ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2012; 8(1):79-92. doi:10.7150/ijbs.8.79 This issue Cite
Research Paper
The Putative Metal Coordination Motif in the Endonuclease Domain of Human Parvovirus B19 NS1 Is Critical for NS1 Induced S Phase Arrest and DNA Damage
Violetta Kivovich 1,2, Leona Gilbert2, Matti Vuento2, Stanley J. Naides3 ![]()
1. Pennsylvania State College of Medicine/ Milton S. Hershey Medical Center, Hershey, PA, U.S.A.;
2. University of Jyväskylä, Jyväskylä, Finland;
3. Quest Diagnostics Nichols Institute, San Juan Capistrano, CA, U.S.A.
Abstract
The non-structural proteins (NS) of the parvovirus family are highly conserved multi-functional molecules that have been extensively characterized and shown to be integral to viral replication. Along with NTP-dependent helicase activity, these proteins carry within their sequences domains that allow them to bind DNA and act as nucleases in order to resolve the concatameric intermediates developed during viral replication. The parvovirus B19 NS1 protein contains sequence domains highly similar to those previously implicated in the above-described functions of NS proteins from adeno-associated virus (AAV), minute virus of mice (MVM) and other non-human parvoviruses. Previous studies have shown that transient transfection of B19 NS1 into human liver carcinoma (HepG2) cells initiates the intrinsic apoptotic cascade, ultimately resulting in cell death. In an effort to elucidate the mechanism of mammalian cell demise in the presence of B19 NS1, we undertook a mutagenesis analysis of the protein's endonuclease domain. Our studies have shown that, unlike wild-type NS1, which induces an accumulation of DNA damage, S phase arrest and apoptosis in HepG2 cells, disruptions in the metal coordination motif of the B19 NS1 protein reduce its ability to induce DNA damage and to trigger S phase arrest and subsequent apoptosis. These studies support our hypothesis that, in the absence of replicating B19 genomes, NS1-induced host cell DNA damage is responsible for apoptotic cell death observed in parvoviral infection of non-permissive mammalian cells.
Keywords: host cell DNA damage, apoptotic cell death, parvoviral infection
INTRODUCTION
The Parvoviridae family includes some of the smallest DNA viruses identified to date with a 4-6 kb linear single-stranded DNA genome (5, 14). A characteristic replication strategy utilizes duplex hairpin telomeres, made up of short palindromic sequences (3, 7). These hairpins serve as priming sequences for strand elongation during DNA replication (12, 58).
The coding region of all parvoviral genomes translates two major protein groups, the non-structural proteins designated NS or Rep, and the structural capsid proteins, designated VP1 through VP2 or VP3. The non-structural proteins of parvoviruses mediate viral gene transcription, genome replication, capsid packaging and egress from host cells (37). The NS/Rep proteins share sequence similarity among parvovirus species (4, 13, 48, 50, 53). The larger NS protein splice products, such as NS1 of MVM, B19 and the Junonia coenia densovirus, or Rep78/68 of AAV, are composed of multiple domains, the functions of which are integrated and temporally controlled by cellular signaling pathways (17, 20, 36, 38, 56, 59, 60). The central region of all large parvoviral NS proteins contains a nucleoside triphosphate (NTP) binding region that contributes to the oligomerization (15, 23, 41) and helicase properties of these proteins (15, 24, 39, 64). Consensus motifs in this characteristic domain place the parvoviral NS proteins into the superfamily 3 (SF3) of helicases (18, 19). The C-terminus bears the least conserved sequence identity among parvoviral NS proteins, thereby encoding some of the more diverse virus and cell specific functions.
Parvovirus NS proteins interact with numerous cellular components. The cytotoxicity of B19, MVM, H1 and bovine parvovirus NS proteins in replication permissive settings has been attributed to NS1 expression, the mechanism being poorly understood (1, 28, 29, 42, 66). However, some insight has been gained by observing the behavior of the protein in laboratory cell lines in the absence of viral replication. The expression of B19 NS1 in laboratory cell lines has been shown to induce cell cycle arrest and apoptotic cell death (32, 44, 46, 65). The route to cell death in the presence of parvoviral NS proteins has been shown to involve the intrinsic apoptotic cascade (44, 46, 49), suggesting that the proteins induce distinct cellular insults such as accumulation of DNA damage, disruption of the mitochondrial membrane or interruption of the cytoskeletal network. In fact, studies have demonstrated that MVM NS1 interferes with cell signaling by interacting with cellular factors such as CKIIa (40) as well as causing cell cycle arrest in both G1 and S phases by potentially inducing chromosomal lesions (42). Further, B19 NS1 expression in HepG2 cells allows cellular DNA adduct formation with NS1 and single strand DNA nicks, activating ATM/ATR and PARP DNA repair pathways (26,44-46).
In this study, we focus our efforts on elucidating the mechanism of B19 NS1 protein cytotoxicity. B19 is a common human pathogen responsible for erythema infectiosum, or fifth disease, a rash illness in children and other syndromes (2, 16, 25, 27, 31, 35, 37). Sequence analysis of the B19 NS1 protein identifies many of the characteristic features described above, such as the N-terminus endonuclease motifs, a central putative NTP binding region and a unique C-terminus. The role of the enzymatic domains in the observed cytotoxicity has been suggested by Momoeda and colleagues, who abolished the cytotoxic nature of B19 NS1 by introducing single amino acid mutations into the putative NTP binding domain of the protein (30). It has been previously suggested that the MVM NS protein induces cell cycle arrest with a potential assault on cellular DNA (42) as a result of its close association with host DNA in the nucleus. B19 NS1 has also been implicated in cell cycle disturbances observed in culture (32, 33). To further elucidate the mechanism of cytotoxicity we focused on the N-terminus endonuclease domain of the B19 NS1 protein. We hypothesized that in the absence of viral DNA replication, which in replication permissive settings binds the majority of NS1, the endonuclease domain of the unoccupied B19 NS1 protein is responsible for an assault on cellular DNA. We test this hypothesis by studying the effect of mutagenesis in the critical metal coordination motif of the endonuclease domain of the NS1 protein on B19 NS1-induced cell cycle disturbances and cytotoxicity. Our results demonstrate that disruptions in the metal coordination motif of B19 NS1 reduce the protein's ability to inflict DNA damage and reduce NS1-associated S phase arrest and apoptotic cell death.
MATERIALS AND METHODS
Cell Culture. Spodoptera frugiperda-derived SF9 cells were cultured in spinner flasks using BioWhittaker® Insect-XPress cell medium (BioWhittaker®, Walkersville, MD) supplemented with 1% Penicillin-Streptomycin (PenStrep) (Gibco [Invitrogen], Carlsbad, CA) at 27oC. HepG2 cells were cultured in Hepatocyte Wash Medium (Gibco) supplemented with 10% Fetal Bovine Serum (FBS) (Gibco), 1% L-glutamine (Gibco) and 1% PenStrep. The cells were incubated at 37oC in 5% CO2.
Cloning and Recombinant Baculovirus Production. A modified pFastBac1 (Invitrogen) vector was used as a backbone for the generation of B19 NS1 expressing baculovirus. The polyhedrin promoter was removed from the pFastBac1 plasmid by SnaB1 (Fermentas, Glen Burnie, MD) and BamH1 (Fermentas) digestion. The Cytomegalovirus (CMV) immediate-early promoter was obtained from pcDNA3.1 (Invitrogen) by NruI (Fermentas) and BamH1 digestion and used to replace the SnaB1-BamH1 fragment in pFastBac1, resulting in pCMVFastBac1. The vector was again digested with Nhe1 (Fermentas) and BamH1 in order to allow insertion of a fragment encoding the Enhanced GFP (EGFP) protein, obtained from pEGFP-C1 (Clontech Laboratories Inc., Saint-Germain-en-Laye, France) using the same restriction enzymes, resulting in pCMVEGFPwoFastBac1. The native B19 NS1 sequence was obtained from the PBDP2 vector (44).
