype isolation
The suspensions of 1 x 107 spleen cells were washed in PBS, resuspended in 100 µL of PBS and separated using the commercial EasySepTM StemCell Technologies, Vancouver, Canada. Briefly, a different tube was used for each phenotype. To each tube containing 1 x 107 total spleen cells 1.5 µL of mouse Fc receptor blocker was added, and the samples were mixed and incubated at room temperature for 15 min with 0.3 µg/mL of each of the following commercial labelling antibodies (BD Pharmingen, San Diego, CA, USA): CD19 FITC for B cells, CD4-FITC for CD4+ cells, CD8-FITC for CD8+ and NK1.1-FITC for NK+ cells. The cells were washed with 10-fold excess buffer (PBS containing 2% FBS (GIBCO) and 1 mM EDTA) and resuspended in 100 µL of the same buffer. Ten microlitres of EasySepTM FITC separation cocktail was added to each cell suspension, and the samples were mixed and incubated at room temperature for 15 min. Five microlitres of EasySepTM magnetic nanoparticles were added, the samples were mixed and incubated at room temperature for 10 min, and the suspensions were brought to 2.5 mL with buffer. Next, each tube was placed onto a magnet and set for 5 min, and the supernatant fraction was poured off. The tubes were removed from the magnet, and 2 mL of buffer was added. After gentle mixing, the tubes were placed back on the magnet and set aside for 5 min. The supernatant fraction was then poured off, and the cells were resuspended in 100 µL of PBS.
2.8 Real Time-Reverse Transcription-PCR (qRT-PCR)
Groups of infected and non-infected mice were treated with AG, LPS or PBS. To evaluate the mRNA levels of TNF-α, IFN-γ, IL-10, or β-actin mRNA was isolated from the spleen using TRIzol (Invitrogen,California, USA). Mice were sacrificed on day 4 post-infection to detect TNF-α or on day 11 post-infection for IFN-γ and IL-10. RNA was digested with DNase I, according to the manufacturer's instructions (Invitrogen). RNA was quantified by spectrophotometry at 260 nm, and 1.5 µg of RNA was retrotranscribed using 1.5 µg of oligo dT (Invitrogen), 0.5 mM dNTPs (Pharmacia, Uppsala, Sweden), 40 U of RNase inhibitor and 200 U of MMLV-RT (Invitrogen). The measurement of mRNA expression by (qRT)-PCR was performed using an ABI 7500 Sequence Detection System (Applied Biosystems, Foster City, CA). The gene primers were as follows: IFN-γ forward 5' CGG TGA GAA GAT GTT CCA TGC CAC [FAM] G; reverse 5' TCT CCT TCA GGA CAA TGT CAA ACA; TNF-α forward 5' CGG CGT TCT TTG AGA TCC ATG C [FAM], reverse 5' CGT CGT AGC AAA CCA CCA AGT G; IL-10 forward 5'CGG TTC TGG ACA ACA TAC TGC TAA C [FAM] C, reverse 5´TGG ATC ATT TCC GAT AAG GCT TG; β-actin forward 5' CGG GTC AGG TAG TCT GTC AGG TCC [JOE] G, reverse 5' CTA TGC TCT CCC TCA CGC CAT C. Each reaction contained 1X PCR master mix buffer (Invitrogen), 10 nM forward and reverse primers, and 1 µL of cDNA. A melting curve was used to verify that there was only one amplicon and no primer dimers. The thermal cycler parameters were 2 min at 50°C, 2 min at 95°C, and 45 cycles involving denaturation at 95°C for 30 sec, annealing at 60°C for 45 sec and extension at 72°C for 45 sec. Standard curves were prepared using serial dilutions of the cDNA. The assays were performed in triplicate. All data were normalised to β-actin (ΔΔCT analysis, User Bulletin #2, ABI Prism 7700 Sequence Detection System).
2.9 Statistical Analysis
Statistical analysis was performed using the Stat Graphs software (version 5.1). The differences between groups were tested for statistical significance using Student's t test. A P value of <0.05 was considered significant. All data are expressed as the mean ± S.D. Each experiment was performed in duplicate.
3. Results
3.1 Increased levels of apoptosis and nitric oxide production and the peak of parasitaemia occur on day 11 post-infection in CBA/Ca mice infected with P. chabaudi AS
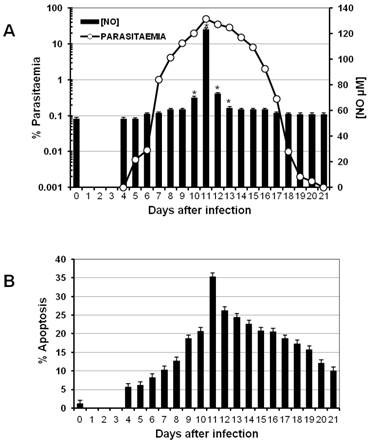
One characteristic of the immune response to malaria is the early strong activation of different lymphocyte populations. Such a response has to be regulated to avoid pathological consequences once the parasite has been eliminated. A typical mechanism to regulate the immune response is apoptosis, which can be modulated by the NO concentration in the serum [25]. To analyse whether parasitaemia levels are associated with both the intensity of apoptosis in splenocytes and the nitric oxide concentration in the serum, a batch of CBA/Ca mice was infected with P. chabaudi AS, and parasitaemia was measured daily (Figure 1A, left axis). On the days indicated, three mice were sacrificed to measure total apoptosis in the spleen (Figure 1B). In addition, nitric oxide levels were measured at the indicated time points during infection (Figure 1A, right axis).
