ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2013; 9(1):94-107. doi:10.7150/ijbs.5246 This issue Cite
Research Paper
AGE-LDL Activates Toll Like Receptor 4 Pathway and Promotes Inflammatory Cytokines Production in Renal Tubular Epithelial Cells
Ao Cheng*, Yuanyuan Dong*, Fengxin Zhu, Youhua Liu, Fan Fan Hou ![]() , Jing Nie
, Jing Nie ![]()
Division of Nephrology, Nanfang Hospital, Southern Medical University, Research Institute of Nephrology Guangdong Province, Key Lab for Organ Failure Research, Ministry of Education, Guangzhou, P.R. China.
* These authors contribute equally to this work.
Abstract
Background/Aims: Accumulation of advanced glycation end-products, the well-recognized pro-inflammatory molecules, has been detected in renal tissues including tubules. The aim of the present study was to investigate the role of advanced glycation end-products modified low density lipoprotein (AGE-LDL) in inflammatory cytokines production in human proximal tubular epithelial cells and the underlying mechanism. Methods: The Interleukin-6 (IL-6) and Interleukin-8 (IL-8) production was examined by real-time PCR and ELISA. The expression of Toll-like receptor 2 and 4 (TLR2/4) was detected by flow cytometry and western blot. The interaction of TLR2/4 with AGE-LDL was examined by co-immunoprecipitation assay. The involvement of MyD88 and the downstream molecules in inflammatory cytokines production was examined by siRNA and pharmacologic inhibitors, respectively. Results: AGE-LDL interacted with TLR2 and TLR4. TLR4 siRNA showed stronger inhibition on AGE-LDL-induced IL-6 and IL-8 production than that of TLR2 siRNA. Silencing MyD88, but not TRIF, inhibited AGE-LDL-induced IL-6 and IL-8 production. AGE-LDL stimulation led to phosphorylation of JNK, p38, Akt and the p65 subunit of nuclear factor-κB (NF-κB). Pharmacologic inhibitor of Akt suppressed AGE-LDL-induced activation of NF-κB, but the inhibitor of JNK, p38 or ERK1/2 had no effect. Blocking MyD88, p38, JNK, Akt or NF-κB attenuated AGE-LDL-triggered IL-6 production. Conclusion: AGE-LDL induced IL-6 and IL-8 production via TLR2/4-MyD88-dependent pathway in tubular epithelial cells. These data suggest that activation of TLRs signaling in tubular epithelial cells by AGE-LDL might be a novel mechanism for the tubulointerstitial inflammation.
Keywords: AGE-LDL, Toll like receptor, MyD88, NF-κB, IL-6.
Introduction
Chronic tubulointerstitial inflammation and subsequent fibrosis is the final common pathway for all kidney diseases leading to chronic renal failure. Many factors contribute to chronic tubulointerstitial inflammation such as proteinuria, hypoxia, oxidant stress and hyperglycemia [1]. Increased formation and accumulation of advanced glycation end-products (AGEs) is common in patients with chronic kidney disease (CKD)[2]. AGEs are a heterogeneous group of proteins and lipids formed by the Malliard reaction occurring in plasma and tissues and present in all renal compartments including tubules and interstitium in diabetic- and non-diabetic CKD [3]. Previous studies have shown that AGE modified albumin initiates inflammatory responses in renal tubular epithelial cells, mainly through interacting with receptor for AGEs (RAGE) [4]. Noteworthy, AGE modification occurs not only in albumin, but also in lipoproteins, lipids and nucleic acids. Accumulation of AGE modified LDL (AGE-LDL) has been detected in renal tubulointerstitium [5]. Although AGE-LDL is known to induce some cellular responses such as induction of foam cell formation [6], it remains unknown whether AGE-LDL plays a role in tubulointerstitial inflammation, and if it is the case, what is the underlying mechanism.
Toll-like receptors (TLRs) are evolutionarily conserved family of pattern recognition receptors that play a critical role in regulating both innate and adaptive immune system [7]. It is becoming increasingly evident that, endogenous ligands released from damaged/stressed tissues can also signal through TLRs to generate inflammation. These ligands include heat-shock proteins, the nonhistone chromatin-binding protein high-mobility group box1 (HMGB1), and extracellular matrix components such as hyaluronan and fibronectin [8]. Recently, TLR4 and/or TLR2 have been implicated in promoting renal injury in renal ischemia-reperfusion injury, acute kidney injury, acute allograft rejection and antibody-mediated glomerulonephritis. The expression of TLR2 has been detected in rat diabetic nephropathy [9]. Knockout TLR2 or TLR4 attenuated tubulointerstitial inflammation and improved renal dysfunction in diabetic nephropathy [10, 11]. It has also been demonstrated that, in type 1 and type 2 diabetic patients, increased TLR2 and TLR4 expression in monocytes is positively correlated with hemoglobin A1c levels and homeostasis model assessment insulin resistance [12]. However, the ligands of TLR2/4 that initiate innate immune response in CKD need further investigation. The present study was set out to test the hypothesis that AGE-LDL promotes local inflammatory response via activating TLRs signaling in tubular epithelial cells.
Materials and Methods
AGE-LDL preparation
LDL was obtained from Yuanyuan Biotech Inc (Guangzhou, China). AGE-LDL was made according to established protocols [13]. In brief, AGE-LDL was prepared by incubation of LDL (2mg protein/ml) with D (+) glucose (0.2M final concentration) at 37ºC under sterile conditions with antioxidants (1mg/ml EDTA and 10μM BHT: 3,5-Di-tert-4-butylhydroxytoluene) for 4 weeks. After preparation, AGE-LDL was treated with DNAse/RNAse and then extensively dialyzed against PBS, pH7.4. Endotoxin levels in the preparation were determined using the amebocyte lysate assay kit (Sigma, St. Louis, MO, USA) and were found to be below 0.025EU/ml.
The free, non-glycated amino groups were determined using the trinitrobenzene sulphonic acid assay (TNBSA) [14]. The extent of LDL protein glycation was evaluated by measuring pentosidine formation spectrofluorometrically (excitation 335 nm, emission 385 nm) [15]. The lipid peroxide level was determined by using thiobarbituric acid reactive substances (TBARS) assay[16]. The electric charge of AGE-LDL and nLDL was determined by agarose gel electrophoresis (0.6%).
AGE-HSA preparation
AGE-HSA was prepared according to established protocols [17]. AGE-HSA contains 109.8U AGE/mg protein by measuring fluorescent AGEs using fluorospectrometry [17], while native albumin contains 1.9 U AGE/mg protein.
