ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2012; 8(7):1075-1084. doi:10.7150/ijbs.4742 This issue Cite
Research Paper
Reduced Activity of Protein Kinase C in the Frontal Cortex of Subjects with Regressive Autism: Relationship with Developmental Abnormalities
Lina Ji1,2, Abha Chauhan1, Ved Chauhan1 ![]()
1. NYS Institute for Basic Research in Developmental Disabilities, 1050 Forest Hill Road, Staten Island, New York 10314, USA.
2. The State Key Lab of Pharmaceutical Biotechnology, College of Life Sciences, Nanjing University, 22 HanKou Rd., Nanjing, People's Republic of China 210093.
Abstract
Autism is a neurodevelopmental disorder with unknown etiology. In some cases, typically developing children regress into clinical symptoms of autism, a condition known as regressive autism. Protein kinases are essential for G-protein-coupled receptor-mediated signal transduction, and are involved in neuronal functions, gene expression, memory, and cell differentiation. Recently, we reported decreased activity of protein kinase A (PKA) in the frontal cortex of subjects with regressive autism. In the present study, we analyzed the activity of protein kinase C (PKC) in the cerebellum and different regions of cerebral cortex from subjects with regressive autism, autistic subjects without clinical history of regression, and age-matched control subjects. In the frontal cortex of subjects with regressive autism, PKC activity was significantly decreased by 57.1% as compared to age-matched control subjects (p = 0.0085), and by 65.8% as compared to non-regressed autistic subjects (p = 0.0048). PKC activity was unaffected in the temporal, parietal and occipital cortices, and in the cerebellum in both autism groups, i.e., regressive and non-regressed autism as compared to control subjects. These results suggest brain region-specific alteration of PKC activity in the frontal cortex of subjects with regressive autism. Further studies showed a negative correlation between PKC activity and restrictive, repetitive and stereotyped pattern of behavior (r= -0.084, p = 0.0363) in autistic individuals, suggesting involvement of PKC in behavioral abnormalities in autism. These findings suggest that regression in autism may be attributed, in part, to alterations in G-protein-coupled receptor-mediated signal transduction involving PKA and PKC in the frontal cortex.
Keywords: Autism, behavior, protein kinase C, protein kinases, regression, signal transduction.
INTRODUCTION
Autism spectrum disorders (ASDs) are neurodevelopmental disorders characterized by impairment in social interactions, verbal and non-verbal communication skills, and restricted, repetitive and stereotyped patterns of behavior [1]. Recently, Centers for Disease Control and Prevention reported the prevalence of ASDs to be 1 in 88 children in the United States [2]. The symptoms of ASDs usually start before the age of 3 years, and are often accompanied by abnormalities in cognitive functioning, learning, attention, and sensory processing. The cause of ASDs is not known. However, ASDs are considered as heterogeneous and multifactorial disorders that are influenced by genetic and environmental factors [3-5]. Several lines of evidence from our and other groups have suggested increased oxidative stress [3; 6-14], mitochondrial dysfunctions [8; 10; 15; 16], immune dysfunction and inflammation [10; 17-21] in autism.
The onset of autism is gradual in many children. However, in regressive autism, children first show sign of normal social and language development but lose these developmental skills at 15-24 months and develop autistic behavior [22]. The reported incidence of regressive autism varies from 15% to 62% of cases in different studies [23-26]. In few cases, regression may significantly affect language with lesser impact on other domains such as social interaction or imaginative play [23; 27]. On the other hand, some children may regress particularly in social functions and not in language [28].
The cause of regression in autism is not understood. Protein kinases are known to play important roles in cellular signaling pathways, and are involved in neurodevelopment [29-31]. The brain synapses are the building blocks of memory formation, and synaptic strength contributes to learning and memory [32]. The changes in neurotransmitters release, receptor sensitivity, and gene expression are involved in synaptic strength, structure and function. Because protein kinases-mediated phosphorylation modifies the functions of proteins, altered activities of protein kinases affect the synaptic efficacy.
Recently, we reported that the activity of cAMP-dependent protein kinase A (PKA) is decreased in the frontal cortex of subjects with regressive autism as compared to age-matched control subjects and autistic subjects without clinical history of regression [33]. Protein Kinase C (PKC), a ubiquitous phospholipid-dependent serine/threonine kinase, is another G-protein-coupled receptor-mediated kinase. PKC is known to be involved in signal transduction associated with the control of brain functions, such as ion channel regulation, receptor modulation, neurotransmitters release, synaptic potentiation/depression, and neuronal survival [34]. It also plays crucial roles in cell proliferation, differentiation and apoptosis. Neuronal tissues have high activity of PKC.
Genetic studies have suggested an involvement of PKC in autism [35; 36]. The analysis of genome-wide linkage and candidate gene association showed PKCβ gene (PRKCB1) linkage to a region on chromosome 16p in the neocotex of subjects with autism [35;36]. High-resolution single-nucleotide polymorphism genotyping and analysis of this region showed strong association of haplotypes in the PKCβ gene with autism. The present study was undertaken to compare the activity of PKC in the cerebellum and different regions of cerebral cortex from subjects with regressive and non-regressive autism and their age-matched control subjects. The relationship between PKC activity and behavioral abnormalities was also studied in autism.