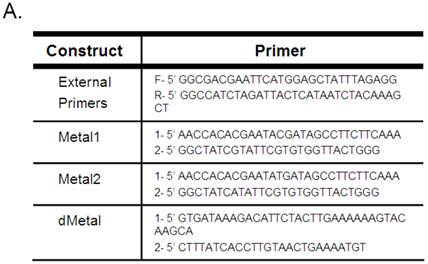
Mutagenesis to the native sequence was performed using the sequential PCR technique (11). Briefly, two overlapping fragments of the desired mutant construct were produced using unique internal oligonucleotides encoding a collection of point mutations and deletions and B19 NS1 generic external primers (Figure 1A). The two fragments were then annealed by mutually primed synthesis using the EcoR1 (Fermentas) and Xba1 (Fermentas) encoding B19 NS1 generic external primers (Figure 1A). The following mutants were generated: Metal1, a double amino acid replacement, H81R and H83R; Metal2, a single amino acid replacement, H83R; and dMetal, a 33 residue deletion of amino acids 74-106. The NS1 constructs were ligated into the EcoR1-Xba1 digested pCMVEGFPwoFastBac1 vector backbone to produce the desired EGFP-NS1 fusion protein and its mutations under the CMV immediate-early promoter. Mutation sequences were confirmed by direct sequencing.
Recombinant baculoviruses were prepared using the Bac-to-Bac® Baculovirus Expression system (Invitrogen). Third-generation virus was collected by centrifugation at 3800x g for 10 min. Sterile inactivated FBS (Gibco) was added to each virus to make a 10% solution, after which the stock was filtered through a 0.2 um filter (Millipore, Billerica, MA) and stored at 4oC.
Baculovirus Transduction of HepG2 Cells. HepG2 cells were seeded in culture flasks and grown overnight in supplemented medium as described above. The following day, the media was removed, cells were washed once with sterile phosphate-buffered saline (PBS), and the baculovirus solution was added to the culture flask. A multiplicity of infection (moi) equivalent to ~100-200 was used for each experiment, resulting in a transduction efficiency of ~25% for each virus at 24 hrs post-transduction. The treated HepG2 cells were kept on ice and remained immersed in the baculovirus solution for 1 hr to promote viral binding to the cellular membrane, after which the virus was removed, the cells were washed once with sterile PBS, and pre-warmed supplemented medium was added back into the culture flasks. Transduced HepG2 cells were stored at 37oC in 5% CO2.
Flow Cytometry. HepG2 cells were collected by trypsinization (0.5% Trypsin EDTA in PBS [Gibco]) at indicated times, washed once with fresh medium (500 μg) and counted to prepare required cell numbers per sample for each experiment. For transduction efficiency analysis, 5 x 105 HepG2 cells were resuspended in 500 μl of ice-cold PBS and immediately analyzed by flow cytometry. The percentage of cells expressing the EGFP marker, as well as the distribution of signal intensity for each construct transduced, was established. This ensured that a comparable amount of protein was being expressed in each construct transduction, for the comparison of different NS1 mutant groups. For cell cycle experiments, 3 x 106 cells were washed once with ice-cold PBS and fixed in 70% ice-cold ethanol (900 μl) for 24-48 hrs at 4oC. The fixed cells were washed twice with PBS to rehydrate the sample and incubated in propidium iodide 40 μg/ml (Molecular Probes [Invitrogen]) and RNase 40 μg/ml (Roche, Basel, Switzerland) in PBS at 37oC for 20 min. The cells were centrifuged, resuspended in ice-cold PBS, and immediately analyzed by flow cytometry. For AnnexinV binding analysis, 5 x 105 cells were washed once with PBS and resuspended in 500 μl AnnexinV Binding Buffer and 5 μl AnnexinV-PE (AMS Biotechnology LTD., Abingdon, OX.), incubated for 15 min at room temperature in the dark and immediately analyzed by flow cytometry.
All samples were analyzed on the FACSCALIBUR flow cytometer (Becton-Dickinson), data collected using Cell-Quest software (Becton-Dickinson) and statistical analysis completed by FlowJo software (Tree Star, Inc., Ashland, OR, USA).
Comet Assay. The comet assay was performed using a reagent kit for single-cell gel electrophoresis (Trevigen, Inc., Gathersburg, MD, USA). Briefly, HepG cells were collected by trypsinization (trypsin/EDTA in PBS) at indicated times, washed once with fresh medium (500 μl), counted to prepare a suspension of 200,000 cells/ml and kept on ice. Etoposide treated HepG2 cells were used as positive control for this assay. The cells were immobilized in a 1:10 bed of low melting point agarose and applied to a CometSlideTM. The cells were gently lysed and the DNA in the resulting nucleoids subjected to electrophoresis under either neutral or alkaline conditions. For the neutral comet assay, the slides were immersed in TBE (1L 10X TBE solution containing 108 g Tris Base, 55g Boric Acid and 9.3 g EDTA) and run for 10 minutes at 1 Volt/cm in an electrophoresis tank. For the alkaline comet assay, the slides were immersed in alkaline solution (pH >13) (1L solution containing 12 g NaOH pellet, 2 ml 500 mM EDTA pH 8, dissolved in dH2O) and run for 30 minutes at 1 Volt/cm with a current of 300 mA, while the electrophoresis tank was maintained on ice for temperature control. The electrophoresis solution was then washed off and slides fixed in 70% ethanol for 5 min, dehydrating the agarose bed and bringing all nucleoids into a single plain. The slides were thus stored until analysis. At analysis, comet slides were stained with SYBR®Green (Trevigen, Inc., Gaithersburg, MD, USA) and studied using a fluorescent microscope. Total cell nucleoids in a field were counted and the number of nucleoids exhibiting comet tail formation was identified. Results were quantified as the number of comet nuclei out of the total number of nuclei observed.
Live Cell Microscopy. Live cell imaging was performed using confocal microscopy (Olympus, Tokyo, Japan). HepG2 cells were grown overnight in 3.5 cm glass-bottom culture dishes (MatTek Corporation, Ashland, MA, USA) and transduced with recombinant baculovirus as described above. Before visualization the cells were treated with Hoecst333258 (0.5 g/ml in HepG2 cell medium) for 15 minutes at 37oC. The flasks were then placed on the pre-warmed (37oC) sample stage, visualized with a 60x APO oil immersion objective and analyzed with FV-10ASW software (Olympus, Tokyo, Japan). Confocal images were processed with ImageJ software (ImageJ, National Institutes of Health, Bethesda, Maryland, USA). Each image was then evaluated for the following: number of cells present, number of cells expressing EGFP or EGFP tagged NS1 constructs, intracellular distribution of green signal (cytoplasmic, semi-nuclear, fully-nuclear) and presence of cellular condensation due to cell death. A minimum of 120 cells were counted for each construct described.