Cell Culture
The immortalized HK-2 (American Type Culture Center, Manassas, VA, USA) was cultured at 37°C in 5% CO2 in DMEM/Ham's F12 (GibcoBRL, Grand Island, NY, USA) with 10% fetal bovine serum. After serum-starved in serum-free medium for 24h, HK-2 cells incubated with 10μg/ml LPS (E. coli 055: B5, Sigma, St. Louis, MO, USA) or various concentrations of AGE-LDL for indicated time period. TLR4 neutralization antibody (HTA125, eBioscience, San Diego, CA, USA), TLR2 neutralization antibody (Abcam, Cambridge, MA, USA), MAPK inhibitors (SP600125/SB203508/UO126, Calbiochem, San Diego, CA, USA), Akt inhibitor LY294002 (Cell Signaling Technology Inc. Danvers, MA, USA) or NF-κB inhibitor Bay 11-7082 (Sigma, St. Louis, MO, USA) was added in the culture medium 2 h before AGE-LDL treatment.
Flow cytometry
After incubating with a monoclonal mouse anti-human TLR2 antibody or a polyclonal rabbit anti-human TLR4 antibody for 1 h at 4°C, HK-2 cells were then washed and incubated with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse secondary IgG (Santa Cruz Biotechnology Inc. CA, USA) or FITC-conjugated goat anti-rabbit secondary (Santa Cruz Biotechnology Inc. CA, USA) for 45 min and analyzed by flow cytometry using FACS Calibur and CellQuest software (BD FACS Calibur System, Franklin Lakes, NJ, USA).
Co-immunoprecipitation and western blot
Co-Immunoprecipitation was performed as previously reported [18]. In brief, after AGE-LDL treatment, HK-2 cell were lysed with lysis buffer (50mM Tris-HCl, pH 7.5, 150mMgt NaCl, 1% v/v Protease and Phosphatase Inhibitor cocktails). Cell lysates were preabsorbed with protein A/G agarose beads at 4°C for 1 h. After centrifuging, the supernatants were incubated with polyclonal AGE antibody (BD Biosciences, San Jose, CA ,USA) overnight at 4°C followed by incubating with Protein A/G agarose beads (Santa Cruz Biotechnology Inc. CA, USA) for 2 h at 4°C.
For western blot, the immunocomplexes or cell lysates were resolved with SDS-PAGE and transferred to nitrocellulose membranes. Membranes were incubated with indicated primary antibody overnight at 4°C. After washing with TBST, membranes were incubated with horseradish peroxidase (HRP)-linked secondary antibodies at room temperature for 1 h. Images were developed using ECL and exposed on X-ray films. Band density was analyzed using NIH Image J software.
Immunofluorescence staining
Immunofluorescence staining was performed as previously reported [19]. Briefly, cells were cultured to 60-70% confluence. After washing with cold PBS, cells were treated with 100% methanol at -20°C for 10 min followed by 0.1% Triton-X treatment at room temperature for 15 min. After washing with PBS, cells were incubated with rabbit polyclonal antibody against NF-κB p65 (Cell Signaling Technology, USA) overnight at 4°C followed by incubating with Alex488-conjugated goat anti-rabbit IgG for 1 h. Nuclei were counter-stained with Hochest 33258 (Sigma, St. Louis, MO, USA). Images were taken by confocal microscopy (FluoView™ FV1000, Olympus, Tokyo, Japan).
ELISA
Concentrations of IL-6 and IL-8 in the supernatants were measured using mouse-specific ELISA kits (R&D, Minneapolis, MN, USA) according to the manufacturer's protocols. Results were normalized by cellular protein levels.
Real time PCR
HK-2 cell were homogenized with Trizol reagent (Invitrogen, Carlsbad, CA, USA), and total RNA was extracted according to the manufacturer's instructions. RNA concentration was calculated using a Nanodrop ND1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). Aliquots of each RNA extraction were reverse-transcribed simultaneously into cDNA using One-Step RT-PCR kit (TaKaRa, Tokyo, Japan) according to the manufacturer's protocol. Each quantitative real-time PCR was performed in a total volume of 25μl in duplicate by using the SYBR®Premix Ex Taq™ kit (TaKaRa, Kyoto, Japan) and the Fast Real-Time PCR system 7500 (Applied Biosystems Inc., Foster City, CA, USA). The following sets of primers were used:
IL-6: 5'-GTGGAGATTGTTGCCATCAACG-3' (sense); 5'-CAGTGGATGCAGGGATGATGTTCTG-3' (antisense);
IL-8: 5'-GACATACTCCAAACCTTTCCAC-3' (sense); 5'-AACTTCTCCACAACCCTCTGC-3' (antisense);
IFN-β: 5'-TTGTGCTTCTCCACTACAGC-3' (sense); 5'-CTGTAAGTCTGTTAATGAAG-3' (antisense);
GAPDH: 5'-GCACCGTCAAGGTGAGAAC-3' (sense); 5'-TGGTGAAGACGCCAGTGGA-3' (antisense).
Briefly, the thermal cycling conditions comprised a 30 seconds step at 95°C, followed by 95°C for 5 seconds and 60°C for 34 seconds for 40 cycles with melting curve analysis. Relative quantification of each gene was calculated after normalization to GAPDH mRNA by using the 2-∆∆CT method.
RNA interference
Small interfering RNAs used in this study were obtained from Shanghai GenePharma Co. LTd. (Shanghai, China). HK-2 cells were transfected with indicated siRNA oligonucleotides using LipofectamineTM 2000 transfection reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol.
TLR4-siRNA: 5'-CCAGGUGCAUUUAAAGAAATT-3';
TLR2-siRNA: 5'-GCACUUUAUAUUCACUUCATT-3';
MyD88-siRNA: 5'-CUGGAACAGACAAACUAUCTT-3';
TRIF-siRNA: 5'-GACCAGACGCCACUCCAACTT-3';
NF-κB p65-siRNA: 5'-CCUCCUUUCAGGAGAUGAATT-3';
Scramble siRNA: 5'-UUCUCCGAACGUGUCACGUTT-3'.
Statistical analysis
Data were presented as mean ± standard deviation (SD). Difference in mean values between groups was tested with the one-way ANOVA. To identify significant differences between the two groups, comparisons were made by independent sample t-test. p≤0.05 was considered statistically significant. Statistical analysis was performed with SPSS 13.0 for Windows.