MATERIALS AND METHODS
Autism and control subjects. Samples of postmortem frozen brain regions, i.e., the cerebellum, and cortices from the frontal, temporal, parietal and occipital lobes from autistic (N= 7-10 for different brain regions) and age-matched typically developed, control subjects (N= 9-10) were obtained from the National Institute of Child Health and Human Development (NICHD) Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, MD. The age (mean ± S.E.) for autistic subjects was 12.6 ± 3.2 years, and for control subjects, 12.4 ± 3.3 years. All brain samples were stored at -70°C. This study was approved by the Institutional Review Board (IRB) of the New York State Institute for Basic Research in Developmental Disabilities.
Diagnostic classification. The case history and clinical characteristics for the autism and control subjects is summarized in Table 1. Donors with autism had met the diagnostic criteria of the Diagnostic and Statistical Manual-IV for autism. Autism Diagnostic Interview-Revised (ADI-R) test was performed for UMB # 4671, 4849, 1174, 797, 1182, 4899 and 1638 (Table 2). According to the results of ADI-R diagnostic algorithm, the donor's impairments in social interaction, qualitative abnormalities in communication and restricted, repetitive and stereotyped patterns of behavior are consistent with diagnosis of autism. All exceeded cut off score in each of these parameters. Diagnosis of autism was assigned to UMB # 1349 after extensive evaluation of behavioral tests, including Autism Diagnostic Observation Schedule (ADOS), Vineland Adaptive Behavioral Scale (VABS), and Bayley Scales for Infant Development-II, (BSID-II). In addition to ADIR, UMB # 4849 was also evaluated by BSID-II and Childhood Autism Rating Scale (CARS), which indicated moderate to severe autism, and autism in UMB # 4671 was also verified by VABS. Table 3 shows VABS test which assesses adaptive behavior in four domains: communication, daily living skills, socialization, and motor skills.
In this study, the subjects with autism were divided into two subgroups: regressive autism and non-regressive autism, depending on the pattern of onset of symptoms for autism. Regressive autism refers to a child where parents report an early history of normal development, which is followed by a loss of previously acquired skills. Language regression is the most common form of regression but it can also be accompanied by more global regression involving loss of social skills and interest. On the other hand, in non-regressive autism, the child never gains normal language and social skills, and initial symptoms are delayed speech development, and/or delay in development of social skills and nonverbal communication. These children do not demonstrate regression in terms of loss of language or social skills.
Case history and clinical characteristics of autism and control donors of brain tissue samples.
| Brain tissue (UMB #) | Diagnosis | Autism Diagnostic tests | Age (y) | Sex | PMI (h) | Regressive autism | Other medical conditions | Medications | Cause of death |
|---|---|---|---|---|---|---|---|---|---|
| 4671 | Autism | ADIR, VABS, BSID-II | 4.5 | F | 13 | No | Multiple injuries from fall | ||
| 1349 | Autism | ADOS, VABS, BSID-II | 5.6 | M | 39 | Yes | Drowning | ||
| 4849 | Autism | ADIR, BSID-II, CARS | 7.5 | M | 20 | Yes | Lead poisoning | Drowning | |
| 1174 | Autism | ADIR, VABS | 7.8 | F | 14 | No | Seizures | Depakote, Tegretol | Multiple-system organ failure |
| 4231 | Autism | 8.8 | M | 12 | No | Hyperactivity | Zyprexia, Reminyl | Drowning | |
| 797 | Autism | ADIR | 9.3 | M | 13 | No | Attention deficit disorder, migraine headache | Desipramine | Drowning |
| 1182 | Autism | ADIR | 10.0 | F | 24 | Yes | Smoke inhalation | ||
| 4899 | Autism | ADIR | 14.3 | M | 9 | Yes | Seizures | Trileptal, Zoloft,Clonidine, Melatonin | Drowning |
| 1638 | Autism | ADIR | 20.8 | F | 50 | Yes | Seizures, Attention deficit hyperactivity disorder | Zoloft, Zyprexa, Mellaril, Depoprovera | Seizure-related |
| 5027 | Autism | WISC-R, Bender-Gestalt | 38.0 | M | 26 | No | Respirdal, Luvox | Obstruction of bowel | |
| 4670 | Control | 4.6 | M | 17 | Commotio Cordis from an accident | ||||
| 1185 | Control | 4.7 | M | 17 | Drowning | ||||
| 1500 | Control | 6.9 | M | 18 | Motor vehicle accident | ||||
| 4898 | Control | 7.7 | M | 12 | Hyperactive disorder | Concerta, Clonidone | Drowning | ||
| 1708 | Control | 8.1 | F | 20 | Motor vehicle accident | ||||
| 1706 | Control | 8.6 | F | 20 | Congenital heart disease with heart transplant | Rejection of cardiac allograft transplantation | |||
| 1407 | Control | 9.1 | F | 20 | Asthma allergies | Albuterol, Zirtec, Alegra, Rodact, Flovent, Flonase | Asthma | ||
| 4722 | Control | 14.5 | M | 16 | Motor vehicle accident | ||||
| 1846 | Control | 20.6 | F | 9 | Motor vehicle accident | ||||
| 4645 | Control | 39.2 | M | 12 | Arteriosclerotic heart disease |
ADI-R: Autism Diagnostic Interview Revised.
ADOS: Autism Diagnostic Observation Scale.
VABS: Vineland Adaptive Behavioral Scale.
BSID-II: Bayley Scales of Infant Development-Second Edition.
CARS: Childhood Autism Rating Scale.
WISC-R: Wechsler Intelligence Scale for Children-Revised.