P. chabaudi AS infection in CBA/Ca mice is not lethal. Parasitaemia began to increase on day 4 following infection (day 0) and reached its peak on day 11. Thereafter, parasitaemia decreased until it was resolved on day 21 (Figure 1A). Interestingly, the maximal level of total apoptosis in the spleen, the peak of parasitaemia and the highest concentration of NO were all detected on day 11 post-infection (Figures 1A and 1B).
Time course of parasitaemia, NO serum levels and apoptosis during P. chabaudi AS infection in CBA/Ca mice. CBA/Ca mice were infected with P. chabaudi AS. Parasitaemia, apoptosis versus serum NO levels were monitored daily. The graphs present parasitaemia compared to NO serum levels (A) and the percentage of total spleen cells undergoing apoptosis during the infection (B). Each measurement represents the average of 6 values obtained from 6 mice. The bars represent the standard deviation. * indicates statistical significance compared to control mice treated with PBS. In (B) apoptosis is significantly increased from day 4 to 21 post-infection.
3.2 Nitric oxide is not involved in controlling parasitaemia in CBA/Ca mice infected with P. chabaudi AS
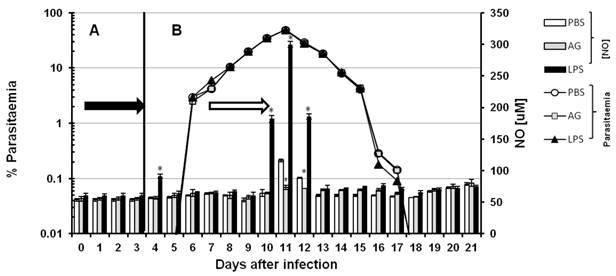
Although both RNI (reactive nitrogen intermediaries) and ROI (reactive oxygen intermediaries) have been reported to kill Plasmodium falciparum and murine malaria parasites in vitro [26, 27] it has been difficult to probe this function in vivo [28, 29]. To evaluate whether serum levels of NO contribute to the control of parasitaemia in vivo, we modulated NO production. Thus, we interfered with nitric oxide synthesis using AG and we induced NO production with LPS in CBA/Ca mice infected with P chabaudi AS. The inhibitory effect of AG is transitory [30]; hence, the experimental schemes were developed to maintain the inhibition of iNOS for 3 days. The 21 days of infection were divided into two phases. In each phase, we injected the mice i.p. with AG for three consecutive days. Figure 2 shows the time course of parasitaemia (white circles, grey squares and black triangles) and the serum levels of NO (bars). Panels A and B represent two independent sets of experiments. In panel A, the mice were infected on day zero and treated with PBS, AG or LPS on days one to three (black arrow). Parasitaemia and serum NO levels were recorded daily from day 0 to 7. In panel B, the mice were infected on day zero, and PBS, AG or LPS was administered from days 9 to 11 (empty arrow). Parasitaemia and serum levels of NO were recorded daily from day 0 to 21. The detected NO levels remained constant throughout the experiment (approximately 65 µM). Statistically significant increases were only observed on day 4 (panel A) in the first regimen and on days 10, 11 and 12 (panel B) in the second. On days 11 and 12, the NO concentrations in the control mice reached 116 and 85 µM, respectively, and on days 4, 10, 11 and 12, the LPS treatment elicited NO levels of 88, 175, 300 and 180 µM, respectively. In the group treated with AG, the NO levels remained within the basal range throughout the experiment, except on days 11 and 12 when they reached 73 and 71 µM, respectively. Despite the reduction in serum levels of NO in the mice treated with AG, the time course of parasitaemia remained unchanged in these mice compared to mice treated with PBS. Likewise, the dramatic increase in serum levels of NO in response to LPS on days 10, 11 and 12 did not alter the kinetics of parasitaemia compared to mice treated with PBS. Our attempts to manipulate NO levels with AG or LPS did not affect parasitaemia, indicating that NO does not play a role in the direct elimination of this parasite (Figure 2).
Time course of parasitaemia and nitric oxide levels in P. chabaudi AS-infected mice treated with aminoguanidine (AG) or LPS. Parasitaemia (left axis) and serum NO concentrations (right axis) were monitored as described in the Materials and Methods in two experimental groups (A and B). Each group contained 6 mice. All animals were infected with P. chabaudi AS and injected i.p. with PBS, aminoguanidine (AG) or with lipopolysaccharide (LPS) at different time points. In group A, after infection with P. chabaudi AS, the mice were injected daily from day 1 to day 3 (black arrow in panel A). In group B, the treatment began 8 days after infection, and the mice were injected daily from day 8 to day 11 (empty arrow in panel B). Each measurement is the average of 6 values obtained from six mice. The bars represent the standard deviation. * indicates statistical significance compared to control mice treated with PBS.