Results
AGE-LDL interacted with TLR4 and TLR2 in human proximal tubular cells
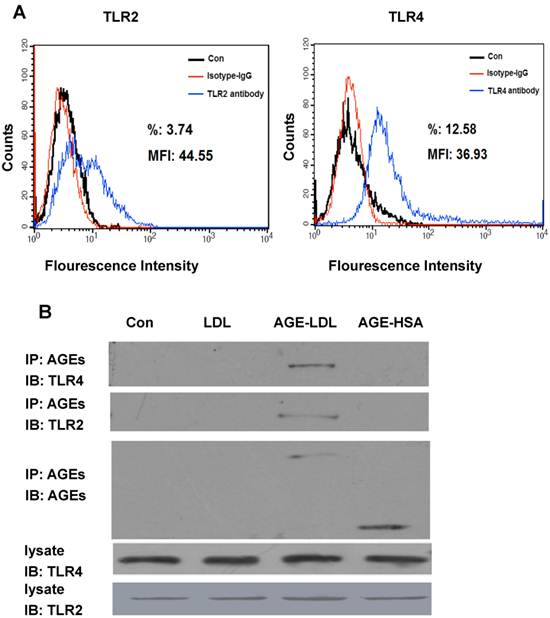
To investigate the expression of TLRs in human proximal tubular cells (HK-2), we first performed FACS analysis. As shown in Figure 1A, when HK-2 cells were incubated with anti-TLR2 antibody or anti-TLR4 antibody, 12.58% of HK-2 cells were detected as positive for TLR4 and 3.74% of cells were detected as positive for TLR2, suggesting TLR4 is expressed more frequently and with higher intensity than TLR2 on HK-2 cells. The expression of TLR4 and TLR2 in HK-2 cells was further confirmed by western blot (Figure 1B). To test whether AGE-LDL interacts with TLR4/2, HK-2 cells treated with AGE-LDL were lysed and cell lysates were incubated with anti-AGE antibody. The immunoprecipitates were then immunoblotted with anti-TLR4 or anti-TLR2, respectively. As shown in Figure 1B, precipitation of AGE resulted in the co-immunoprecipitation of TLR4 and TLR2, suggesting that AGE-LDL interacts with both TLR4 and TLR2 in HK-2 cells. Neither native LDL nor AGE modified human serum albumin (AGE-HSA) was co-immunoprecipated with anti-TLR4 antibody, indicating that interaction of AGE-LDL with TLRs is a specific property of AGE-LDL.
TLR2 and TLR4 were expressed on HK-2 cells and interacted with AGE-LDL. (A) Protein expression of TLR2 and TLR4 on the surface of HK-2 cells was detected by flow cytometry analysis. (B) Co-immunoprecipitation of AGE-LDL and TLR2/4 in HK-2 cells. HK-2 cells were treated with AGE-LDL, native LDL, or AGE-BSA, respectively, the interaction between AGE-LDL and TLR2 or TLR4 was analyzed by immunoprecipitation using anti-AGE antibody and detected by immunoblotting using anti-TLR2 or anti-TLR4 antibody, respectively. The protein level of TLR2 and TLR4 in HK-2 cell lysate was examined by western blot.
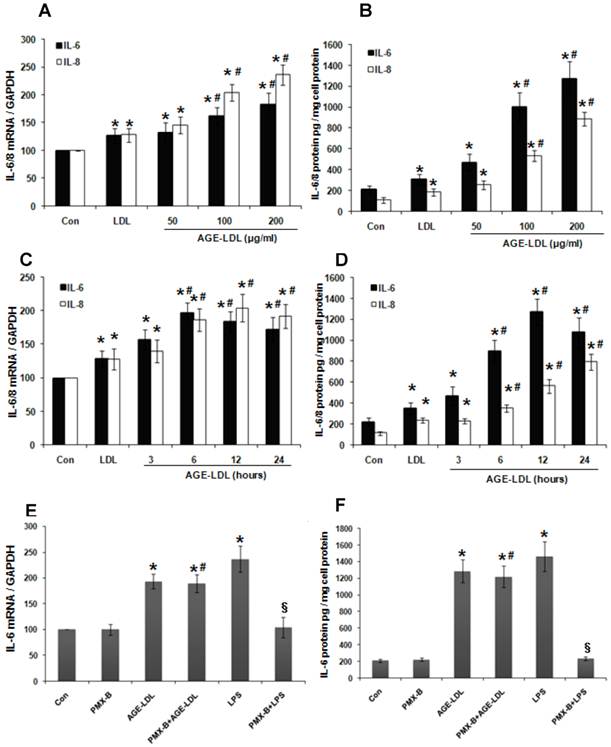
AGE-LDL-induced IL-6 expression was mainly mediated by TLR4
Since IL-6 production is tightly regulated by TLRs signaling [20], we next examined whether AGE-LDL stimulation lead to IL-6 production. As shown in Figure 2A-D, Real time PCR (Figure 2A, 2C) and ELISA (Figure 2B, 2D) demonstrated that, compared with native LDL, AGE-LDL significantly induced IL-6 production in HK-2 cells in a time- and dose-dependent manner. Likewise, AGE-LDL significantly increased the production of IL-8, another downstream target of TLRs signaling (Figure 2A-D). We further treated HK-2 cells with Polymyxin B (PMX-B), a specific inhibitor for lipopolysaccharide (LPS). As shown in Figure 2E-F, PMX-B dramatically inhibited LPS-induced IL-6 production at both mRNA and protein level, but had no obvious effect on AGE-LDL-induced IL-6 production, indicating that the stimulatory effect of AGE-LDL on IL-6 production was not caused by LPS contamination.
AGE-LDL induced IL-6 and IL-8 expression in a time- and dose-dependent manner. (A-B) HK-2 cells were treated with native LDL (100μg/ml) or indicated amount of AGE-LDL for 12 h. The mRNA level of IL-6 and IL-8 was examined by real time PCR (A) and the protein level of IL-6 and IL-8 was measured by ELISA (B). (C-D) HK-2 cells were treated with native LDL (100μg/ml) or AGE-LDL (100μg/ml) for indicated time period. The mRNA level of IL-6 and IL-8 was examined by real time PCR (C) and the protein level of IL-6 and IL-8 was measured by ELISA (D). (E-F) AGE-LDL-induced IL-6 production was not caused by LPS contamination. HK-2 cells were pretreated with PMX-B (10μg/ml) for 2 h before adding AGE-LDL or LPS. The mRNA level of IL-6 was examined by real time PCR (E) and the protein level of IL-6 was measured by ELISA (F). *P<0.05 versus untreated cells. #P>0.05 versus AGE-LDL treated cells. §P<0.05 versus LPS treated cells. Data are expressed as mean±SD of three independent experiments.