According to the medical histories for UMB-4231 and UMB-5027, the donors had psychological evaluation, and met the criteria for a diagnosis of autism. Detailed information is not available.
Autism Diagnostic Interview-Revised test scores in donors of brain tissue samples.
| Autism Diagnostic Interview-Revised (ADI-R)a | |||||||
|---|---|---|---|---|---|---|---|
| Diagnostic Algorithm | Cutoff score for autism | UMB 4671 | UMB 4849 | UMB 1174 | UMB 797 | UMB 4899 | UMB 1638 |
| Abnormalities in reciprocal social interaction (Scores:0-30) | 10 | 26 | 22 | 22 | 24 | 22 | 21 |
| Abnormalities in communication: | |||||||
| Verbal (Scores:0-26) | 8 | - | 18 | - | 20 | - | - |
| Non-verbal (Scores: 0-14) | 7 | 13 | N/A | 11 | 13 | 14 | 11 |
| Restricted, repetitive and stereotyped patterns of behavior (Scores: 0-12) | 3 | 3 | 8 | 6 | 6 | 8 | 7 |
| Abnormalities of development evident at or before 36 months | 1 | 5 | 3 | 5 | - | 4 | 5 |
a: Higher score represents greater impairment.
UMB 1182: ADI-R was conducted but the scores are not available. The donor met the criteria for a diagnosis of autism.
Vineland Adaptive Behavioral Scales diagnostic test for autism in donors of brain tissue samples.
| Vineland Adaptive Behavioral Scales (VABS)a | |||||||
|---|---|---|---|---|---|---|---|
| UMB 1349 | UMB 4671 | UMB 1174 | |||||
| At age: 25 months | At age: 33 months | At age: 39 months | At age: 6.4 y | ||||
| Domain (Scores:20-160) | Standard Score | Age equivalent performance | Standard Score | Age equivalent performance | Standard Score | Age equivalent performance | Standard score |
| Communication | 57 | 9 months | 69 | 18 months | 52 | 10 months | 41 |
| Daily living skills | 65 | 16 months | 62 | 16 months | 54 | 14 months | 22 |
| Socialization | 60 | 9 months | 71 | 17 months | 51 | 4 months | 52 |
| Motor skills | - | - | - | - | 65 | 24 months | - |
| Composite | - | - | - | - | 51 | 13 months | 35 |
a: Higher score represents better function.
Preparation of brain homogenates. The postmortem brain tissue samples from regressive autism, non-regressive autism, and control subjects were homogenized (10% w/v) in cold buffer containing 50 mM Tris-HCl (pH 7.4), 8.5% sucrose, 2 mM EDTA, 10 mM β-mercaptoethanol and protease inhibitor cocktail at 4°C. For extraction of protein kinases, the homogenates were mixed with an equal volume of extraction buffer containing 40 mM Tris-HCl (pH 7.4), 300 mM NaCl, 2 mM EDTA, 2 mM EGTA, 2% Triton, 5 mM sodium pyrophosphate, 2 mM β-glycerophosphate, 2 mM Na3VO4, 100 mM NaF, and 2 μg/ml leupeptin. The samples were allowed to stand on ice for 10 minutes, and then centrifuged at 135,000 g for 20 minutes at 4°C. The supernatants were collected, and stored at -70ºC. The concentrations of total proteins in the supernatants were measured by the biocinchoninic acid protein assay kit (Thermo Scientific, Rockford, IL).
Assay of PKC activity. The activity of PKC in the brain supernatants was measured by solid phase enzyme-linked immuno-absorbent assay (ELISA) kit from Enzo Life Sciences International, Inc. The assay is designed for the analysis of PKC activity in the solution phase. In this assay, microplates pre-coated with PKC substrate were used. The microplate wells were soaked with dilution buffer and were emptied after 10 minutes. An equal volume of the brain supernatants was added to the wells, followed by the addition of ATP to initiate the reaction. After incubation for 90 minutes at 30°C, the kinase reaction was terminated by emptying the contents of each well. The phosphopeptide substrate thus obtained was immunodetected by using phospho-substrate specific primary antibody and peroxidase-conjugated secondary antibody as per manufacturer's instructions. The mean absorbance (x103) of samples was divided by the quantity of total protein (μg) used per assay, and the data is represented as relative PKC activity.
Statistical analysis. Initially, autistic and control cases were collected as age-matched pairs. As data for both members of a pair were not available in all cases, and data were approximately normally distributed, unpaired two-tailed t-tests were employed to make comparisons of PKC activity in various brain regions between autistic vs. control cases. The data was considered significant if 'p' was < 0.05. Comparisons among controls and autistic cases showing or not showing clinical signs of regression in function were made using one-way analysis of variance (ANOVA). To guard against Type I error, a Bonferroni adjustment for multiple comparisons was made to the t-tests of multiple brain regions, and for the pair wise post-hoc t- tests comparing each pair of the three groups that were compared in the overall ANOVA. Data is presented as Mean ± S.E.
Pearson's correlation coefficient (r) was used to evaluate if there was relationship between PKC activity in autism and behavioral abnormalities (ADI-R score).
RESULTS
PKC activity in the cerebellum and different cerebral regions of brain from subjects with regressive autism, non-regressed autism and their age-matched controls.