3.3 Effect of AG on apoptosis of CD4+ T, CD8+ T and B220+ splenic cells in P. chabaudi AS infected CBA/Ca mice
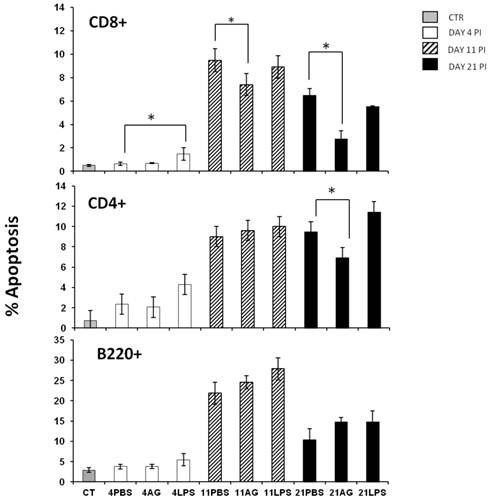
Apoptosis is an important regulator of the immune response [31-33] and is relevant during malaria infection both in murine experimental models and in humans [34-36]. During malaria infection, splenic lymphocytes undergo a high degree of activation that can promote apoptosis [37]. In addition, it has been shown that nitric oxide can prevent or induce apoptosis in many cell types [38]. Because maximal serum NO levels and the peak of splenic apoptosis occurred on the same day, we determined whether these phenomena were related. We wanted to know if different populations of splenic lymphocytes manifested differential susceptibility to NO levels. We analysed three different subpopulations of splenic lymphocytes during early (day 4), middle (day 11) and late (day 21) phases of infection in: control-, AG- or LPS-treated mice. Apoptosis was evaluated in CD4+ T, CD8+ T and B220+ cells from the spleen using flow cytometry as described in the Materials and Methods section. The B220+ cells exhibited the highest levels of apoptosis on day 11 post-infection, but treatment with AG or LPS did not significantly influence apoptosis (Figure 3 A). The incidence of B220+ cell apoptosis was twice as high as that of CD8+ T or CD4+ T cells. The treatment with AG or LPS did not alter the levels of CD4+ T cell apoptosis, with the exception of a significant decrease (-30%) in apoptosis on day 21 post-infection in the AG-treated group compared to the PBS-treated mice (Figure 3 middle panel). Interestingly, infection with P. chabaudi AS significantly increased apoptosis of CD8+ T cells. On days 11 and 21 post-infection, we observed a similar pattern of the CD8+ T cell population as in the CD4+ T cells, in which apoptosis fell by 25% on day 11 and by 56% on day 21 in the AG treated group. Similar to the CD4+ T cells from LPS-treated mice, no significant increase in CD8+ T cells apoptosis was observed on days 11 or 21 (Figure 3C). These findings suggest that NO levels can modulate apoptosis only at specific time points and in specific subsets of splenic lymphocytes. It should be noted that the increase in apoptosis of CD8+ splenocytes after P. chabaudi infection indicates that these cells were stimulated during the blood stages of malaria infection and the fact that apoptosis is reduced by AG treatment suggests a role for NO in this process.
Apoptosis in three different splenic lymphocyte populations from P. chabaudi AS infected mice treated with AG or LPS. The mice were infected and treated as described in Figure 2. At the indicated days, the mice were sacrificed and the cells were isolated from the spleen and stained for lymphocytic markers (B220+, CD4+ and CD8+) and for apoptosis before analysis by flow cytometry. The histograms represent the percentage of apoptosis in each cell subpopulation. Each bar represents the average number of cells from three independent mice ± standard deviation. * indicates statistical significance compared to control mice treated with PBS on the indicated days. CT: control; AG: aminoguanidine; LPS: lipopolysaccharide. Each result is representative of at least two different experiments.
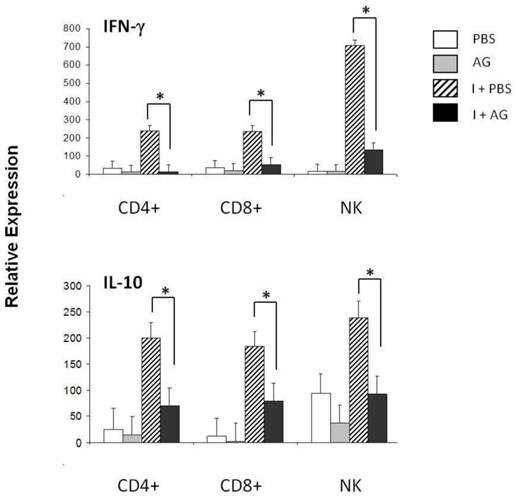
qRT-PCR of IFN-γ and IL-10 in three subpopulations of spleen cells on day 11 of P. chabaudi AS infection. Eleven days after the infection, CD4+, CD8+ or NK.1 (NK) positive cells were isolated from the total spleen cells from control (PBS) or P. chabaudi AS-infected mice (I) treated with or without aminoguanidine (AG). cDNA from each population was used to perform qRT-PCR using primers specific for IFN- γ, IL-10 or β-actin. All of the procedures were performed as described in the Material and Methods. The results are expressed as the average value ± the standard deviation of the mean of the relative signals for each cytokine normalised to β-actin. n = 6, * indicates statistically significant differences.