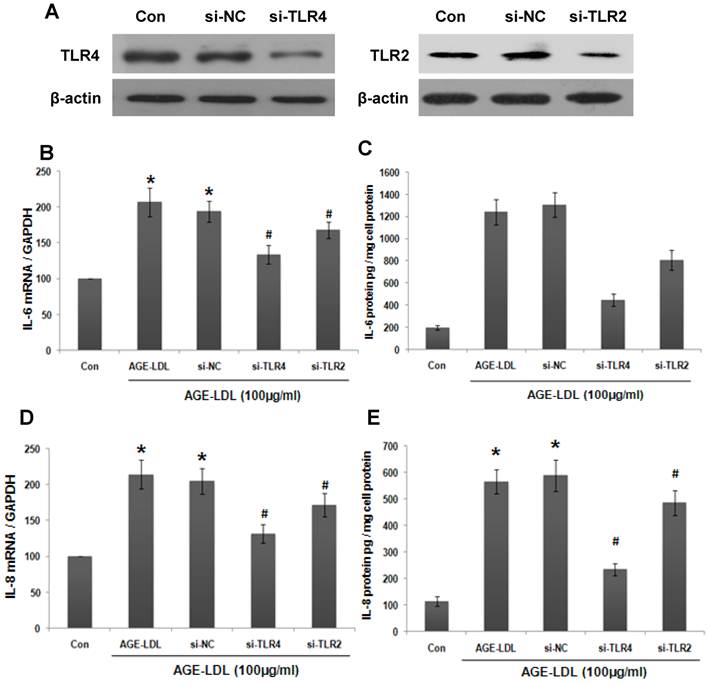
We next investigated whether the effect of AGE-LDL on IL-6 production was mediated by TLR2/4. To address this issue, we knocked down endogenous TLR4 or TLR2 in HK-2 cells using siRNA and the efficiency of TLR4/2 siRNA was examined by western blot (Figure 3A). The mRNA level of IL-6 was measured by real-time PCR. As shown in Figure 3B, TLR4 siRNA resulted in a more dramatic decrease of AGE-LDL-induced IL-6 production than that of TLR2 siRNA (68% and 33% decrease of the IL-6 expression by TLR4 and TLR2 siRNA, respectively). Consistent with this data, ELISA showed that knockdown TLR4 led to 65% decrease of IL-6 secretion, whereas, TLR2 siRNA caused 29% decrease of IL-6 secretion (Figure 3C). Altogether, these data indicated that AGE-LDL induced IL-6 expression was mainly mediated by TLR4 in tubular epithelial cells. Similarly, TLR4 and TLR2 siRNA lowered AGE-LDL induced-IL-8 production as that of IL-6 (Figure 3D-E).
AGE-LDL induced IL-6 and IL-8 production was mainly mediated by TLR4. (A) HK-2 cells were transfected with TLR2 siRNA, TLR4 siRNA or scramble siRNA, respectively. Western blot was performed to analyze the expression of endogenous TLR2/4 in HK-2 cells. GAPDH was used to verify equivalent loading. The effect of TLR2/4 siRNA on AGE-LDL-induced IL-6 and IL-8 expression was determined by real-time PCR (B, D) and ELISA (C, E). *P<0.05 versus untreated cells. #P<0.05 versus scramble siRNA transfected cells. Data are expressed as mean±SD of three independent experiments.
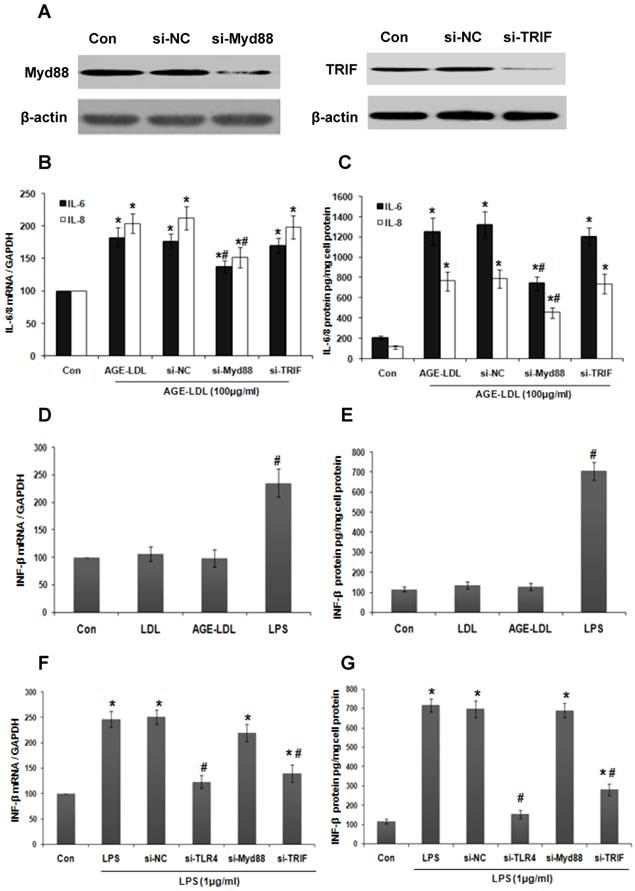
MyD88 mediated AGE-LDL-induced-IL-6 production
Since MyD88 and TRIF are adaptor proteins downstream of TLR4 involved in the stimulation of the inflammatory response [21], we next examined which one is responsible for AGE-LDL-induced IL-6 and IL-8 expression by knockdown endogenous MyD88 or TRIF using siRNA. The efficiency of siRNA was confirmed by western blot (Figure 4A). MyD88 siRNA effectively lowered the level of AGE-LDL-induced IL-6 and IL-8 mRNA (Figure 4B) and protein expression (Figure 4C). However, TRIF siRNA had no influence on IL-6/8 expression. To further confirm that AGE-LDL specifically activates MyD88 pathway but not TRIF pathway, we examined the effect of AGE-LDL on the expression of IFN-β, a downstream target of TRIF pathway. As expected, LPS significantly augmented IFN-β expression at both mRNA and protein level, but AGL-LDL had no obvious effect (Figure 4D-E). TLR4 and TRIF siRNA dramatically inhibited LPS-induced IFN-β production, but MyD88 siRNA had no effect (Figure 4F-G). Altogether, these results indicated that MyD88 is responsible for mediating AGE-LDL-induced IL-6/8 production in tubular epithelial cells.