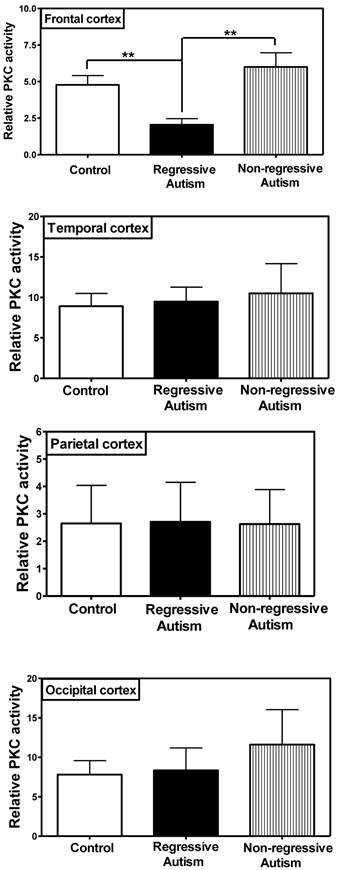
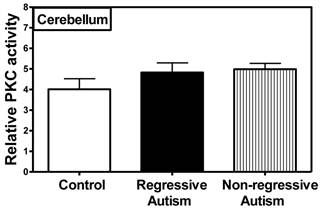
PKC activity was assayed in the frontal, temporal, occipital and parietal cortices (Fig. 1), and cerebellum (Fig. 2) from subjects with autism (regressive and non-regressive) and their age-matched control subjects. As shown in Fig. 1, PKC activity was significantly decreased by 65.8% (p = 0.0048) in the frontal cortex of subjects with regressive autism (Mean ± S.E.; 2.05 ± 0.41) as compared to non-regressive autistic subjects (Mean ± S.E.; 6.00 ± 0.97), and by 57.1% (p = 0.0085) as compared to age-matched control subjects (Mean ± S.E.; 4.78 ± 0.63). On the other hand, PKC activity was similar between non-regressive autism and age-matched control groups. We also analyzed the data with one-way ANOVA test using Bonferroni adjustment for multiple comparison, and observed that data was significant (p = 0.0057). Alteration of PKC activity in the frontal cortex of subjects with regressive autism was brain regions-specific. PKC activity was not affected in other brain regions i.e., cerebellum, and in the temporal, occipital and parietal cortices from autism subjects (regressive and non-regressive autism) as compared to age-matched controls (Figs. 1 and 2).
Protein kinase C activity in different regions of cerebral cortex, i.e., frontal, temporal, occipital and parietal cortex from subjects with regressive autism, non-regressed autism and their age-matched controls. The mean absorbance (x103) of samples was divided by the quantity of total protein (μg) used per assay, and the data is represented as relative PKC activity. **p < 0.01 as compared to control and non-regressed autism groups.
Protein kinase C activity in the cerebellum from subjects with regressive autism, non-regressed autism and their age-matched control subjects. The mean absorbance (x103) of samples was divided by the quantity of total protein (μg) used per assay, and the data is represented as relative PKC activity.
Postmortem interval (PMI) and age of the subjects. There was no significant difference in age (mean ± S.E.) of the subjects among the regressive autism (11.6 ± 2.7 years), non-regressive autism (13.7 ± 6.1 years) and control groups (12.4 ± 3.3 years). Our results also suggest that PMI was not a contributing factor to the observed alteration in PKC activity in the frontal cortex of individuals with regressive autism because PKC activity in the cerebellum and the temporal, parietal, and occipital cortices was not affected in subjects with regressive autism in comparison with control subjects, while it was affected only in the frontal cortex from these individuals with regressive autism.
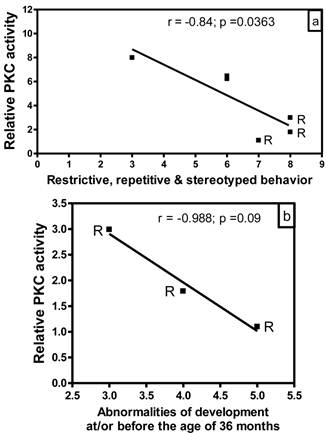
Correlation of PKC activity with behavioral abnormalities in autism. In order to evaluate whether there is any correlation between reduced PKC activity and behavioral abnormalities in subjects with autism, we analyzed the data of PKC activities in the frontal cortex as a function of ADI-R scores for different behavioral parameters (Fig. 3). In this study, we had ADI-R scores of only six subjects with autism, which included three regressive autism subjects (UMB # 4849, 4899, 1638) and three non-regressive autism subjects (UMB # 4671, 1174, 797). Fig. 3a shows the correlation between PKC activity in the frontal cortex and ADI-R score for restrictive, repetitive and stereotyped behavior in regressive and non-regressive autistic subjects. It was observed that ADI-R test score for restrictive, repetitive and stereotyped pattern of behavior was higher in regressive autism as compared to non-regressive autistic subjects. Interestingly, linear regression analysis showed a negative significant correlation between PKC activity in the frontal cortex and restrictive, repetitive and stereotyped behavior (r = -0.84, p =0.0363). A comparison of PKC activity in combined regressive and non-regressive autism group with ADI-R scores for abnormalities of development before the age of 36 months did not show a correlation between these two parameters (data not shown). However, a negative correlation (r = -0.988) in subjects with regressive autism was observed between PKC activity and abnormalities of development before the age of 36 months, though it did not reach significance (p =0.09, n=3) (Fig. 3b). On the other hand, there was no correlation between reduced PKC activity and impairments in reciprocal social interaction in regressive or non-regressive autistic subjects (data not shown). Abnormalities in communication had two types of scores: verbal and non-verbal. Only two scores were available in verbal category, which were not sufficient for analysis. Therefore, we analyzed the PKC data in the frontal cortex with respect to non-verbal score, and did not find any significant correlation between these parameters in regressive or non-regressive autism (data not shown).