3.4 Effect of AG on IFN-γ expression by CD4+ T, CD8+ T and B220+ splenic cells in P. chabaudi AS infected CBA/Ca mice
The increase in CD8+ T cells apoptosis on days 11 and 21 post-infection together with the decrease in apoptosis observed in response to AG suggests that NO participates in the regulation of CD8+ cells during P. chabaudi AS infection. An additional measurement of cell function is the production of cytokines, such as IFN-γ or IL-10. In particular, NK cells have been shown to be a major source of IFN-γ in P. chabaudi AS malaria [39, 40]. Therefore, we assessed whether changes in NO levels could modulate IFN-γ expression. First, we measured IFN-γ mRNA levels in the total spleen on days 4, 11 and 21 post-infection (data not shown). A significant increase in the levels of IFN-γ was detected on day 11 post-infection. We thus isolated CD4+ T, CD8+ T or NK+ cells from the spleens of P. chabaudi AS-infected mice treated with AG using negative selection to analyse the mRNA levels of IFN-γ (Figure 4A). Infection with P. chabaudi AS resulted in a significant upregulation of IFN-γ mRNA levels in CD4+ T and NK cells (I+PBS). Interestingly, CD8+ T cells also manifested significantly increased levels of IFN-γ mRNA. Furthermore, when iNOS was inhibited by the AG treatment, the expression of the IFN-γ gene in CD4+ T, CD8+ T and NK+ cells fell by 83, 84 and 92 %, respectively (the differences are indicated in brackets). These results suggest that NO is required for the upregulation of IFN-γ mRNA expression in CD4+ T, CD8+ T and NK+ spleen cells in P. chabaudi AS infected CBA/Ca mice.
3.5 Effect of AG on the expression of IL-10 by CD4+ T, CD8+ T and NK+ splenic cells in P. chabaudi AS-infected CBA/Ca mice
IL-10 plays a protective role against cerebral malaria during Plasmodium berghei (ANKA) infection [41], and is well known to suppress the inflammatory responses induced by IFN-γ [42] Because IFN-γ mRNA expression was downregulated when NO synthesis was inhibited in CD4+, CD8+ and NK+ cells, we further investigated whether there was also an effect on IL-10 expression in these cell subpopulations. The mRNA levels of this cytokine were assessed on day 11 after P. chabaudi AS infection in mice treated with AG (Figure 4 B). An upregulation of IL-10 was detected on day 11 post-infection. Treatment with AG led to reduced IL-10 mRNA levels in all three subpopulations analysed, falling 64, 61 and 56 % in CD4+, CD8+ and NK+ cells, respectively. The effect of AG on IL-10 expression suggests that NO is involved in regulating IL-10 mRNA levels in splenic lymphocytes from CBA/Ca mice that have been infected with P. chabaudi AS.
3.6 AG has no effect on the mRNA levels of TNF-α in CD4+, CD8+ and NK+ splenic cells in P. chabaudi AS-infected CBA/Ca mice
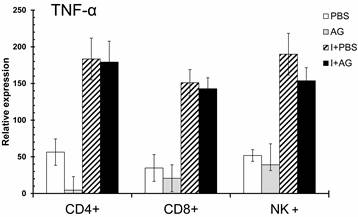
We have previously observed an increase in TNF-α mRNA levels in the total spleens of P. chabaudi AS-infected CBA/Ca mice on day 4 (data not shown). In addition, TNF-α is recognised as an important modulator associated with both the development of immunity and the pathogenesis of severe malaria [43-45]. The levels of IFN-γ and IL-10 were influenced by AG treatment during the mid-phase of infection (day 11), when NO levels in the serum reached their highest levels. Consequently, we evaluated the effect of AG on TNF-α mRNA levels during the early phase of infection (day 4), when parasitaemia begins to increase and no change in serum NO levels were detected (Figure 5). Infection with P. chabaudi AS significantly increased the expression of TNF-α mRNA in all of the cells analysed. However, TNF-α expression was not significantly affected by AG treatment in any of the three cell populations analysed. In support of the idea that CD8+ cells are activated during the early phase of P. chabaudi AS infection (day 4), we observed a marked increase in TNF-α mRNA levels in CD8+ cells. These results were anticipated because the levels of NO did not change between the PBS control and AG treated groups at day 4 post-infection. This finding suggests that NO is not relevant for the upregulation of TNF-α mRNA during the early phase of infection with P. chabaudi AS.
qRT-PCR of TNF-α in three subpopulations of spleen cells at day 4 of P. chabaudi AS infection. Four days after the infection, CD4+, CD8+ or NK+ cells were isolated from total spleen cells as described in the Material and Methods section. The control animals were injected with PBS and the experimental groups were infected with P. chabaudi AS (I) and treated with or without aminoguanidine (AG). cDNA from each population was used to perform qPCR using primers specific for TNF-α or β-actin. All of the procedures were performed as described in the Materials and Methods section. The results are expressed as the average values ± the standard deviation of the mean of the relative TNF-α signal normalised to β-actin. n = 6.