AGE-LDL-induced phosphorylation of JNK, p38 and Akt was dependent on MyD88
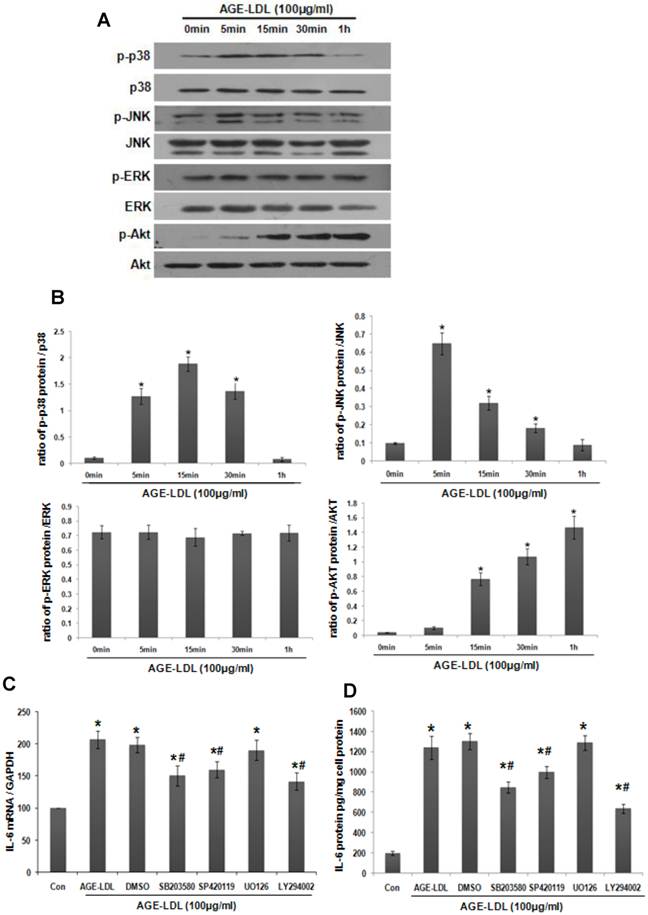
Activation of mitogen-activated protein kinases (MAPKs) or Akt is the downstream signaling of MyD88 [20]. As shown in Figure 5A-B, AGE-LDL stimulation resulted in rapid phosphorylation of Akt, p38 and JNK, but not ERK1/2. To explore their relative importance in AGE-LDL-augmented IL-6 production, specific pharmacologic inhibitors SP600125 (SP), UO126 (UO), SB203580 (SB) and LY294002 (LY) were used to inhibit JNK, ERK, p38 MAPK and Akt, respectively. As shown in Figure 5C-D, pretreatment with SB, SP or LY significantly suppressed AGE-LDL induced IL-6 expression and secretion, but UO had no obvious effect.
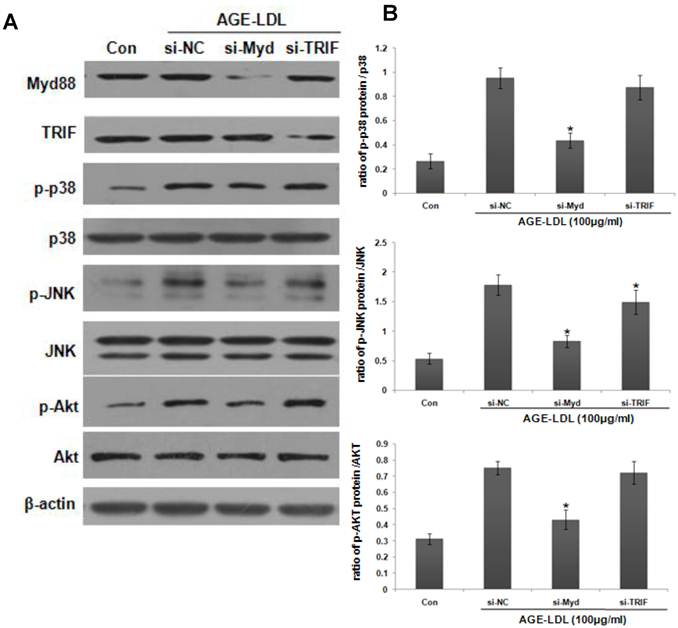
To examine the involvement of MyD88 in AGE-LDL-induced p38, JNK and Akt activation, we knocked down MyD88 using siRNA. As shown in Figure 6, MyD88 siRNA significantly suppressed AGE-LDL-induced phosphorylation of p38, JNK and Akt. On the other hand, TRIF siRNA had no obvious effect. Taken together, these results defined JNK, p38 and Akt, but not ERK, as the downstream mediators that propagate MyD88 activation into downstream pro-inflammatory responses induced by AGE-LDL in tubular epithelial cells.
AGE-LDL induced NF-kB activation via MyD88-Akt signaling
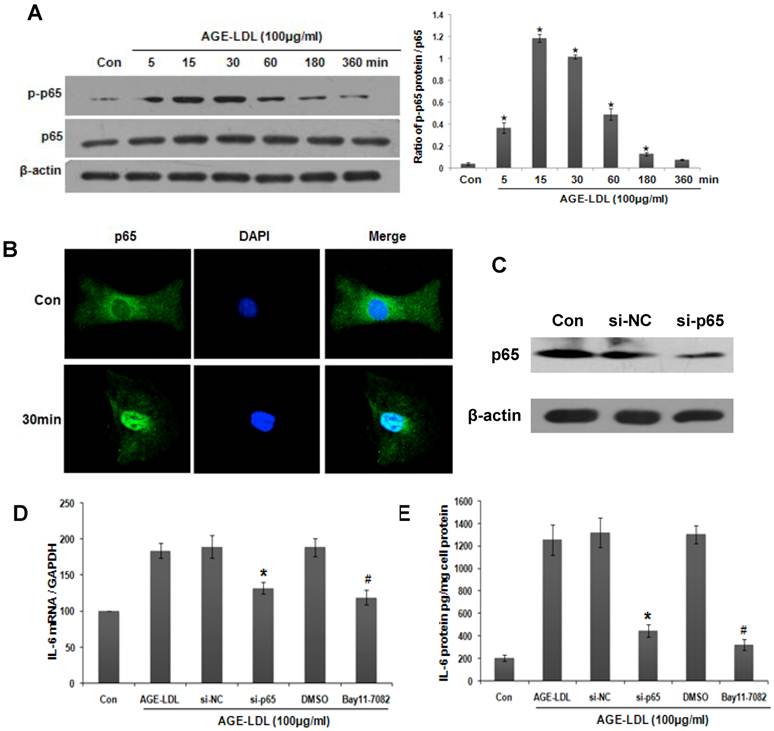
NF-κB activation is a signature downstream event for TLRs-ligand interaction [20]. We thus examined the effect of AGE-LDL on NF-κB activation. As shown in Figure 7A, phosphorylation of NF-κB subunit p65 was increased 5 min after AGE-LDL stimulation. Maximal phosphorylation occurred at 15 to 30 min after AGE-LDL stimulation. The translocation of NF-κB into nucleus after AGE-LDL treatment was detected by immunofluorescence staining (Figure 7B).
To explore the role of NF-κB activation in AGE-LDL induced IL-6 expression, we knocked down NF-κB subunit p65 using siRNA. The efficiency of p65 siRNA was examined by western blot (Figure 7C). Real time PCR and ELISA showed that IL-6 expression (Figure 7D) and secretion (Figure 7E) was decreased in p65 siRNA transfected cells compared with scramble siRNA transfected cells. Pre-incubating HK-2 cells with NF-κB inhibitor Bay11-7082 showed similar inhibitory effect on IL-6 production (Figure 7D-E). These data suggest that NF-κB activation is responsible for AGE-LDL-induced IL-6 production.