Western Blot Analysis. HepG2 cells were collected by trypsinization (trypsin / EDTA in PBS) at indicated times, washed once with fresh medium (500 μl) and counted. Actinomycin D treated HepG2 cells were used as positive control for H2A.X western blot analysis. Histone extraction for Histone H2A.X analysis was performed using a slightly modified protocol from that previously described (www.abcam.com/technical). HepG2 cells were washed twice with ice-cold PBS and resuspended in Triton Extraction Buffer (TEB) consisting of PBS containing 0.5% Triton X 100 (v/v), 2 mM phenylmethylsulfonyl fluoride (PMSF), 0.02% (w/v) NaN3 at a concentration of 1 x 107 cells/ml and incubated on ice for 20 min. The cells were collected by centrifugation at 8000x g for 10 min at 4oC, washed once with ice-cold TEB and centrifuged for another 10 min. The resulting pellet was resuspended in ice-cold 0.2 N HCl at a cell density of 4 x 107/ml and left at 4oC overnight, shaking vigorously in order to extract the acid soluble protein. The samples were then centrifuged and the supernatant containing the acid soluble fraction analyzed for protein concentration by near UV absorbance (A280) spectroscopy (Nanodrop 1000, Thermo Fisher Scientific Inc., Waltham, MA). Six volumes of acetone were then added and the solution incubated at -20oC overnight to precipitate the acid soluble protein. The precipitated protein was collected by centrifugation and resuspended in running buffer (6.25 mM TrisHCl pH 6.8, 4 M urea, 10% glycerol, 2% SDS, 5% 2-mercaptoethanol, 0.003% bromophenol blue) to a concentration of 20 µg/µl. A total of 100 µg of protein was separated by SDS-PAGE and transferred onto nitrocellulose membrane. A mouse monoclonal anti-H2A.X antibody (Biolegend, San Diego, CA) was used to probe for the presence of double strand breaks in the samples. The antibody was detected with an HRP conjugated anti-mouse antibody, developed using the ECL reagent kit (Thermo Fisher Scientific Inc.) and analyzed using the ChemidocTM XRC and Quantity One® Software (BioRad Laboratories Inc., Hercules, CA.).
Cytotoxicity Assay. Trypan blue exclusion was used to determine cell viability. Adherent and loose HepG2 cells were collected by trypsinization (trypsin EDTA in PBS) at indicated times, as described above, and washed once in sterile PBS. The cells were then resuspended 1:10 in trypan blue solution (%) and incubated at room temperature 2-5 min. The cells were counted using a hemocytometer and trypan blue stained cells were identified. The results were quantified as number of trypan blue staining cells out of total cells counted.
Statistical Analysis. Student's t-test was used to identify statistically significant differences among samples in cell cycle, comet and cytotoxicity assays, comparing means of 3 separate experiments. p<0.05 was considered significant.
RESULTS
Mutations in the putative metal coordination motif of B19 NS1 do not effect
Cellular distribution of the protein. Live HepG2 cells transduced with baculoviruses carrying the above-described constructs (Figure 1) were visualized by confocal microscopy. HepG2 cells transduced with the non-engineered baculovirus lacking in recombinant protein expression showed no cytological changes compared to mock-transduced cells, while cells transduced with EGFP expressing baculovirus displayed even green signal distribution throughout both the cytoplasm and nucleus as early as 4 hrs post-transduction (data not shown). Maximal cellular transduction efficiency was observed at 24 hrs post-transduction (Figure 2A). No cytopathic changes were observed as late as 54 hrs post-transduction in the EGFP expressing cells. HepG2 cells transduced with NS1 and expressing baculovirus showed protein expression at 4 hrs post-transduction and continued to demonstrate increasing protein expression until 24 hrs post-transduction (Figure 2A). Cytopathic changes such as nuclear fragmentation and cytoplasmic condensation, both signs of apoptotic cell death, began to be observed around 24 hrs post-transduction and peaked around 48 hrs post-transduction (data not shown).
Along with observing the consequence of recombinant baculovirus transduction on HepG2 cells, live cell imaging was also used to examine the influence of the introduced mutations on NS1 distribution inside the cells. In an effort to quantify the sub-cellular distribution analysis, the recombinant protein in each imaged cell was scored as fully-nuclear, semi-nuclear (showing some nuclear penetration), cytoplasmic or condensed (displaying cell death). Cellular signal distribution can be seen in Figure 2A with the quantitative demonstration of the microscopy data in the adjacent bar graph (Figure 2B). No fully-nuclear distribution was observed with any of the constructs examined. Unlike EGFP alone, the NS1 expressing cells did not show even distribution of green signal throughout the cells. The NS1 expressing constructs demonstrated a predominantly cytoplasmic signal concentration with approximately 60% of imaged cells displaying some nuclear penetration. Nuclear sequestration of the NS1 signal was not observed in HepG2 cells transduced with NS1 in contrast to that previously described using plasmid based expression systems (43). Mutations in the putative metal coordination site of the B19 NS1 protein did not change the distribution of the recombinant protein inside HepG2 cells.
B19 NS1 Constructs. (A) Mutagenesis to the native sequence was performed using the primers described in A. (B) Diagrams of the NS1 fusion (top) and deletion mutant dMetal (bottom) constructs with the relative organization of the relevant sequence motifs. Metal, putative metal coordination site. ASY, active site tyrosine. NTP, nucleoside-triphosphate-binding motifs.
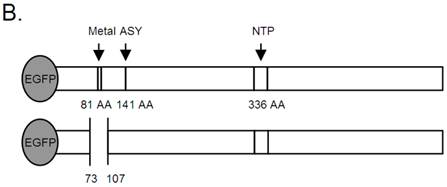
Intracellular Localization. (A) Confocal imaging of live HepG2 cells transduced with EGFP, NS1, Metal1, Metal2 and dMetal 24 hrs post-transduction. Blue panels represent the Hoecst33258 nuclear stain, green panels demonstrate the EGFP or EGFP-fusion protein signal, and the gray panels are the DIC micrographs of imaged cells. The overlay images exhibit EGFP or EGFP-fusion protein penetration into the nuclear region of transduced HepG2 cells. (B) Quantitative analysis of sub-cellular distribution of the recombinant protein in transduced HepG2 cells. Semi-nuclear describes HepG2 cells with some nuclear penetration of EGFP signal, cytoplasmic describes HepG2 cells with no apparent nuclear EGFP penetration and condensed describes dying HepG2 cells which appear small and contracted with concentrated nuclear staining. Minimum of 120 cells were counted for each construct described. Results shown are the mean of three independent experiments with bars demonstrating the standard deviation between the individual runs.
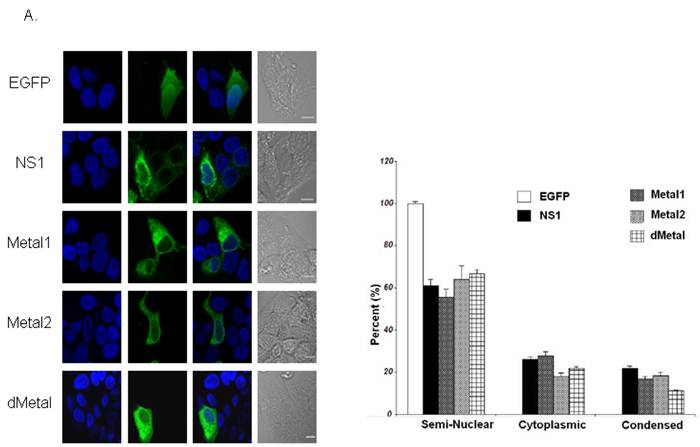
Mutations in the putative metal coordination motif of B19 NS1 interfere with NS1 induced cell cycle disruptions. The effect of NS1 transduction on HepG2 cell cycle progression was examined using propidium iodide staining and subsequent flow cytometry analysis. Non-transduced HepG2 cells, HepG2 cells transduced with moi 400 of un-engineered baculovirus, and HepG2 cells transduced with EGFP expressing virus to 95% transduction efficiency at 24 hrs post-transduction were used to establish HepG2 cell cycle distribution (negative controls). G0/G1, S, G2/M and SubG1 (representing cells undergoing apoptotic cell death) phases were identified on the linear fluorescence intensity plots for propidium iodide emission (Figure 3A). All three negative control conditions displayed the same pattern of cell cycle progression with approximate distribution of cells at 12 hrs as follows: 60% G1, 20% S, 19% G2 and 1% SubG1. For clarity only one representative negative control condition, labeled “Cell,” is presented in Figure 3. After 12 hrs, the population of cells in the G1 phase of the cell cycle slowly grew, reaching approximately 72% at 48 hrs post-transduction. This accumulation was accompanied by a proportional decrease in the S and G2 phase fractions while no significant increase in the SubG1 population was observed. The slow rise in the G1 phase population represented the effect of both nutrient depletion and contact inhibition on HepG2 cells kept in culture for 72 hrs after seeding. The lack of significant changes in the SubG1 content of the cell cycle demonstrated the absence of apoptosis and the innocuous nature of baculovirus transduction in this system.