4. Discussion
The participation of CD8+ T cells and nitric oxide in the blood stages of malaria infection has been a matter of controversy [46]. In this work, we provide evidence that splenic CD8+ cells undergo apoptosis at increased frequency and upregulate the expression of TNF-α mRNA on day 4 and IFN-γ and IL-10 mRNA on day 11 post-infection. Interestingly, the expression of IFN-γ and IL-10 in CD8+ cells appeared to be dependent on nitric oxide levels because inhibition of iNOS by AG decreased the mRNA levels of these cytokines.
The adaptive immune response to blood-stage malaria parasites is dependent on both cell- and antibody-mediated immune mechanisms. Several authors have shown that activated CD4+ cells release factors, including IFN-γ, which drive parasite-killing mechanisms [47]. Detection of fragmented DNA and condensed chromatin in P. falciparum suggest that apoptotic and/ or cytotoxic mechanisms are responsible for the killing of blood-stage malaria parasites [48]. Both reactive oxygen intermediates and reactive nitrogen intermediates, especially NO, have been implicated as possible malaria parasite-killing agents [49, 50].
Most ROIs, including hydrogen peroxide (H2O2) and superoxide ions (O2-), can function independently as cytotoxic agents or mediate the formation of other toxic molecules in the presence of NO, including the hydroxyl radical OH-, hypochlorous acid (HOCl) and peroxynitrite (ONOO-) [51]. It is interesting to note that all these molecules are associated with the regulation of apoptosis.
Although there is no doubt that NO is produced during Plasmodium infection, the precise role of NO during the blood stages has been questioned [30, 52]. We show that three events, an increase in serum nitric oxide levels, parasitaemia and apoptosis in the spleen, all reached peak levels on day 11 post-infection. In an attempt to study whether nitric oxide is involved in the regulation of the immune response against malaria parasites by inducing apoptosis in lymphocytes during the peak of parasitaemia, we manipulated NO levels in vivo. We interfered with NO synthesis using AG, or alternatively, we increased NO levels with LPS. Our findings indicate that variations in NO levels had no effect on the destruction of blood-stage P. chabaudi AS parasites in vivo. This result is consistent with previous reports [52-54]. However, our results contrast with the earlier studies of Taylor-Robinson [50], who demonstrated that both mortality and the levels of parasitaemia were associated with the modification of nitric oxide levels by the administration of L-NAME in NIH mice that were infected with P. chabaudi AS. One probable explanation for the difference between our study and that of Taylor-Robinson is that the effect of NO on cell death could differ between various strains of mice, considering that he used the NIH strain whereas we used CBA/Ca mice.
Because NO and O2- are extremely reactive molecules, they can form ONOO- under normal physiological conditions [55, 56], ONOO- can protonate to generate OH• and nitrogen dioxide radicals (.NO2), which are both more reactive than the peroxynitrite precursors [51]. Unlike NO, which has antioxidant, anti-inflammatory, and tissue-protective properties, ONOO- is capable of damaging lipids [57], proteins [58], and nucleic acids [59] and facilitating redox-induced apoptosis [60], Peroxynitrite has a critical role in the immune defence mechanisms against numerous pathogens, including Mycoplasma pulmonis, Rhodococcus equi [61, 62], Trypanosoma and Salmonella spp, most of which have developed defence mechanisms to nullify the toxic effect of ONOO- [63, 64]. Although the effect of ONOO- on blood-stage malaria parasites is not clear, it has been suggested that ONOO- may play a role in the pathogenesis of cerebral malaria by inducing the apoptosis of endothelial cells [65].
The immune system is strongly activated during malarial infection. Apoptosis is an important physiological process for removing cells, including those that are generated in excess that have already completed their specific functions or that are harmful to the host [33]. When NO is oxidised to reactive nitrogen species, it elicits various biological effects, including the regulation of apoptosis [66, 67]. Parasite destruction is supposed to occur in the spleen, but NO levels are usually measured in the serum. Therefore, in this work, we determined whether variations in serum NO levels affect cytokine production by splenic lymphocytes in malaria-infected mice. Our results suggest that while AG prevents an increase in serum NO, it has a limited efficacy in reducing apoptosis in the total splenic cell population. In contrast, the AG treatment had a profound effect on the production of IFN-γ and IL-10 by CD4+ T, CD8+ T and NK+ splenic lymphocytes. This suggests that splenic NO levels rather than serum NO levels, might be relevant during lymphocyte activation in response to blood-stage plasmodium infection. Consequently, a more detailed analysis of splenic NO production may be a better parameter to evaluate the role of NO during plasmodium infection.
In support of our observations, a few groups have recently demonstrated the expansion of splenic CD8+ T cells during the blood stages of malaria infection [68-70]. In addition, using bromodeoxyuridine, it has been shown that CD8+ T cells are activated during blood-stage malaria infection and that activated CD8+ T cells express different markers such as IL-7R, PD-1 and CD62L [46]. Other authors have suggested that CD8+ T cells producing IFN-γ, perforin and granzyme B are involved in protective immunity against murine blood-stage infection with Plasmodium yoelii 17XL [70].
Taken together, our results indicate that NO could be involved in the regulation of CD8+ T cells during the blood stages of Plasmodium infection. This finding extends our understanding of the role of both NO and CD8+ T cells in the immune responses against Plasmodium.