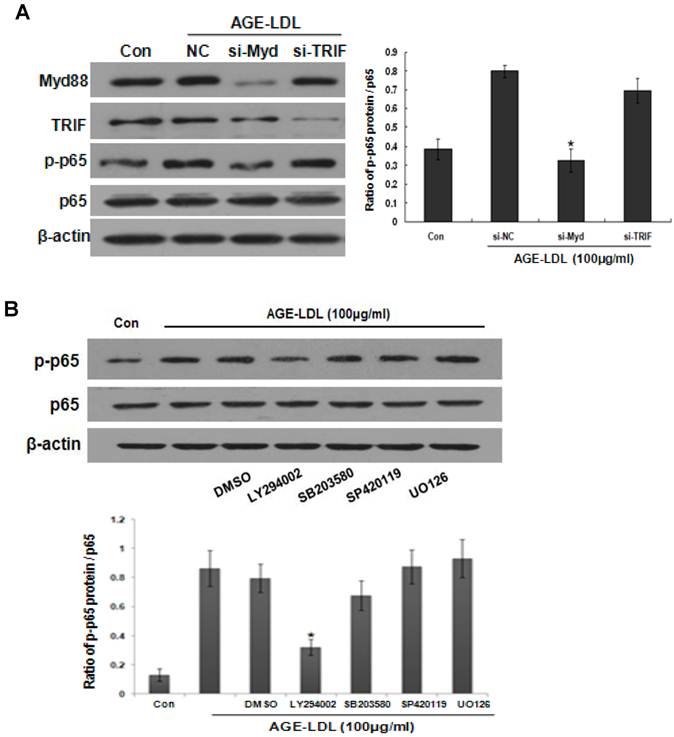
To decipher the signaling events involved in AGE-LDL induced-NF-κB activation, we first knocked down MyD88 or TRIF by siRNA. As shown in Figure 8A, MyD88 siRNA significantly abolished AGE-LDL-induced phosphorylation of p65, but TRIF siRNA had no obvious effect. We next pre-incubated HK-2 cells with SB, SP, UO or LY before AGE-LDL stimulation, respectively. As shown in Figure 8B, Akt inhibitor LY dramatically attenuated AGE-LDL-induced phosphorylation of p65, but inhibition of p38, JNK and ERK1/2 had no obvious effect, indicating that AGE-LDL-induced NF-κB activation is specifically mediated by Akt.
MyD88 mediated AGE-LDL induced IL-6 and IL-8 production. (A) HK-2 cells were transfected with MyD88 siRNA, TRIF siRNA or scramble siRNA, respectively. Western blot was performed to verify the efficacy of siRNA on knocking down the expression of endogenous MyD88 and TRIF. (B-C) HK-2 cells were transfected with MyD88 siRNA, TRIF siRNA or scramble siRNA, respectively. 24 h after transfection, HK-2 cells were treated with 100μg/ml AGE-LDL or native LDL. IL-6 and IL-8 expression was measured by real-time PCR (B) and ELISA (C). *P<0.05 versus untreated control cells. #P<0.05 versus scramble siRNA transfected cells. Data are expressed as mean±SD of three independent experiments. (D-E) HK-2 cells were treated with native LDL, AGE-LDL or LPS, respectively. The expression of IFN-β was examined by real time PCR (D) and ELISA (E). #P<0.05 versus untreated cells. Data are expressed as mean±SD of three independent experiments. (F-G) HK-2 cells were transfected with TLR4 siRNA, MyD88 siRNA, TRIF siRNA or scramble siRNA, respectively. 24 h after transfection, HK-2 cells were treated with 1μg/ml LPS. INF-β expression was measured by real-time PCR (F) and ELISA (G). *P<0.05 versus untreated control cells. #P<0.05 versus scramble siRNA transfected cells. Data are expressed as mean±SD of three independent experiments.
AGE-LDL-induced activation of JNK, p38 and Akt was responsible for IL-6 expression. (A) HK-2 cells were treated with AGE-LDL (100μg/ml) for indicated time period. The phosphorylation level and the total protein level of JNK, p38, ERK1/2 and Akt were examined by western blot. (B) Graphic representation of the ratio of phosphorylated JNK, p38, ERK1/2 and Akt to total JNK, p38, ERK1/2 or Akt. *P<0.05 versus untreated cells. Data are expressed as mean±SD of three independent experiments. (C-D) HK-2 cells were pretreated with vehicle (DMSO), 10μM of JNK inhibitor (SP600125), p38 inhibitor (SB203580), ERK1/2 inhibitor (UO126) or Akt inhibitor (LY294002) before AGE-LDL stimulation. The mRNA level of IL-6 was determined by real time PCR (C) and the protein level of IL-6 was measured by ELISA (D). *P<0.05 versus untreated control cells. #P<0.05 versus AGE-LDL treated vehicle control cells. Data are expressed as mean±SD of three independent experiments.
Activation of JNK, p38 and Akt was the downstream signaling event of MyD88. HK-2 cells were transfected with MyD88 siRNA, TRIF siRNA or scramble siRNA before AGE-LDL treatment. (A) The phosphorylation level and the total protein level of p38, JNK and Akt were examined by western blot analysis. (B) Graphic representation of the ratio of phosphorylated p38, JNK and Akt to total p38, JNK or Akt. *P<0.05 versus AGE-LDL stimulated scramble siRNA trasnfected cells. Data are expressed as mean±SD of three independent experiments.
Activation of NF-κB was responsible for AGE-LDL induced IL-6 expression. (A) HK-2 cells were treated with 100μg/ml AGE-LDL for indicated time period. The phosphorylation level and the total protein level of p65 were determined by western blot. *P<0.05 versus control cells. Data are expressed as mean±SD of three independent experiments. (B) The translocation of p65 into nuclei was examined by immunofluorescence staining. HK-2 cells were treated with 100μg/ml AGE-LDL for 30 min, and then stained with antibody against p65 (green). Nuclei were stained with DAPI (blue). Images were taken by confocal microscopy. Magnification 400x. (C) HK-2 cells were transfected with p65 siRNA and the protein level of endogenous p65 was determined by western blot. (D-E) HK-2 cells were either transfected with p65 siRNA or treated with NF-κB inhibitor (Bay11-7082, 10μM) before AGE-LDL stimulation. IL-6 production was measured by real time PCR (D) and ELISA (E). *P<0.05 versus scramble siRNA transfected cells. #P<0.05 versus DMSO vehicle control cells. Data are expressed as mean±SD of three independent experiments.