Cell Cycle Analysis. (A) Representative fluorescence intensity plots for propidium iodide emission demonstrating the cell cycle distribution of mock-transduced control (top) and NS1 (bottom) expressing HepG2 cells at 12, 24, 36 and 48 hrs post-transduction. (B-E) Quantitative graphic representation of HepG2 cell cycle distribution for mock, NS1, Metal1, Metal2 and dMetal transduced HepG2 cells. (B) G1 phase. (C) S phase. (D) G2 phase. (E) SubG1 phase. The data represents three independent experiments with bars demonstrating the standard deviation between the individual runs. One star (*) indicates statistically significant (p < 0.05) change between Cell and the marked set. Two stars (**) - statistically significant (p< 0.05) change between NS1 and the marked set.
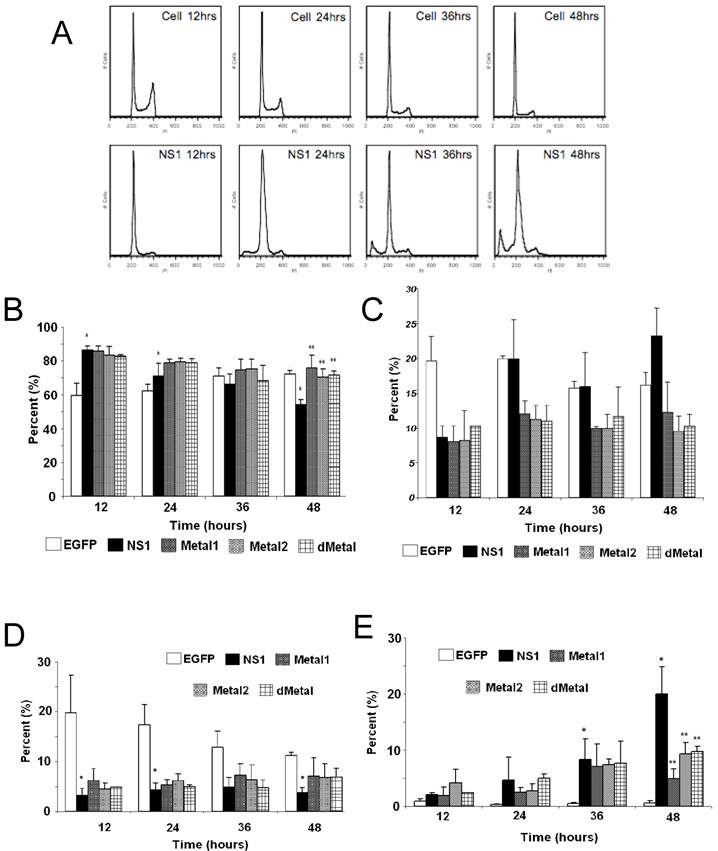
HepG2 cells transduced with baculovirus carrying the NS1 construct were identified by flow cytometry and subsequent cell cycle analysis performed. The pattern of cell cycle distribution for NS1 expressing cells differed significantly from that seen for the negative controls (Figure 3A, bottom panels). At 12 hrs post-transduction, 86% of cells were in the G1 phase of the cell cycle, with 8.7% in S, 3.2% in G2 and 2.1% in SubG1, suggesting a significant early G1/S checkpoint activation by B19 NS1 in HepG2 cells (Cell vs. NS1, p = 0.009) (Figure 3B, 3C, 3D, 3E respectively). The significant accumulation of cells in the G1 phase did not continue into later time points, but instead declined as the S phase and SubG1 populations increased (Figure 3B, 3C, 3E, respectively). At 48 hrs post-transduction, 54.3% of cells were in the G1 phase with 23.3% in S and 20% in SubG1 (Figure 3B, 3C, 3E respectively). The SubG1 accumulation demonstrated the cytotoxic nature of B19 NS1, which has been previously shown to cause apoptotic cell death in HepG2 cells. However, the fraction of cells undergoing apoptotic cell death did not fully compensate for the significant decrease of over 30% in G1 from the 12 hr time point, suggesting that some of the cells were able to overcome the G1/S checkpoint and move into the DNA synthesis phase of the cell cycle. The S phase content of NS1 expressing HepG2 cells was significantly increased compared to the negative controls (p = 0.047), suggesting another point of arrest induced by B19 NS1 in HepG2 cells. No significant changes were observed for G2 phase content, suggesting that the cell cycle aberrations prevent normal progression of HepG2 cells into cell division. The cell cycle observations for NS1 expressing cells were then compared to cells expressing the Metal1, Metal2 and dMetal mutations. At 12 hrs post-transduction, all mutant NS1 proteins exhibited a similar effect on HepG2 cells as the wild-type NS1 protein with approximately 84% of the cells in G1, 8% in S, 5% in G2 and 3% in SubG1 (Figure 3B, 3C, 3D, 3E, respectively). However, unlike the wild-type protein, which demonstrated a decrease of G1 content over time, the mutated constructs continued to maintain a significantly larger proportion of the cells in the G1 phase of the cell cycle (p < 0.02). As late as 48 hrs post-transduction, approximately 72% of transduced HepG2 cells were in the G1 phase of the cell cycle, 10% in S phase, 6% in G2 phase, and 12% in the SubG1 population (Figure 3B, 3C, 3D, 3E, respectively). At 48 hrs post-transduction the 12% loss of G1 content from early observations can be attributed to apoptotic cell death, which can be observed in the SubG1 population growth over time. No significant S phase arrest was seen with any of the putative metal coordination site mutant constructs compared to the negative control, unlike that observed with the wild-type NS1 transduction.
Mutations in the putative metal coordination motif of B19 NS1 reduce NS1 induced DNA damage. Single-cell neutral and alkaline gel electrophoresis (comet) assay was used to directly examine HepG2 cells for the presence of DNA damage. The assay requires a dilution of a sample to form a single cell suspension, which is then embedded in an agarose film and treated to remove all cytoplasmic components before denaturing the remaining nucleoids containing the cellular DNA. Because the embedded nucleoids can no longer be identified as EGFP-tagged protein expressing cells after processing, the percentage of cells expressing the recombinant protein was measured for each sample by flow cytometry before the cells were embedded. Mock-transduced cells served as the negative control and were compared to EGFP transduced HepG2 cells in preliminary experiments. All subsequent experiments and results were normalized to EGFP expressing cells. The results are reported as the percentage of comet positive nucleoids out of the recombinant protein expressing HepG2 cells in the sample.