Relationship between PKC activity of frontal cortex and Autism Diagnostic Interview Revised (ADI-R) test scores in subjects with autism. PKC activity was plotted against individual ADI-R scores for (a) restricted, repetitive and stereotyped patterns of behavior, and (b) abnormalities of development evident before the age of 36 months. R represents subjects with regressive autism.
DISCUSSION
Autism is a multifactorial disorder with variability in many domains. The variability of domains includes high or low functioning autism, regressive or non regressive autism, and comorbidities such as epilepsy. No single factor can explain variability observed with different domains of autism. Our results are suggestive of reduced PKC activity in the frontal cortex as one of the factors contributing to regression in autism. Recently, we reported that PKA is also affected in the frontal cortex of subjects with regressive autism [33]. Collectively, our present results and previous report suggest that regression in autism may be the result of alterations in protein kinases-mediated signal transduction. It is possible that both of these kinases (PKC and PKA) are affected by a common pathway because both protein kinases are activated by G-protein-coupled receptors. PKA gets activated by G-protein-coupled adenyl cyclase that converts ATP to c-AMP, an activator of PKA. On the other hand, activation of PKC is associated with G-protein-coupled phospholipase C-mediated cleavage of phosphoinositides into two intracellular messengers, i.e., diacylglycerol (DG) (activator of PKC) and inositol trisphosphate (IP3) (a Ca2+ mobilizer).
In the brain, the signals that control cognition vary depending on type of G-protein-coupled signal input. Several receptors such as glutamatergic receptors [37], cholinergic receptors [38], serotonergic receptors [39] and dopaminergic receptors [40] regulate the functions of PKC. PKC is a key regulator of neuronal signal transduction pathways that are crucial to learning and memory consolidation [41-45]. Neuronal plasticity and synaptic connections are important for information processing in the brain. Activation of PKC facilitates synaptic plasticity that includes responses such as Ca2+ influx, neurotransmitters release, and a decrease in Ca2+-activated K current in the brain, leading to the enhancement of neuronal excitability and potentiation of synaptic response [46; 47]. Li et al. [48] also reported effect of chronic treatment with staurosporine (PKC-inhibitor) on acquisition and expression of contextual fear conditioning in rats. Considering the importance of PKC in neuronal functions, decreased PKC activity in subjects with regressive autism may result in decreased neuronal plasticity, thus affecting neuronal excitability and synaptic response.
The formation of functional neuronal synapse requires various molecular players in presynaptic and postsynaptic growth. Dysfunction of proteins such as neuroligins, neurexin and SHANK that are required for synaptic development have been reported in ASDs [49; 50]. Neural disconnection leading to abnormalities in cortical networks has been suggested in autism. Different isozymes of PKC are known to have important roles at various stages of brain development. Purkayastha et al. [51] reported that serotonin 1A receptor-mediated signaling during neonatal hippocampal development initially requires PKCε to boost neuronal proliferation, and then uses PKCα to promote synaptogenesis.
Our results also suggest a relationship of reduced PKC activity in the frontal cortex with some behavioral abnormalities in autism. According to ADI-R diagnostic algorithm criteria, higher score reflects greater behavioral impairment. A negative significant relationship was observed between PKC activity in the frontal cortex and restrictive, repetitive and stereotyped behavioral score in autistic subjects (r= -0.84). A negative correlation was also observed between PKC activity in the frontal cortex and abnormalities of development before the age of 36 months in regressive autistic subjects (r= -0.988). However, later correlation did not reach significance, which may be due to small sample size in this study. The correlation between reduced PKC activity in the frontal cortex and behavioral abnormalities in autism needs further validation with larger sample size.
The prefrontal cortex has been implicated in autism to explain deficits in brain functions related to cognition, language, sociability and emotion [52]. Our findings of decreased activities of PKC and PKA in the frontal cortex of subjects with regressive autism suggest defective phosphorylation/dephosphorylation of proteins. Both PKC and PKA are involved in neuronal signal transduction. Chronic treatment with carbamazepine (a mood-stabilizer drug) has been reported to increase phosphorylation of myristoylated alanine-rich C kinase substrate (MARKS) in the rat cerebral cortex, suggesting involvement of PKC -mediated phosphorylation of MARKS in behavioral changes [53]. A recent study showed that PKA inhibitor could induce behavioral and neurological antidepressant-like effects in rats [54]. Since both PKC and PKA are activated by G-proteins-coupled receptors and are extensively involved in brain functions, we suggest that inhibition of these kinases in the cerebral cortex may have significant role in regressive autism.