Acknowledgements
We thank Dr. Armando Cervantes for his help with the statistical analyses.
This work was supported in part by the grants from DGAPA, UNAM, PAPIIT IN220310, PAPIME PE200910 and CONACyT 52782.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Sauerwein RW, Roestenberg M Moorthy VS. Experimental human challenge infections can accelerate clinical malaria vaccine development. Nat Rev Immunol. 2011;11:57-64
2. Langhorne J, Albano FR Hensmann M. et al. Dendritic cells, pro-inflammatory responses, and antigen presentation in a rodent malaria infection. Immunol Rev. 2004;201:35-47
3. Li C, Sanni LA Omer F, Riley E Langhorne J. Pathology of Plasmodium chabaudi chabaudi infection and mortality in interleukin-10-deficient mice are ameliorated by anti-tumor necrosis factor alpha and exacerbated by anti-transforming growth factor beta antibodies. Infect Immun. 2003;71:4850-4856
4. Savill J, Dransfield I Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965-975
5. Riley EM, Wahl S Perkins DJ, Schofield L. Regulating immunity to malaria. Parasite Immunol. 2006;28:35-49
6. Bogdan C. Nitric oxide and the immune response. Nat Immunol. 2001;2:907-916
7. Legorreta-Herrera M, Meza RO Moreno-Fierros L. Pretreatment with Cry1Ac protoxin modulates the immune response, and increases the survival of Plasmodium-infected CBA/Ca mice. J Biomed Biotechnol. 2010;2010:198921
8. Rockett KA, Awburn MM Cowden WB, Clark IA. Killing of Plasmodium falciparum in vitro by nitric oxide derivatives. Infect Immun. 1991;59:3280-3283
9. Hollenstein U, Looareesuwan S Aichelburg A. et al. Serum procalcitonin levels in severe Plasmodium falciparum malaria. Am J Trop Med Hyg. 1998;59:860-863
10. Remick DG, Villarete L. Regulation of cytokine gene expression by reactive oxygen and reactive nitrogen intermediates. J Leukoc Biol. 1996;59:471-475
11. Hanum PS, Hayano M Kojima S. Cytokine and chemokine responses in a cerebral malaria-susceptible or -resistant strain of mice to Plasmodium berghei ANKA infection: early chemokine expression in the brain. Int Immunol. 2003;15:633-640
12. Hildeman DA, Mitchell T Teague TK. et al. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity. 1999;10:735-744
13. Van Parijs L, Abbas AK. Homeostasis and self-tolerance in the immune system: turning lymphocytes off. Science. 1998;280:243-248
14. Villeval JL, Lew A Metcalf D. Changes in hemopoietic and regulator levels in mice during fatal or nonfatal malarial infections. I. Erythropoietic populations. Exp Parasitol. 1990;71:364-374
15. Jacobs P, Radzioch D Stevenson MM. In vivo regulation of nitric oxide production by tumor necrosis factor alpha and gamma interferon, but not by interleukin-4, during blood stage malaria in mice. Infect Immun. 1996;64:44-49
16. Asensio VC, Oshima H Falanga PB. Plasmodium berghei: is nitric oxide involved in the pathogenesis of mouse cerebral malaria?. Exp Parasitol. 1993;77:111-117
17. Nahrevanian H. Immune effector mechanisms of the nitric oxide pathway in malaria: cytotoxicity versus cytoprotection. Braz J Infect Dis. 2006;10:283-292
18. Good MF, Doolan DL. Immune effector mechanisms in malaria. Curr Opin Immunol. 1999;11:412-419
19. Jayawardena AN, Murphy DB Janeway CA, Gershon RK. T cell-mediated immunity in malaria. I. The Ly phenotype of T cells mediating resistance to Plasmodium yoelii. J Immunol. 1982;129:377-381
20. Mogil RJ, Patton CL Green DR. Cellular subsets involved in cell-mediated immunity to murine Plasmodium yoelii 17X malaria. J Immunol. 1987;138:1933-1939
21. Suss G, Eichmann K Kury E, Linke A Langhorne J. Roles of CD4- and CD8-bearing T lymphocytes in the immune response to the erythrocytic stages of Plasmodium chabaudi. Infect Immun. 1988;56:3081-3088
22. van der Heyde HC, Manning DD, Roopenian DC, Weidanz WP. Resolution of blood-stage malarial infections in CD8+ cell-deficient beta 2-m0/0 mice. J Immunol. 1993;151:3187-3191
23. Misko TP, Moore WM Kasten TP. et al. Selective inhibition of the inducible nitric oxide synthase by aminoguanidine. Eur J Pharmacol. 1993;233:119-125
24. Ramos-Avila A, Ventura-Gallegos JL Zentella-Dehesa A. et al. Immunomodulatory role of chloroquine and pyrimethamine in Plasmodium yoelii 17XL infected mice. Scand J Immunol. 2007;65:54-62
25. Trubiani O, Salvolini E Vignini A, D'Arcangelo C Di Primio R, Mazzanti L. NF-kappaB and NOS may play a role in human RPMI-8402 cell apoptosis. Cell Biol Int. 2005;29:529-536