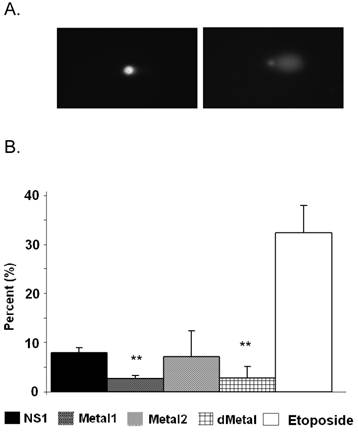
No comet formation was observed with the neutral comet assay as late as 36 hrs post-transduction (data not shown). Therefore all subsequent comet analysis was completed using alkaline assay conditions. Approximately 8% of NS1 expressing cells demonstrated comet formation, displaying the presence of cleaved DNA fragments that migrate freely in an electric field at 24 hrs post-transduction (Figure 4). Such fragments form when the cell nucleoids contain single-strand DNA breaks, double-strand DNA breaks and/or other alkali labile sites. The mutant Metal1 and dMetal expressing HepG2 cells exhibited a significantly (p < 0.0005 and p = 0.036, respectively, Figure 4B) reduced number of comet forming cells compared to the wild-type protein expressing cells, suggesting a reduced level of DNA damage formation by NS1 proteins with severe disruptions in their putative metal coordination site. HepG2 cells expressing the Metal2 construct did not show a significant reduction in comet tail formation compared to the wild-type NS1 protein expressing cells, although the variability in the results did not show a significantly increased level of DNA damage compared to the other mutant constructs either. This observation suggests that the one amino acid replacement in the Metal2 mutant did not fully obliterate the enzymatic activity of the site, with some fraction of the mutated protein still able to perform the endonuclease activity attributed to the mutated region of the protein.
Comet Assay. (A) Fluorescent microscopy images of comet assay demonstrating the intact cellular DNA (left) and comet formation (right) in the presence of DNA damage. (B) Quantitative representation of comet assay for NS1, Metal1, Metal2 and dMetal expressing HepG2 cells 24hrs post-transduction and Etoposide (positive control) treated HepG2 cells. The results have been normalized for background comet formation using EGFP expressing cells. The data represents three independent experiments with bars demonstrating the standard deviation between the individual experiments. One star (*), statistically significant (p < 0.05) change between NS1 and the marked set.
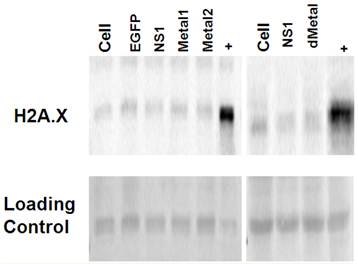
Double strand breaks do not contribute to initial NS1 induced DNA damage. Western blot analysis for histone H2A phosphorylation was used to probe for the presence of double strand DNA breaks in HepG2 cells transduced with EGFP, NS1, or the mutant Metal1, Metal2 and dMetal expressing viruses. Histone H2A is phosphorylated on serine 139, becoming H2A.X within minutes after double strand break formation (52). Cells undergoing apoptotic cell death are also positive for H2A.X as the cellular DNA becomes fragmented in the process of cellular digestion. When tested at 12-24 hrs post-transduction, HepG2 cells expressing the NS1 construct did not show increased histone H2A phosphorylation when compared to the mock transduced (Cell) or EGFP expressing HepG2 cells (Figure 5). At later time points, however, as more of the NS1 expressing cells began to undergo apoptosis, stronger H2A.X staining was observed in the NS1 expressing samples compared to the negative controls (data not shown). The same pattern of histone H2A.X phosphorylation was seen for the HepG2 cells transduced with the mutant constructs (Metal1, Metal2, dMetal) of the B19 NS1 fusion protein. These results demonstrated that double strand breaks are not a major early contributor to B19 NS1 induced DNA damage.
Histone 2A Phosphorylation Analysis. Histone extractions were performed on mock (cell), EGFP, NS1, Metal1, Metal2 and dMetal transduced HepG2 cells 24 hrs post-transduction. Figure demonstrates western blot analysis of acid soluble fraction . Top row demonstrates two individual blots probed for H2A.X. Bottom row represents the Ponceau red loading control of the accompanying blots. + - Actinomycin D treated HepG2 cells. Data are representative of three independent experiments.
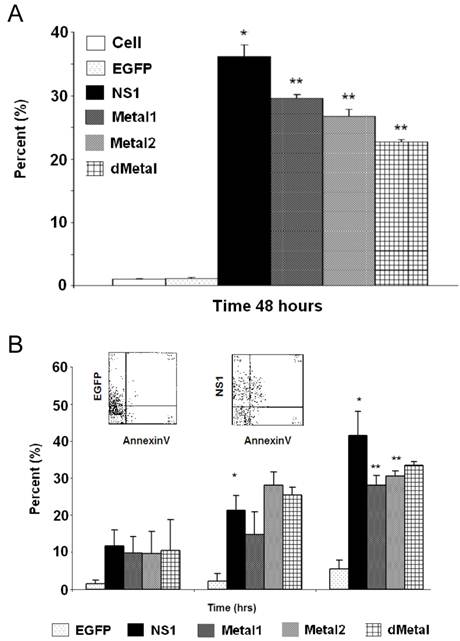
Mutations in the putative metal coordination motif of B19 NS1 interfere with NS1 induced apoptotic cell death. In an effort to understand whether the putative metal coordination region of the B19 NS1 protein contributes to NS1 associated cytotoxicity, we studied the fate of the transiently transduced HepG2 cells by the trypan blue dye exclusion assay for generalized cell viability (Figure 6A) and AnnexinV binding to identify apoptotic cell death (Figure 6B). At 24 hrs post transduction, some early death could be observed in cells expressing NS1 by both trypan blue uptake and AnnexinV binding. However, the variability in the data made the accumulation statistically insignificant. These values significantly increased as the cells continued to bear the burden of NS1 expression. At 48 hrs post transduction, both trypan blue uptake and AnnexinV binding demonstrated 36% (Figure 6A) and 42% (Figure 6B) loss of viability, respectively. HepG2 cells transduced with constructs expressing mutations in the putative metal coordination site (Metal1, Metal2, dMetal) exhibited a similar pattern of cell loss, with continued decline in transduced cell viability until 48 hrs post-transduction. However, unlike wild-type B19 NS1 transduced cells, approximately 10% fewer putative metal coordination site mutant transduced HepG2 cells underwent apoptotic cell death as late as 48 hrs post-transduction (Figure 6B). This significant reduction in cytotoxicity by the putative metal coordination site mutated constructs corresponds to the parallel reductions in the DNA damage observed by the comet assay for Metal1 and dMetal mutants (Figure 4B) as well as the SubG1 population seen in the cell cycle analysis (Figure 3E).
Cytotoxicity Assays. HepG2 cells were mock, EGFP, NS1, Metal1, Metal2 or dMetal transduced, collected at indicated times post-transduction and analyzed by either trypan blue exclusion (A) or AnnexinV binding (B). (A) Data representing the mean of three independent experiments demonstrating the percentage of total cells in the sample permeable to trypan blue dye. (B) Graphic representation of the means of three independent experiments, demonstrating the percentage of EGFP or EGFP-fusion protein expressing HepG2 cells that bind AnnexinV-PE, as identified by flow cytometry analysis. The AnnexinV results have been normalized for background AnnexinV-PE binding using mock-transduced cells. Histograms show AnnexinV staining on EGFP and NS1, respectively, at 48 hours (insert). Bars demonstrate the standard deviation between individual runs. One star (*), statistically significant (p < 0.05) change between Cell and marked set. Two stars (**), statistically significant (p< 0.05) change between NS1 and marked set.