Autism belongs to a group of neuropsychiatric disorders. The roles of PKC and PKA have also been suggested in other neuropsychiatric disorders. Decreased protein expression of PKCβ1, PKCξ and PKA regulatory Iα subunit and PKA catalytic subunits (α and β) has been reported in the postmortem brain samples from major depressive subjects as compared to controls [55]. Alterations in PKC activity were reported in manic depression, and antimanic agents (lithium carbonate and sodium valproate) inhibited PKC-associated signaling in brain tissue [56]. In other studies, prenatal and postnatal exposures to valproic acid (antiepileptic drug) have been used for animal model of autism to induce behavioral and neuropathological abnormalities similar to those observed in individuals with autism [57-61]. In pediatric bipolar disorder, decreased expression of specific PKC isozymes and decreased PKC activity in the platelets were reported [62]. PKC has also been suggested as a molecular target in pathogenic and therapeutic mechanisms of mood disorders in which electroconvulsive seizure (ECS) is effective [63]. This group reported phosphorylation of PKC substrates, including GAP-43, myristoylated alanine-rich C-kinase substrate, and neurogranin in the brain of rats after ECS. Another study showed significant decrease in the activities of phospholipase C and PKC in the membrane and cytosolic fractions of platelets from patients with bipolar disorder, suggesting that PKC may be involved in the pathophysiology of bipolar disorder [64].
The involvement of PKC has also been reported in other conditions such as inflammation [65], immune disorders [66], and oxidative stress [67]. These studies have suggested inhibitors of PKC theta as anti-inflammatory therapeutic agents [65], and PKC isozymes as potential therapeutic targets in immune disorders [66]. Abnormalities in inflammation, immune system and oxidative stress have been observed in autism [7]. Several lines of evidence from our and other groups have shown increased oxidative stress damage coupled with reduced antioxidant defense in blood [3; 6; 11; 14], brains [8-10; 13] and urine [12] of subjects with autism. We and others have also reported increased inflammatory markers in autism [17-21]. Therefore, PKC may also have a role in inflammation, immune defects and oxidative stress observed in autistic individuals.
Our results suggest that PMI and age cannot account for the observed alteration in PKC activity in subjects with regressive autism. Other factors, such as comorbidity with seizure disorder, reported for three of 10 autism cases (of which two had regressive autism, and one had non-regressive autism), and medications, reported for two regressive autism cases, four non-regressive autism cases, and two control cases, do not seem to be contributing factors to the altered activity of PKC in regressive autism. Furthermore, PKC activity was affected only in the frontal cortex but not in other brain regions of subjects with regressive autism. However, further studies with a larger autistic group should be done to explore this issue.
Considering the central role played by PKC in cellular signaling, the present findings on reduced PKC activity in subjects with regressive autism may result in disruption of neuronal signal transduction pathways in the frontal cortex, which may, in part, be responsible for regression in autism. It will be interesting to conduct a detailed study on the relationship between PKC activity and behavioral abnormalities with larger number of samples from subjects with autism. In conclusion, our study suggests that brain region-specific reduced PKC activity in the frontal cortex of individuals may be associated with regressive autism.
Acknowledgements
This work was supported by funds from Department of Defense Autism Spectrum Disorders Research Program AS073224P2, Autism Research Institute, Autism Collaboration (autism.org), and NYS Office of People with Developmental Disabilities. Human brain tissues were obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, MD.
COMPETING INTERESTS
The authors have declared that no competing interest exists.
References
1. Lord C, Cook EH, Leventhal BL. et al. Autism spectrum disorders. Neuron. 2000;28:355-363
2. Autism and Developmental Disabilities Monitoring Network Surveillance Year 2008 Principal Investigators. Prevalence of autism spectrum disorders--Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008. MMWR Surveill Summ. 2012;61:1-19
3. Chauhan A, Chauhan V. Oxidative stress in autism. Pathophysiology. 2006;13:171-181
4. Deth R, Muratore C, Benzecry J. et al. How environmental and genetic factors combine to cause autism: A redox/methylation hypothesis. Neurotoxicology. 2008;29:190-201
5. Herbert MR. Contributions of the environment and environmentally vulnerable physiology to autism spectrum disorders. Curr Opin Neurol. 2010;23:103-110
6. Chauhan A, Chauhan V, Brown WT, Cohen I. Oxidative stress in autism: increased lipid peroxidation and reduced serum levels of ceruloplasmin and transferrin--the antioxidant proteins. Life Sci. 2004;75:2539-2549
7. Chauhan A, Chauhan V, Brown WT. Autism: Oxidative stress, inflammation and immune abnormalities. Florida: CRC Press, Taylor and Francis Group. 2009