26. Ockenhouse CF, Schulman S Shear HL. Oxidative killing of malaria parasites by mononuclear phagocytes. Prog Clin Biol Res. 1984;155:93-108
27. Rockett KA, Awburn MM Cowden WB, Clark IA. Killing of Plasmodium falciparum in vitro by nitric oxide derivatives. Infect Immun. 1991;59:3280-3283
28. Greve B, Lehman LG Lell B, Luckner D Schmidt-Ott R, Kremsner PG. High oxygen radical production is associated with fast parasite clearance in children with Plasmodium falciparum malaria. J Infect Dis. 1999;179:1584-1586
29. Perlmann P, Troye-Blomberg M. Malaria and the immune system in humans. Chem Immunol. 2002;80:229-242
30. Taylor-Robinson AW, Smith EC. A dichotomous role for nitric oxide in protection against blood stage malaria infection. Immunol Lett. 1999;67:1-9
31. Yolcu ES, Ash S Kaminitz A, Sagiv Y Askenasy N, Yarkoni S. Apoptosis as a mechanism of T-regulatory cell homeostasis and suppression. Immunol Cell Biol. 2008;86:650-658
32. Brenner D, Krammer PH Arnold R. Concepts of activated T cell death. Crit Rev Oncol Hematol. 2008;66:52-64
33. Birge RB, Ucker DS. Innate apoptotic immunity: the calming touch of death. Cell Death Differ. 2008;15:1096-1102
34. Foller M, Bobbala D Koka S, Huber SM Gulbins E, Lang F. Suicide for survival--death of infected erythrocytes as a host mechanism to survive malaria. Cell Physiol Biochem. 2009;24:133-140
35. Pino P, Taoufiq Z Nitcheu J, Vouldoukis I Mazier D. Blood-brain barrier breakdown during cerebral malaria: suicide or murder?. Thromb Haemost. 2005;94:336-340
36. Kapoor G, Bagai U Banyal HS. Plasmodium berghei induces apoptotic changes in splenic and peripheral blood cells. Trop Biomed. 2011;28:119-124
37. Helmby H, Jonsson G Troye-Blomberg M. Cellular changes and apoptosis in the spleens and peripheral blood of mice infected with blood-stage Plasmodium chabaudi chabaudi AS. Infect Immun. 2000;68:1485-1490
38. Perrotta C, De Palma C Falcone S, Sciorati C Clementi E. Nitric oxide, ceramide and sphingomyelinase-coupled receptors: a tale of enzymes and messengers coordinating cell death, survival and differentiation. Life Sci. 2005;77:1732-1739
39. Muxel SM, Freitas do Rosario AP Sardinha LR. et al. Comparative analysis of activation phenotype, proliferation, and IFN-gamma production by spleen NK1.1(+) and NK1.1(-) T cells during Plasmodium chabaudi AS malaria. J Interferon Cytokine Res. 2010;30:417-426
40. Mohan K, Moulin P Stevenson MM. Natural killer cell cytokine production, not cytotoxicity, contributes to resistance against blood-stage Plasmodium chabaudi AS infection. J Immunol. 1997;159:4990-4998
41. Kossodo S, Monso C Juillard P, Velu T Goldman M, Grau GE. Interleukin-10 modulates susceptibility in experimental cerebral malaria. Immunology. 1997;91:536-540
42. Linke A, Kuhn R Muller W, Honarvar N Li C, Langhorne J. Plasmodium chabaudi chabaudi: differential susceptibility of gene-targeted mice deficient in IL-10 to an erythrocytic-stage infection. Exp Parasitol. 1996;84:253-263
43. Clark K, Kulk N Amante F, Haque A Engwerda C. Lymphotoxin alpha and tumour necrosis factor are not required for control of parasite growth, but differentially regulate cytokine production during Plasmodium chabaudi chabaudi AS infection. Parasite Immunol. 2007;29:153-158
44. Palmer RM, Ashton DS Moncada S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature. 1988;333:664-666
45. Richards AL. Tumour necrosis factor and associated cytokines in the host's response to malaria. Int J Parasitol. 1997;27:1251-1263
46. Chandele A, Mukerjee P Das G, Ahmed R Chauhan VS. Phenotypic and functional profiling of malaria-induced CD8 and CD4 T cells during blood-stage infection with Plasmodium yoelii. Immunology. 2011;132:273-286
47. Li C, Langhorne J. Tumor necrosis factor alpha p55 receptor is important for development of memory responses to blood-stage malaria infection. Infect Immun. 2000;68:5724-5730
48. Picot S, Burnod J Bracchi V, Chumpitazi BF Ambroise-Thomas P. Apoptosis related to chloroquine sensitivity of the human malaria parasite Plasmodium falciparum. Trans R Soc Trop Med Hyg. 1997;91:590-591
49. Clark IA, Hunt NH. Evidence for reactive oxygen intermediates causing hemolysis and parasite death in malaria. Infect Immun. 1983;39:1-6
50. Taylor-Robinson AW, Phillips RS Severn A, Moncada S Liew FY. The role of TH1 and TH2 cells in a rodent malaria infection. Science. 1993;260:1931-1934