DISCUSSION
Like other parvoviral non-structural proteins, B19 NS1 is a multi-domain replication mediator that performs most of its tasks on strands of replicating DNA. As part of its function in creating complete viral genomes, the B19 NS1 endonuclease domain must bind and nick the unresolved double stranded viral replicating intermediates. Previous work in our laboratory has demonstrated that B19 NS1 co-precipitates with cellular DNA in transient transfection experiments with HepG2 cells (45). The same studies demonstrated that apoptotic cell death in such settings could be reduced by treatment with DNA damage sensor inhibitors such as caffeine (45). We suggest that in the absence of viral genome replication, the nicking capacity of B19 NS1 is directed at the cellular DNA, resulting in accumulation of DNA damage and the activation of DNA repair and apoptotic pathways (6). In this study, we undertook a mutagenic analysis of the putative metal coordination motif (HuHuu) in the endonuclease domain of the B19 NS1 protein, previously shown to be critical for MVM and AAV NS protein nickase function (15). We demonstrate that mutations within the metal coordination motif of B19 NS1 disrupt the protein's ability to induce DNA damage in HepG2 cells, thereby significantly reducing the cytotoxicity of the protein.
Cellular distribution of wild-type NS1 suggested that the protein can enter the nucleus, but is not sequestered there (Figure 2). Previous work with B19 NS1 in HepG2 cells demonstrated that the protein is predominantly localized to the nucleus (47). The expression of NS1 in our previous studies was controlled by the insect steroid inducible ecdysone promoter, a weak promoter in mammalian cells, the introduction of which required dual plasmid transfection, and resulted in a low transfection efficiency (2%). Although low transfection efficiency was adequate for qualitative microscopic analysis as previously reported, the studies described in this report required more robust expression; therefore, we turned to the baculovirus delivery system for introducing the recombinant protein into HepG2 cells. This novel gene delivery vehicle has been previously studied in mammalian cells, shown to be non-cytopathic, and capable of transducing HepG2 cells with a variety of recombinant proteins at greater than 25% transduction efficiency when expressed from the immediate-early CMV promoter (8, 9, 54). In our hands, both the NS1 fusion protein and the mutant NS1 fusion constructs were all efficiently introduced by baculovirus transduction into HepG2 cells. Although slight variability was observed between each new batch of virus produced, generally a multiplicity of infection (MOI) of 100-200 resulted in a transduction efficiency of 25% at 24 hrs post-transduction.
The significantly improved transduction efficiency and strong promoter activity increased not only the number of cells expressing the protein, but also the concentration of protein expressed in each individual HepG2 cell. As members of the SF3 helicase family, parvoviral non-structural proteins oligomerize into either dimers or hexamers (22). In order to perform ATP-dependent DNA unwinding, hexameric rings must surround the DNA strands to function as unwinding motors (63) and therefore the protein must exist in hexameric form in the nucleus. Parvoviral non-structural protein entry into the nucleus has been shown to require some homo-oligomerization (41, 47). However, whether dimers or hexamers are required for nuclear entry is unclear. We propose that the high concentration of B19 NS1 in the cytoplasm of HepG2 cells transduced with baculovirus expressing NS1 from the immediate-early CMV promoter shifts homo-oligomerization of the protein toward hexameric rings instead of homo-dimers, decreasing the efficiency of nuclear entry. For the current work, the presence of B19 NS1 in the nucleus suggested that some nuclear penetration was maintained in our baculovirus expression system, thereby allowing us to examine the potential genotoxic nature of the B19 NS1 protein.
In this study, we demonstrate that the expression of NS1 in HepG2 cells induces significant changes in normal cell cycle progression. An early G1 arrest in the presence of B19 NS1 has been previously observed in UT7/Epo-S1 cells, a parvovirus B19 replication permissive cell line, and this arrest was suggested to be the mediating step in B19 NS1 induced apoptotic cell death (32). Here we support this observation in HepG2 cells, but also show an additional later cell cycle disruption, an S phase arrest, for cells that overcome the G1/S checkpoint. Arrest in the S phase slows the synthesis of novel DNA and activates a similar stress response to that seen at the G1/S checkpoint (34), with mobilized repair pathways (61) and possible signaling for programmed cell death in the presence of un-repairable insults. The S phase arrest observed for cells expressing the wild type NS1 was significantly reduced in cells expressing the putative metal coordination site mutants of the protein. This disruption in the nickase function reduced the protein's genotoxicity. However, no changes in the G1 arrest were observed, suggesting that the G1 arrest may not be due to B19 NS1 endonuclease activity, but a possible second mechanism. Since the G1/S checkpoint can be activated by numerous cell stress scenarios, cytoplasmic effects of the NS1 protein cannot be discounted. MVM NS1 cytotoxicity has been suggested to be mediated by interaction with cytoplasmic cell signaling cascades causing cytoskeletal disruptions (40). It is therefore reasonable to suggest that more than one mechanism is active in B19 NS1 induced cytotoxicity.
DNA damage in HepG2 cells expressing NS1, suggested by the S phase disruption, was directly demonstrated by the alkaline comet assay results of this study. The comet assay is a sensitive and specific assay for the presence of DNA damage. The conditions of the assay can be varied for closer discernment of the nature of the damage by inserting an alkaline unwinding step into the neutral conditions protocol. The neutral conditions are conducive for identifying the presence of double strand breaks, while the alkaline method allows for the identification of single-strand breaks and other alkali-labile sites introduced into the DNA by excision repair mechanisms (55). We first performed the neutral comet assay on HepG2 cells expressing NS1 but were unable to identify any comet formation before 36 hrs post-transduction. Since a significant amount of apoptosis is present at 36 hrs, a process that includes DNA digestion and double-strand break accumulation, the observed comet formation at 36 hours could not be attributed to B19 NS1 induced DNA damage alone. Introduction of the alkaline unwinding step into the protocol allowed us to identify a fraction of cells with accumulated DNA damage as early as 12 hrs post-transduction. The absence of comet formation at 24 hrs post-transduction in neutral conditions and its appearance with alkaline treatment supports the hypothesis that the DNA damage introduced by B19 NS1 is in the form of single-strand breaks. This hypothesized accumulation of single-strand nicks induced by the endonuclease activity of B19 NS1 is further supported by the significant reduction of comet formation in HepG2 cells expressing the double amino acid replacement mutant of the putative metal coordination site (Metal1) as well as the deletion mutant of the entire metal coordination region (dMetal) (Figure 4B). The failure of the single amino acid replacement mutant (Metal2) to reduce DNA damage suggests that some residual nicking capacity may remain in this construct. Since the Metal2 mutant showed significant reduction in S phase arrest as well as in cytotoxicity, we propose that the residual enzymatic activity is compensated for by the cellular DNA repair machinery, which corrects the nicks and allows the cell to progress into the G2 phase and cell division.
We confirmed that apoptotic digestion of cellular DNA did not contribute to the early DNA damage observed in the comet assay by probing the cells for Histone 2A phosphorylation (H2A.X). No early H2A.X staining could be observed in HepG2 cells expressing NS1 or the mutant constructs of the protein. A well accepted marker for identifying the presence of double-strand breaks and apoptotic cell death, the H2A.X data further suggest that the initial DNA damage observed by the comet assay is composed of single-strand breaks and/or other alkali-labile damage such as adduct formation.