AGE-LDL induced NF-κB activation was mediated by MyD88-Akt pathway. (A) HK-2 cells were transfected with MyD88 or TRIF siRNA before AGE-LDL stimulation, the phosphorylation level and the total protein level of p65 were measured by western blot. *P<0.05 versus AGE-LDL stimulated scramble siRNA transfected cells. Data are expressed as mean±SD of three independent experiments. (B) HK-2 cells were pre-treated with vehicle (DMSO), 10μM of JNK inhibitor (SP600125), p38 inhibitor (SB203580), ERK1/2 inhibitor (UO126) or AKt inhibitor (LY294002) before AGE-LDL treatment. The phosphorylation level of p65 and the total protein level of p65 were measured by western blot. *P<0.05 versus AGE-LDL treated vehicle control cells. Data are expressed as mean±SD of three independent experiments.
Discussion
Accumulating evidence indicates that immunologic and inflammatory elements play a significant role in initiating and extending tubular injury in chronic kidney diseases, but the mechanisms are not fully understood. In the present study, we detected the expression of TLR2 and TLR4 on the surface of human proximal tubular cells which is consistent with previous reports [9], but the expression level of TLR4 is higher than that of TLR2. Since previous study has shown that AGE-LDL interacts with TLR4 in endothelial cells [18], we therefore examined the interaction of AGE-LDL with TLR2/4 in proximal tubular cells. Co-immunoprecipitation assay revealed that AGE-LDL interacted with both TLR4 and TLR2. However, silencing TLR4 resulted in a more dramatic decrease of IL-6/8 expression than that of TLR2 siRNA, suggesting that TLR4 plays a more critical role in AGE-LDL-induced inflammatory response than that of TLR2. In support of our data, previous study reported the increased expression of TLR4 in the renal tubules in human kidney with diabetic nephropathy, but not of TLR2 [12].
We next sought to identify the signaling components involved in AGE-LDL-induced IL-6 production in renal tubular epithelial cells. Since all the TLRs except TLR3 activate the MyD88 pathway [21] and TRIF is the MyD88-independent adaptor protein of TLR4 involved in LPS-induced inflammatory response [22], we first analyzed their roles in AGE-LDL-induced IL-6 production. Knocking down MyD88 dramatically inhibited AGE-LDL-induced IL-6 production, but TRIF siRNA had no effect, suggesting that, unlike LPS, AGE-LDL did not activate TRIF pathway. To further confirm that AGE-LDL indeed activated MyD88 pathway, we examined the expression of IL-8, another downstream target gene of MyD88. Real time PCR and ELISA assay revealed that AGE-LDL significantly upregulated IL-8 expression. Moreover, we showed that AGE-LDL did not affect IFN-β production, a proven downstream target of TRIF pathway [20].
We next investigated the effect of AGE-LDL on the activation of MyD88-dependent downstream signaling. LPS has been shown to activate p38, JNK and ERK1/2 all three MAPKs [23]. HMGB1, the endogenous ligand of TLR4, enhanced the phosphorylation of ERK1/2 and p38, but no effect on the phosphorylation of JNK [24]. In the present study, we showed that AGE-LDL selectively activated JNK and p38, but not ERK1/2.
NF-κB activation is a signature downstream event for TLRs-ligand interaction [20]. Studies with MyD88-null mice suggested that LPS activates a MyD88-independent pathway mediating NF-κB activation [25]. However, we observed that silencing MyD88 almost completely inhibited AGE-LDL-triggered NF-κB activation, but silencing TRIF had no effect. When we further dissected the downstream signaling of MyD88, we found that Akt plays a central role in AGE-LDL-induced NF-κB activation, whereas p38 and ERK1/2 apparently were not involved. Previous studies showed that p38 contributes to HMGB1-induced nuclear translocation of NF-κB [26]. It has also been shown that p38 participates in LPS-induced nuclear accumulation of NF-κB in neutrophils and is involved in modulating NF-κB-dependent transcription in macrophages [26]. Although the mechanism underlying this difference remains to be investigated, these findings suggest that various ligands of TLR4 may lead to the activation of different downstream pathways and cause diverse phathophysiological events. Supporting of this hypothesis, a previous study has shown that interaction of TLR4 with an early form of oxidized LDL induces polymerization and spreading of macrophages via activation of spleen tyrosine kinase [27]. Also, the interaction of TLR4 with oxidized LDL results in the activation of the NF-κB-ERK1/2 pathway and upregulated the expression of bone morphogenetic protein-2 [28].
AGEs modified proteins have been increasingly scrutinized as immunological provocateurs in CKD [29]. In vitro studies demonstrate that AGEs initiate a range of inflammatory responses, including chemotaxis and synthesis of pro-inflammatory cytokines [29]. In vivo, the relevance of AGEs in CKD is supported by the observations that AGEs formation occurs at an accelerated rate in CKD, and AGEs correlate with neopterin and TNF-α levels in patients with CKD [29]. Mounting evidence indicates that AGEs induce tissue damage by several mechanisms, including cross-linking of proteins, binding to the receptors for inducing oxidative stress, chemotaxis, synthesis of pro-inflammatory cytokines, and cellular dysfunction [2]. In renal tubular epithelial cells, AGE modified albumin initiates inflammatory responses mainly through interacting with scavenger receptors such as RAGE [4]. However, the receptor mediating the effect of AGE modified lipids on these cells remains largely unknown. The present study showed that AGE-LDL specifically interacted with TLR4 and TLR2 in human proximal tubular cells, indicating that AGE-LDL may initiate innate immune response via activation of TLRs signaling. Most recent study demonstrated that RAGE binds to TIRAP and MyD88 thus mediate the activation of TLRs signaling by AGE-BSA [30]. Taken together with our findings, these data indicate that AGE modified molecules could activate TLRs signaling through binding to either TLR receptors on the cell surface or the intracellular adaptor proteins of TLRs. AGEs accumulation is prevalent in various forms of CKD especially in diabetic nephropathy [2]. Therefore, activation of TLRs signaling by AGEs implies a novel mechanism for tubulointerstitial inflammation in diabetes associated CKD.