51. Beckman JS, Beckman TW Chen J, Marshall PA Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620-1624
52. van der Heyde HC, Gu Y, Zhang Q, Sun G, Grisham MB. Nitric oxide is neither necessary nor sufficient for resolution of Plasmodium chabaudi malaria in mice. J Immunol. 2000;165:3317-3323
53. Yoneto T, Yoshimoto T Wang CR. et al. Gamma interferon production is critical for protective immunity to infection with blood-stage Plasmodium berghei XAT but neither NO production nor NK cell activation is critical. Infect Immun. 1999;67:2349-2356
54. Stevenson MM, Tam MF Wolf SF, Sher A. IL-12-induced protection against blood-stage Plasmodium chabaudi AS requires IFN-gamma and TNF-alpha and occurs via a nitric oxide-dependent mechanism. J Immunol. 1995;155:2545-2556
55. Goldstein S, Czapski G. The reaction of NO. with O2.- and HO2.: a pulse radiolysis study. Free Radic Biol Med. 1995;19:505-510
56. Huie RE, Padmaja S. The reaction of no with superoxide. Free Radic Res Commun. 1993;18:195-199
57. Radi R, Beckman JS Bush KM, Freeman BA. Peroxynitrite-induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide. Arch Biochem Biophys. 1991;288:481-487
58. Ischiropoulos H, Zhu L Chen J. et al. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys. 1992;298:431-437
59. Epe B, Ballmaier D Roussyn I, Briviba K Sies H. DNA damage by peroxynitrite characterized with DNA repair enzymes. Nucleic Acids Res. 1996;24:4105-4110
60. Vouldoukis I, Sivan V Vozenin MC. et al. Fc-receptor-mediated intracellular delivery of Cu/Zn-superoxide dismutase (SOD1) protects against redox-induced apoptosis through a nitric oxide dependent mechanism. Mol Med. 2000;6:1042-1053
61. Darrah PA, Hondalus MK Chen Q, Ischiropoulos H Mosser DM. Cooperation between reactive oxygen and nitrogen intermediates in killing of Rhodococcus equi by activated macrophages. Infect Immun. 2000;68:3587-3593
62. Hickman-Davis J, Gibbs-Erwin J Lindsey JR, Matalon S. Surfactant protein A mediates mycoplasmacidal activity of alveolar macrophages by production of peroxynitrite. Proc Natl Acad Sci U S A. 1999;96:4953-4958
63. Chakravortty D, Hansen-Wester I Hensel M. Salmonella pathogenicity island 2 mediates protection of intracellular Salmonella from reactive nitrogen intermediates. J Exp Med. 2002;195:1155-1166
64. Thomson L, Denicola A Radi R. The trypanothione-thiol system in Trypanosoma cruzi as a key antioxidant mechanism against peroxynitrite-mediated cytotoxicity. Arch Biochem Biophys. 2003;412:55-64
65. Pino P, Vouldoukis I Dugas N, Hassani-Loppion G Dugas B, Mazier D. Redox-dependent apoptosis in human endothelial cells after adhesion of Plasmodium falciparum-infected erythrocytes. Ann N Y Acad Sci. 2003;1010:582-586
66. Jun CD, Park SJ Choi BM. et al. Potentiation of the activity of nitric oxide by the protein kinase C activator phorbol ester in human myeloid leukemic HL-60 cells: association with enhanced fragmentation of mature genomic DNA. Cell Immunol. 1997;176:41-49
67. Bogdan C, Rollinghoff M Diefenbach A. Reactive oxygen and reactive nitrogen intermediates in innate and specific immunity. Curr Opin Immunol. 2000;12:64-76
68. Miyakoda M, Kimura D Yuda M. et al. Malaria-specific and nonspecific activation of CD8+ T cells during blood stage of Plasmodium berghei infection. J Immunol. 2008;181:1420-1428
69. Lundie RJ, de Koning-Ward TF Davey GM. et al. Blood-stage Plasmodium infection induces CD8+ T lymphocytes to parasite-expressed antigens, largely regulated by CD8alpha+ dendritic cells. Proc Natl Acad Sci U S A. 2008;105:14509-14514
70. Imai T, Shen J Chou B. et al. Involvement of CD8+ T cells in protective immunity against murine blood-stage infection with Plasmodium yoelii 17XL strain. Eur J Immunol. 2010;40:1053-1061
Author contact
![]() Corresponding author: Martha Legorreta-Herrera: Laboratorio de Inmunología Molecular, Facultad de Estudios Superiores Zaragoza, Universidad Nacional Autónoma de México, Batalla 5 de mayo esq. Fuerte de Loreto, Iztapalapa 09230, México, D.F. Tel and Fax: +52 55 56230736; E-mail: marthalunam.mx
Corresponding author: Martha Legorreta-Herrera: Laboratorio de Inmunología Molecular, Facultad de Estudios Superiores Zaragoza, Universidad Nacional Autónoma de México, Batalla 5 de mayo esq. Fuerte de Loreto, Iztapalapa 09230, México, D.F. Tel and Fax: +52 55 56230736; E-mail: marthalunam.mx
Received 2011-9-1
Accepted 2011-10-1
Published 2011-11-1