Significant accumulation of un-repairable DNA lesions results in the activation of cellular stress-signaling and the apoptotic cell death pathway (10). In this study, we confirmed previous observations that B19 NS1 induces apoptosis in mammalian cells. We also demonstrated that the apoptotic cell death induced by B19 NS1 is partially due to the accumulation of DNA damage inflicted upon the cellular DNA by the endonuclease domain of the protein by studying the cytotoxic nature of the putative metal coordination site mutant constructs of B19 NS1. Approximately 10% reduction in cytotoxicity, as shown by trypan blue dye exclusion and AnnexinV binding at 48 hrs post-transduction, was observed in HepG2 cells expressing the endonuclease deficient mutants of B19 NS1. The reduced cytotoxicity in HepG2 cells transduced with the mutant constructs paralleled the level of reduction in DNA damage observed by the comet assay. The reduced assault on cellular DNA by the nicking capacity of the protein increased cell survival, supporting the hypothesis that DNA damage is one of the mechanisms of B19 NS1 induced cytotoxicity in mammalian cells (21, 45, 51).
Acknowledgements
We thank Pirjo Kauppinen and Eija Korhonen for technical assistance. This study was supported by the Academy of Finland (Contract # 122061).
CONFLICT OF INTERESTS
The authors have declared that no conflict of interest exists.
References
1. Abdel-Latif L, Murray BK, Renberg RL, O'Neill KL, Porter H, Jensen JB, Johnson FB. Cell death in bovine parvovirus infected embryonic bovine tracheal cells is mediated by necrosis rather than apoptosis. J Gen Virol. 2006;87:2539-48
2. Anderson M.J, Jones SE, Fisher-Hoch SP, Lewis E, Hall SM, Bartlett CL, Cohen BJ, Mortimer PP, Pereira MS. Human parvovirus, the cause of erythema infectiosum (fifth disease)?. Lancet. 1983;1:1378
3. Astell C.R, Chow MB, Ward DC. Sequence analysis of the termini of virion and replicative forms of minute virus of mice DNA suggests a modified rolling hairpin model for autonomous parvovirus DNA replication. J Virol. 1985;54:171-7
4. Astell C.R, Mol CD, Anderson WF. Structural and functional homology of parvovirus and papovavirus polypeptides. J Gen Virol. 1987;68(Pt 3):885-93
5. Berns K.I, Rose JA. Evidence for a single-stranded adenovirus-associated virus genome: isolation and separation of complementary single strands. J Virol. 1970;5:693-9
6. Blank M, Shiloh Y. Programs for cell death: apoptosis is only one way to go. Cell Cycle. 2007;6:686-95
7. Bohenzky R.A, LeFebvre RB, Berns KI. Sequence and symmetry requirements within the internal palindromic sequences of the adeno-associated virus terminal repeat. Virology. 1988;166:316-27
8. Boyce F.M, Bucher NL. Baculovirus-mediated gene transfer into mammalian cells. Proc Natl Acad Sci U S A. 1996;93:2348-52
9. Cheng T, Xu CY, Wang YB, Chen M, Wu T, Zhang J, Xia NS. A rapid and efficient method to express target genes in mammalian cells by baculovirus. World J Gastroenterol. 2004;10:1612-18
10. Cimprich K.A, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616-27
11. Cormack B. Current Protocols in Molecular Biology, Supplement 37 ed, vol 1. US: John Wiley & Sons Inc. 1997
12. Cotmore S.F, Tattersall P. Structure and organization of the viral genome. In: (ed.) Kerr JR. et al. Parvoviruses. London: Edward Arnold Publishers Ltd. 2006:73-94
13. Cotmore S.F, Tattersall P. The autonomously replicating parvoviruses of vertebrates. Adv Virus Res. 1987;33:91-174
14. Crawford L.V, Follett EA, Burdon MG, McGeoch DJ. The DNA of a minute virus of mice. J Gen Virol. 1969;4:37-46
15. Davis M.D, Wu J, Owens RA. Mutational analysis of adeno-associated virus type 2 Rep68 protein endonuclease activity on partially single-stranded substrates. J Virol. 2000;74:2936-42
16. Diaz F, Collazos J. Glomerulonephritis and Henoch Schoenlein purpura associated with acute parvovirus B19 infection. Clin Nephrol. 2000;53:237-8
17. Ding C, Urabe M, Bergoin M, Kotin RM. Biochemical characterization of Junonia coenia densovirus nonstructural protein NS-1. J Virol. 2002;76:338-45
18. Gorbalenya A.E, Koonin EV. Helicases: amino acid sequence comparison and structure-function relationships. Curr Opin Struct Biol. 1993;3:419-429
19. Gorbalenya A.E, Koonin EV, Wolf YI. A new superfamily of putative NTP-binding domains encoded by genomes of small DNA and RNA viruses. FEBS Lett. 1990;262:145-8
20. Han S.I, Kawano MA, Ishizu K, Watanabe H, Hasegawa M, Kanesashi SN, Kim YS, Nakanishi A, Kataoka K, Handa H. Rep68 protein of adeno-associated virus type 2 interacts with 14-3-3 proteins depending on phosphorylation at serine 535. Virology. 2004;320:144-55
21. Harley J.B, Harley IT, Guthridge JM, James JA. The curiously suspicious: a role for Epstein-Barr virus in lupus. Lupus. 2006;15:768-77
22. Hickman A.B, Dyda F. Binding and unwinding: SF3 viral helicases. Curr Opin Struct Biol. 2005;15:77-85
23. James J.A, Aggarwal AK, Linden RM, Escalante CR. Structure of adeno-associated virus type 2 Rep40-ADP complex: insight into nucleotide recognition and catalysis by superfamily 3 helicases. Proc Natl Acad Sci U S A. 2004;101:12455-60
24. Jindal H.K, Yong CB, Wilson GM, Tam P, Astell CR. Mutations in the NTP-binding motif of minute virus of mice (MVM) NS-1 protein uncouple ATPase and DNA helicase functions. J Biol Chem. 1994;269:3283-9
25. Kerr J.R, Mattey DL, Thomson W, Poulton KV, Ollier WE. Association of symptomatic acute human parvovirus B19 infection with human leukocyte antigen class I and II alleles. J Infect Dis. 2002;186:447-52
26. Kivovich V, Gilbert L, Vuento M, Naides SJ. Parvovirus B19 Genotype Specific Amino Acid Substitution in NS1 Reduces the Protein's Cytotoxicity in Culture. Int J Med Sci. 2010;7:110-9
27. Lunardi C, Tinazzi E, Bason C, Dolcino M, Corrocher R, Puccetti A. Human parvovirus B19 infection and autoimmunity. Autoimmun Rev. 2008;8:116-20
28. Moens U, Seternes OM, Hey AW, Silsand Y, Traavik T, Johansen B, Rekvig OP. In vivo expression of a single viral DNA-binding protein generates systemic lupus erythematosus-related autoimmunity to double-stranded DNA and histones. Proc Natl Acad Sci U S A. 1995;92:12393-7
29. Moffatt S, Yaegashi N, Tada K, Tanaka N, Sugamura K. Human parvovirus B19 nonstructural (NS1) protein induces apoptosis in erythroid lineage cells. J Virol. 1998;72:3018-28
30. Momoeda M, Wong S, Kawase M, Young NS, Kajigaya S. A putative nucleoside triphosphate-binding domain in the nonstructural protein of B19 parvovirus is required for cytotoxicity. J Virol. 1994;68:8443-6
31. Moore T.L, Bandlamudi R, Alam SM, Nesher G. Parvovirus infection mimicking systemic lupus erythematosus in a pediatric population. Semin Arthritis Rheum. 1999;28:314-8