Acknowledgements
This study is supported by National 973 program (No. 2012CB517700 and 2011CB504005), National Nature and Science Grants (No. U0932002) to Dr. Fan Fan Hou. National Nature and Science Grants (No. 81288001 and 81070554) to Dr. Jing Nie.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Schlondorff DO. Overview of factors contributing to the pathophysiology of progressive renal disease. Kidney Int. 2008;74:860-866
2. Busch M, Franke S, Ruster C, Wolf G. Advanced glycation end-products and the kidney. Eur J Clin Invest. 2010;40:742-755
3. Miyata T, Iida Y, Horie K, Cai Z, Sugiyama S, Maeda K. Pathophysiology of advanced glycation end-products in renal failure. Nephrol Dial Transplant. 1996;11(Suppl 5):27-30
4. D'Agati V, Schmidt AM. RAGE and the pathogenesis of chronic kidney disease. Nat Rev Nephrol. 2010;6:352-360
5. Nosadini R, Tonolo G. Role of oxidized low density lipoproteins and free fatty acids in the pathogenesis of glomerulopathy and tubulointerstitial lesions in type 2 diabetes. Nutr Metab Cardiovasc Dis. 2011;21:79-85
6. Jinnouchi Y, Sano H, Nagai R, Hakamata H, Kodama T, Suzuki H, Yoshida M, Ueda S, Horiuchi S. Glycolaldehyde-modified low density lipoprotein leads macrophages to foam cells via the macrophage scavenger receptor. J Biochem. 1998;123:1208-1217
7. Anders HJ, Banas B, Schlondorff D. Signaling danger: toll-like receptors and their potential roles in kidney disease. J Am Soc Nephrol. 2004;15:854-867
8. Tsan MF, Gao B. Endogenous ligands of Toll-like receptors. J Leukoc Biol. 2004;76:514-519
9. Li F, Yang N, Zhang L, Tan H, Huang B, Liang Y, Chen M, Yu X. Increased expression of toll-like receptor 2 in rat diabetic nephropathy. Am J Nephrol. 2010;32:179-186
10. Devaraj S, Tobias P, Kasinath BS, Ramsamooj R, Afify A, Jialal I. Knockout of toll-like receptor-2 attenuates both the proinflammatory state of diabetes and incipient diabetic nephropathy. Arterioscler Thromb Vasc Biol. 2011;31:1796-1804
11. Devaraj S, Tobias P, Jialal I. Knockout of toll-like receptor-4 attenuates the pro-inflammatory state of diabetes. Cytokine. 2011;55:441-445
12. Lin M, Yiu WH, Wu HJ, Chan LY, Leung JC, Au WS, Chan KW, Lai KN, Tang SC. Toll-like receptor 4 promotes tubular inflammation in diabetic nephropathy. J Am Soc Nephrol. 2012;23:86-102
13. Sima AV, Botez GM, Stancu CS, Manea A, Raicu M, Simionescu M. Effect of irreversibly glycated LDL in human vascular smooth muscle cells: lipid loading, oxidative and inflammatory stress. J Cell Mol Med. 2010;14:2790-2802
14. Nivoit P, Wiernsperger N, Moulin P, Lagarde M, Renaudin C. Effect of glycated LDL on microvascular tone in mice: a comparative study with LDL modified in vitro or isolated from diabetic patients. Diabetologia. 2003;46:1550-1558
15. Krishnamurti U, Rondeau E, Sraer JD, Michael AF, Tsilibary EC. Alterations in human glomerular epithelial cells interacting with nonenzymatically glycosylated matrix. J Biol Chem. 1997;272:27966-27970
16. Jinnouchi Y, Sano H, Nagai R, Hakamata H, Kodama T, Suzuki H, Yoshida M, Ueda S, Horiuchi S. Glycolaldehyde-modified low density lipoprotein leads macrophages to foam cells via the macrophage scavenger receptor. J Biochem. 1998;123:1208-1217
17. Hou FF, Miyata T, Boyce J, Yuan Q, Chertow GM, Kay J, Schmidt AM, Owen WF. beta(2)-Microglobulin modified with advanced glycation end products delays monocyte apoptosis. Kidney Int. 2001;59:990-1002
18. Hodgkinson CP, Laxton RC, Patel K, Ye S. Advanced glycation end-product of low density lipoprotein activates the toll-like 4 receptor pathway implications for diabetic atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:2275-2281
19. Rocuts F, Ma Y, Zhang X, Gao W, Yue Y, Vartanian T, Wang H. Carbon monoxide suppresses membrane expression of TLR4 via myeloid differentiation factor-2 in betaTC3 cells. J Immunol. 2010;185:2134-2139
20. Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. 2003;300:1524-1525
21. Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun. 2009;388:621-625
22. Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42:145-151
23. Rao KM. MAP kinase activation in macrophages. J Leukoc Biol. 2001;69:3-10
24. Garcia-Arnandis I, Guillen MI, Gomar F, Pelletier JP, Martel-Pelletier J, Alcaraz MJ. High mobility group box 1 potentiates the pro-inflammatory effects of interleukin-1beta in osteoarthritic synoviocytes. Arthritis Res Ther. 2010;12:R165
25. Shen H, Tesar BM, Walker WE, Goldstein DR. Dual signaling of MyD88 and TRIF is critical for maximal TLR4-induced dendritic cell maturation. J Immunol. 2008;181:1849-1858
26. Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370-7377
27. Bae YS, Lee JH, Choi SH, Kim S, Almazan F, Witztum JL, Miller YI. Macrophages generate reactive oxygen species in response to minimally oxidized low-density lipoprotein: toll-like receptor 4- and spleen tyrosine kinase-dependent activation of NADPH oxidase 2. Circ Res. 2009;104:210-218
28. Su X, Ao L, Shi Y, Johnson TR, Fullerton DA, Meng X. Oxidized low density lipoprotein induces bone morphogenetic protein-2 in coronary artery endothelial cells via Toll-like receptors 2 and 4. J Biol Chem. 2011;286:12213-12220
29. Hou FF, Ren H, Owen WF Jr, Guo ZJ, Chen PY, Schmidt AM, Miyata T, Zhang X. Enhanced expression of receptor for advanced glycation end products in chronic kidney disease. J Am Soc Nephrol. 2004;15:1889-1896
30. Sakaguchi M, Murata H, Yamamoto K, Ono T, Sakaguchi Y, Motoyama A, Hibino T, Kataoka K, Huh NH. TIRAP, an adaptor protein for TLR2/4, transduces a signal from RAGE phosphorylated upon ligand binding. PLoS One. 2011;6:e23132
Author contact
![]() Corresponding author: Dr. Jing Nie and Dr. Fan Fan Hou, Division of Nephrology, Nanfang Hospital, 1838 North Guangzhou Ave, Guangzhou 510515, China, Phone: 86-20-62787972, 86-20-61641591; Fax: 86-20-87281713; E-mail: niejingcom, ffhouguangzhoucom.
Corresponding author: Dr. Jing Nie and Dr. Fan Fan Hou, Division of Nephrology, Nanfang Hospital, 1838 North Guangzhou Ave, Guangzhou 510515, China, Phone: 86-20-62787972, 86-20-61641591; Fax: 86-20-87281713; E-mail: niejingcom, ffhouguangzhoucom.
Received 2012-9-18
Accepted 2012-12-17
Published 2013-1-3