8. Chauhan A, Gu F, Essa MM, Wegiel J, Kaur K, Brown WT, Chauhan V. Brain region-specific deficit in mitochondrial electron transport chain complexes in children with autism. J Neurochem. 2011;117:209-220
9. Chauhan A, Audhya T, Chauhan V. Brain region-specific glutathione redox imbalance in autism. Neurochem Res. 2012;37:1681-1689
10. Fatemi SH, Aldinger KA, Ashwood P. et al. Consensus Paper: Pathological role of the cerebellum in autism. Cerebellum. 2012;11:777-807
11. James SJ, Melnyk S, Jernigan S. et al. Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:947-956
12. Ming X, Stein TP, Brimacombe M. et al. Increased excretion of a lipid peroxidation biomarker in autism. Prostaglandins Leukot Essent Fatty Acids. 2005;73:379-384
13. Sajdel-Sulkowska EM, Xu M, Koibuchi N. Increase in cerebellar neurotrophin-3 and oxidative stress markers in autism. Cerebellum. 2009;8:366-372
14. McGinnis WR. Oxidative stress in autism. Altern Ther Health Med. 2004;10:22-36
15. Gargus JJ, Imtiaz F. Mitochondrial energy-deficient endophenotype in autism. Am J Biochem Biotech. 2008;4:198-207
16. Rossignol DA, Frye RE. Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry. 2012;17:290-314
17. Ashwood P, Krakowiak P, Hertz-Picciotto I. et al. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun. 2011;25:40-45
18. Li X, Chauhan A, Sheikh AM. et al. Elevated immune response in the brain of autistic patients. J. Neuroimmunol. 2009;207:111-116
19. Pardo CA, Vargas DL, Zimmerman AW. Immunity, neuroglia and neuroinflammation in autism. Int Rev Psychiatry. 2005;17:485-495
20. Vargas DL, Nascimbene C, Krishnan C. et al. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005;57:67-81
21. Zimmerman AW, Jyonouchi H, Comi AM. et al. Cerebrospinal fluid and serum markers of inflammation in autism. Pediatr Neurol. 2005;33:195-201
22. Ozonoff S, Williams BJ, Landa R. Parental report of the early development of children with regressive autism: the delays-plus-regression phenotype. Autism. 2005;9:461-486
23. Goldberg WA, Osann K, Filipek PA. et al. Language and other regression: assessment and timing. J. Autism Dev Disord. 2005;33:607-616
24. Hansen RL, Ozonoff S, Krakowiak P. et al. Regression in autism: prevalence and associated factors in the CHARGE Study. Ambul Pediatr. 2008;8:25-31
25. Lord C, Shulman C, DiLavore P. Regression and word loss in autistic spectrum disorders. J Child Psychol Psychiatry. 2004;45:936-955
26. Stefanatos GA. Regression in autistic spectrum disorders. Neuropsychol Rev. 2008;18:305-319
27. Stefanatos GA, Grover W, Geller E. Case study: corticosteroid treatment of language regression in pervasive developmental disorder. J Am Acad Child Adolesc Psychiatry. 1995;34:1107-1111
28. Luyster R, Richler J, Risi S. et al. Early regression in social communication in autism spectrum disorders: a CPEA Study. Dev Neuropsychol. 2005;27:311-336
29. Alcazar A, Fando JL, Azuara C. et al. Protein kinase activities associated with ribosomes of developing rat brain. Identification of eukaryotic initiation factor 2 kinases. Int J Dev Neurosci. 1986;4:525-535
30. Hamada H, Zhang YL, Kawai A. et al. Development-associated myristoylated alanine-rich C kinase substrate phosphorylation in rat brain. Childs Nerv Syst. 2003;19:152-158
31. Turner RS, Raynor RL, Mazzei GJ. et al. Developmental studies of phospholipid-sensitive Ca2+-dependent protein kinase and its substrates and of phosphoprotein phosphatases in rat brain. Proc Natl Acad Sci U.S.A. 1984;81:3143-3147
32. Shobe J. The role of PKA, CaMKII, and PKC in avoidance conditioning: permissive or instructive?. Neurobiol Learn Mem. 2002;77:291-312
33. Ji L, Chauhan V, Flory MJ, Chauhan A. Brain region-specific decrease in the activity and expression of protein kinase A in the frontal cortex of regressive autism. PLoS One. 2011;6:e23751
34. Sun MK, Alkon DL. Pharmacology of protein kinase C activators: cognition-enhancing and antidementic therapeutics. Pharmacol Ther. 2010;127:66-77
35. Lintas C, Sacco R, Garbett K. et al. Involvement of the PRKCB1 gene in autistic disorder: significant genetic association and reduced neocortical gene expression. Mol Psychiatry. 2009;14:705-718
36. Philippi A, Roschmann E, Tores F. et al. Haplotypes in the gene encoding protein kinase c-beta (PRKCB1) on chromosome 16 are associated with autism. Mol Psychiatry. 2005;10:950-960
37. Hasham MI, Pelech SL, Krieger C. Glutamate-mediated activation of protein kinase C in hippocampal neurons. Neurosci Lett. 1997;228:115-118
38. Chen Y, Cantrell AR, Messing RO. et al. Specific modulation of Na+ channels in hippocampal neurons by protein kinase C epsilon. J Neurosci. 2005;25:507-513
39. Carr DB, Cooper DC, Ulrich SL. et al. Serotonin receptor activation inhibits sodium current and dendritic excitability in prefrontal cortex via a protein kinase C-dependent mechanism. J Neurosci. 2002;22:6846-6855
40. Maurice N, Tkatch T, Meisler M. et al. D1/D5 dopamine receptor activation differentially modulates rapidly inactivating and persistent sodium currents in prefrontal cortex pyramidal neurons. J Neurosci. 2001;21:2268-2277
41. Alkon DL, Sun MK, Nelson TJ. PKC signaling deficits: a mechanistic hypothesis for the origins of Alzheimer's disease. Trends Pharmacol Sci. 2007;28:51-60
42. Amadio M, Battaini F, Pascale A. The different facets of protein kinases C: old and new players in neuronal signal transduction pathways. Pharmacol Res. 2006;54:317-325