32. Morita E, Nakashima A, Asao H, Sato H, Sugamura K. Human parvovirus B19 nonstructural protein (NS1) induces cell cycle arrest at G(1) phase. J Virol. 2003;77:2915-21
33. Morita E, Tada K, Chisaka H, Asao H, Sato H, Yaegashi N, Sugamura K. Human parvovirus B19 induces cell cycle arrest at G(2) phase with accumulation of mitotic cyclins. J Virol. 2001;75:7555-63
34. Myung K, Chen C, Kolodner RD. Multiple pathways cooperate in the suppression of genome instability in Saccharomyces cerevisiae. Nature. 2001;411:1073-6
35. Naides S.J, Karetnyi YV, Cooling LL, Mark RS, Langnas AN. Human parvovirus B19 infection and hepatitis. Lancet. 1996;347:1563-4
36. Narasimhan D, Collaco R, Kalman-Maltese V, Trempe JP. Hyper-phosphorylation of the adeno-associated virus Rep78 protein inhibits terminal repeat binding and helicase activity. Biochim Biophys Acta. 2002;1576:298-305
37. Nuesch J.P. Regulation of non-structural protein functions by differential synthesis, modification and traffickng. In: (ed.) Kerr JR. et al. Parvoviruses. London: Edward Arnold Publishers Ltd. 2006:275-289
38. Nuesch J.P, Corbau R, Tattersall P, Rommelaere J. Biochemical activities of minute virus of mice nonstructural protein NS1 are modulated In vitro by the phosphorylation state of the polypeptide. J Virol. 1998;72:8002-12
39. Nuesch J.P, Cotmore SF, Tattersall P. Sequence motifs in the replicator protein of parvovirus MVM essential for nicking and covalent attachment to the viral origin: identification of the linking tyrosine. Virology. 1995;209:122-35
40. Nuesch J.P, Rommelaere J. NS1 interaction with CKII alpha: novel protein complex mediating parvovirus-induced cytotoxicity. J Virol. 2006;80:4729-39
41. Nuesch J.P, Tattersall P. Nuclear targeting of the parvoviral replicator molecule NS1: evidence for self-association prior to nuclear transport. Virology. 1993;196:637-51
42. Op De Beeck A, Caillet-Fauquet P. The NS1 protein of the autonomous parvovirus minute virus of mice blocks cellular DNA replication: a consequence of lesions to the chromatin?. J Virol. 1997;71:5323-9
43. Poole B.D, Gross T, Maier S, Harley JB, James JA. Lupus-like autoantibody development in rabbits and mice after immunization with EBNA-1 fragments. J Autoimmun. 2008;31:362-71
44. Poole B.D, Karetnyi Y.V, Naides SJ. Parvovirus B19-Induced Apoptosis of Hepatocytes. J Virology. 2004;78:7775-83
45. Poole BD, Kivovich V, Gilbert L, Naides SJ. Parvovirus B19 Nonstructural Protein-Induced Damage of Cellular DNA and Resultant Apoptosis. Int. J. Med. Sci. 2011;8:88-96
46. Poole B.D, Zhou J, Grote A, Schiffenbauer A, Naides SJ. Apoptosis of liver-derived cells induced by parvovirus B19 nonstructural protein. J Virol. 2006;80:4114-21
47. Pujol A, Deleu L, Nuesch JP, Cziepluch C, Jauniaux JC, Rommelaere J. Inhibition of parvovirus minute virus of mice replication by a peptide involved in the oligomerization of nonstructural protein NS1. J Virol. 1997;71:7393-403
48. Qiu J, Cheng F, Burger LR, Pintel D. The transcription profile of Aleutian mink disease virus in CRFK cells is generated by alternative processing of pre-mRNAs produced from a single promoter. J Virol. 2006;80:654-62
49. Rayet B, Lopez-Guerrero JA, Rommelaere J, Dinsart C. Induction of programmed cell death by parvovirus H-1 in U937 cells: connection with the tumor necrosis factor alpha signalling pathway. J Virol. 1998;72:8893-903
50. Redemann B.E, Mendelson E, Carter BJ. Adeno-associated virus rep protein synthesis during productive infection. J Virol. 1989;63:873-82
51. Rekvig O. Polyoma induced autoimmunity to DNA; experimental systems and clinical observations in human SLE. Lupus. 1997;6:325-6
52. Rogakou E.P, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858-68
53. Schoborg R.V, Pintel DJ. Accumulation of MVM gene products is differentially regulated by transcription initiation, RNA processing and protein stability. Virology. 1991;181:22-34
54. Shoji I, Aizaki H, Tani H, Ishii K, Chiba T, Saito I, Miyamura T, Matsuura Y. Efficient gene transfer into various mammalian cells, including non-hepatic cells, by baculovirus vectors. J Gen Virol. 1997;78:2657-64
55. Singh N.P, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175:184-91
56. Snyder R.O, Im DS, Muzyczka N. Evidence for covalent attachment of the adeno-associated virus (AAV) rep protein to the ends of the AAV genome. J Virol. 1990;64:6204-13
57. Tanaka A, Sugawara A, Sawai K, Kuwahara T. Human parvovirus B19 infection resembling systemic lupus erythematosus. Intern Med. 1998;37:708-10
58. Tattersall P, Ward DC. Rolling hairpin model for replication of parvovirus and linear chromosomal DNA. Nature. 1976;263:106-9
59. Urabe M, Hasumi Y, Kume A, Surosky RT, Kurtzman GJ, Tobita K, Ozawa K. Charged-to-alanine scanning mutagenesis of the N-terminal half of adeno-associated virus type 2 Rep78 protein. J Virol. 1999;73:2682-93
60. Urcelay E, Ward P, Wiener S.M, Safer B, Kotin R.M. Asymmetric replication in vitro from a human sequence element is dependent on adeno-associated virus Rep protein. J Virol. 1995;69:2038-46
61. van Brabant A.J, Buchanan CD, Charboneau E, Fangman WL, Brewer BJ. An origin-deficient yeast artificial chromosome triggers a cell cycle checkpoint. Mol Cell. 2001;7:705-13
62. Vassin V.M, Wold MS, Borowiec JA. Replication protein A (RPA) phosphorylation prevents RPA association with replication centers. Mol Cell Biol. 2004;24:1930-43
63. Watanabe K, Morishita J, Umezu K, Shirahige K, Maki H. Involvement of RAD9-dependent damage checkpoint control in arrest of cell cycle, induction of cell death, and chromosome instability caused by defects in origin recognition complex in Saccharomyces cerevisiae. Eukaryot Cell. 2002;1:200-12
64. West S.C. DNA helicases: new breeds of translocating motors and molecular pumps. Cell. 1996;86:177-80
65. Wilson G.M, Jindal HK, Yeung DE, Chen W, Astell CR. Expression of minute virus of mice major nonstructural protein in insect cells: purification and identification of ATPase and helicase activities. Virology. 1991;185:90-8
66. Yaegashi N, Niinuma T, Chisaka H, Uehara S, Moffatt S, Tada K, Iwabuchi M, Matsunaga Y, Nakayama M, Yutani C, Osamura Y, Hirayama E, Okamura K, Sugamura K, Yajima A. Parvovirus B19 infection induces apoptosis of erythroid cells in vitro and in vivo. J Infect. 1999;39:68-76
67. Zhou B.B, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433-9
Author contact
![]() Corresponding author: Stanley J. Naides, M.D. Mail: 33608 Ortega Highway, San Juan Capistrano, CA 92675, Phone: 949 728-4578, FAX: 949 728-7852, E-mail: stanley.j.naidescom.
Corresponding author: Stanley J. Naides, M.D. Mail: 33608 Ortega Highway, San Juan Capistrano, CA 92675, Phone: 949 728-4578, FAX: 949 728-7852, E-mail: stanley.j.naidescom.
Received 2011-4-8
Accepted 2011-11-2
Published 2011-11-24