43. Bonini JS, Da Silva WC, Bevilaqua LR. et al. On the participation of hippocampal PKC in acquisition, consolidation and reconsolidation of spatial memory. Neuroscience. 2007;147:37-45
44. Lorenzetti FD, Baxter DA, Byrne JH. Molecular mechanisms underlying a cellular analog of operant reward learning. Neuron. 2008;59:815-828
45. Nelson TJ, Sun MK, Hongpaisan J, Alkon DL. Insulin, PKC signaling pathways and synaptic remodeling during memory storage and neuronal repair. Eur J Pharmacol. 2008;585:76-87
46. Cohen-Matsliah SI, Brosh I, Rosenblum K, Barkai E. A novel role for extracellular signal-regulated kinase in maintaining long-term memory-relevant excitability changes. J Neurosci. 2007;27:12584-12589
47. Zhang GR, Wang X, Kong L. et al. Genetic enhancement of visual learning by activation of protein kinase C pathways in small groups of rat cortical neurons. J Neurosci. 2005;25:8468-8481
48. Li XB, Inoue T, Koyama T. Effect of chronic treatment with the protein kinase C inhibitor staurosporine on the acquisition and expression of contextual fear conditioning. Eur J Pharmacol. 2002;441:151-155
49. Chauhan V, Chauhan A. Abnormalities in membrane lipids, membrane-associated proteins, and signal transduction in autism. Chauhan A, Chauhan V. and Brown W.T. eds. Autism: Oxidative stress, inflammation and immune abnormalities, CRC Press, Taylor and Francis group, Florida. 2009:177-206
50. Mitchell KJ. The genetics of neurodevelopmental disease. Curr Opin Neurobiol. 2011;21:197-203
51. Purkayastha S, Fernando SS, Diallo S. et al. Regulation of protein kinase C isozymes during early postnatal hippocampal development. Brain Res. 2009;1288:29-41
52. Sui L, Chen M. Prenatal exposure to valproic acid enhances synaptic plasticity in the medial prefrontal cortex and fear memories. Brain Res Bull. 2012;87:556-563
53. Hasegawa H, Osada K, Misonoo A. et al. Chronic carbamazepine treatment increases myristoylated alanine-rich C kinase substrate phosphorylation in the rat cerebral cortex via down-regulation of calcineurin A alpha. Brain Res. 2003;994:19-26
54. Liebenberg N, Müller HK, Fischer CW. et al. An inhibitor of cAMP-dependent protein kinase induces behavioural and neurological antidepressant-like effects in rats. Neurosci Lett. 2011;498:158-161
55. Shelton RC, Hal MD, Lewis DA. Protein kinases A and C in post-mortem prefrontal cortex from persons with major depression and normal controls. Int. J Neuropsychopharmacol. 2009;12:1223-1232
56. Yildiz A, Guleryuz S, Ankerst DP. et al. Protein kinase C inhibition in the treatment of mania: a double-blind, placebo-controlled trial of tamoxifen. Arch Gen Psychiatry. 2008;65:255-263
57. Kuwagata M, Ogawa T, Shioda S, Nagata T. Observation of fetal brain in a rat valproate-induced autism model: a developmental neurotoxicity study. Int J Dev Neurosci. 2009;27:399-405
58. Rinaldi T, Perrodin C, Markram H. Hyper-connectivity and hyper-plasticity in the medial prefrontal cortex in the valproic acid animal model of autism. Front Neural Circuits. 2008;2:4
59. Tashiro Y, Oyabu A, Imura Y. et al. Morphological abnormalities of embryonic cranial nerves after in utero exposure to valproic acid: implications for the pathogenesis of autism with multiple developmental anomalies. Int J Dev Neurosci. 2011;29:359-364
60. Wagner GC, Reuhl KR, Cheh M. et al. A new neurobehavioral model of autism in mice: pre- and postnatal exposure to sodium valproate. J Autism Dev Disord. 2006;36:779-793
61. Yochum CL, Dowling P, Reuhl KR. et al. VPA-induced apoptosis and behavioral deficits in neonatal mice. Brain Res. 2008;1203:126-132
62. Pandey GN, Ren X, Dwivedi Y, Pavuluri MN. Decreased protein kinase C (PKC) in platelets of pediatric bipolar patients: effect of treatment with mood stabilizing drugs. J Psychiatr Res. 2008;42:106-116
63. Kim SH, Kim MK, Yu HS. et al. Electroconvulsive seizure increases phosphorylation of PKC substrates, including GAP-43, MARCKS, and neurogranin, in rat brain. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:115-121
64. Pandey GN, Dwivedi Y, SridharaRao J. et al. Protein kinase C and phospholipase C activity and expression of their specific isozymes is decreased and expression of MARCKS is increased in platelets of bipolar but not in unipolar patients. Neuropsychopharmacology. 2002;26:216-228
65. Boschelli DH. Small molecule inhibitors of PKCTheta as potential antiinflammatory therapeutics. Curr Top Med Chem. 2009;9:640-654
66. Lee MR, Duan W, Tan SL. Protein kinase C isozymes as potential therapeutic targets in immune disorders. Expert Opin Ther Targets. 2008;12:535-552
67. Gopalakrishna R, Jaken S. Protein kinase C signaling and oxidative stress. Free Radic Biol Med. 2000;28:1349-1361
Author contact
![]() Corresponding author: Ved Chauhan, NYS Institute for Basic Research in Developmental Disabilities, 1050 Forest Hill Road, Staten Island, New York 10314, USA. Email: ved.chauhanny.gov. Tel: 1-718-494-5257; Fax: 1-718-698-7916
Corresponding author: Ved Chauhan, NYS Institute for Basic Research in Developmental Disabilities, 1050 Forest Hill Road, Staten Island, New York 10314, USA. Email: ved.chauhanny.gov. Tel: 1-718-494-5257; Fax: 1-718-698-7916
Received 2012-6-15
Accepted 2012-8-19
Published 2012-8-30