ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2013; 9(6):564-577. doi:10.7150/ijbs.5425 This issue Cite
Research Paper
Lentiviral miR30-based RNA Interference against Heparanase Suppresses Melanoma Metastasis with Lower Liver and Lung Toxicity
Xiao-yan Liu1, Qiu-su Tang2, Hong-chao Chen1, Xiao-ling Jiang1, Hong Fang1 ![]()
1. Department of Dermatology, the First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang 310003, China
2. Department of Pathology, the First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang 310003, China
Abstract
Aim: To construct short hairpin RNAs (shRNAs) and miR30-based shRNAs against heparanase (HPSE) to compare their safety and their effects on HPSE down-modulation in vitro and in vivo to develop a more ideal therapeutic RNA interference (RNAi) vector targeting HPSE.
Methods: First, we constructed shRNAs and miR30-based shRNAs against HPSE (HPSE-shRNAs and HPSE-miRNAs) and packed them into lentiviral vectors. Next, we observed the effects of the shRNAs on knockdown for HPSE expression, adhesion, migration and invasion abilities in human malignant melanoma A375 cells in vitro. Furthermore, we compared the effects of the shRNAs on melanoma growth, metastasis and safety in xenograft models.
Results: Our data showed that these artificial miRNAs targeting HPSE could be effective RNAi agents mediated by Pol II promoters in vitro and in vivo, although these miRNAs were not more potent than the HPSE-shRNAs. It was noted that obvious lung injuries, rarely revealed previously, as well as hepatotoxicity could be caused by lentivirus-mediated shRNAs (LV shRNAs) rather than lentivirus-mediated miRNAs (LV miRNAs) in vivo. Furthermore, enhanced expression of pro-inflammatory cytokines IL-6 and TGF-β1 and endogenous mmu-miR-21a-5p were detected in lung tissues of shRNAs groups, whereas the expression of mmu-let-7a-5p, mmu-let-7b-5p and mmu-let-7c-5p were down-regulated.
Conclusion: These findings suggest that artificial miRNAs display an improved safety profile of lowered lung injury or hepatotoxicity relative to shRNAs in vivo. The mechanism of lung injuries caused by shRNAs may be correlated with changes of endogenous miRNAs in the lung. Our data here increase the flexibility of a miRNA-based RNAi system for functional genomic and gene therapy applications.
Keywords: RNA interference, microRNA(miRNA), heparanase, metastasis, safety
Introduction
Heparanase (HPSE) is an endo-β-glucuronidase that can cleave heparan sulfate proteoglycans in the basement membrane. It is produced as a latent 65-kD proenzyme that is processed and activated by cathepsin L, yielding an enzymatically active heterodimer composed of 8- and 50-kD subunits [1]. Enzymatic cleavage of heparan sulfate by HPSE leads to disassembly of the extracellular matrix (ECM) and is therefore involved in cancer progression, metastasis, and neovascularization [2]. Although promising results from Phase I/II trials with a specific inhibitor against HPSE have been observed in various tumor types [3], the development of antibody-induced thrombocytopenia or neutropenia has limited the use of HPSE inhibitors in some patients [3, 4]. Thus, it is necessary to develop a more efficient and safer therapeutic method against HPSE.
RNA interference (ribonucleic acid interference, RNAi) is a powerful tool that blocks gene expression in mammalian cells by triggering sequence-specific gene degradation during posttranscriptional gene silencing [5]. In our previous study, we constructed the recombination plasmid pRNATU6.1/HPSE-shRNA and transfected it into A375 melanoma cells, resulting in decreased expression of HPSE and lower invasion ability of A375 cells in vitro [6]. Although the use of shRNA vectors for RNAi analysis under the control of RNA polymerase III (Pol III) (e.g., U6 or H1 promoter) can silence gene expression effectively, long-term suppression using Pol III shRNAs could be problematic [7, 8]. Furthermore, exogenous RNAi mediated by Pol III promoters, which are constitutively expressed in all cell types, can not target tumor tissues effectively in vivo [9, 10].
A class of endogenous non-coding small RNAs, miRNAs, post-transcriptionally regulates gene expression in a perfect or an imperfect sequence-specific manner to exert their RNAi effects [11]. These miRNAs are transcribed predominantly by the Pol II promoter as long primary polyadenylated transcripts (pri-miRNAs) [12]. Designing such shRNAs in the context of naturally occurring Pol II-driven miRNA transcripts increases the flexibility of this approach, allowing for conditional and cell type-specific expression [13, 14]. Researchers from several laboratories have mimicked diverse miRNA scaffolds, such as miR-30, miR-21, or miR-155, to develop potent shRNAs [15-17]. Such miRNA-based RNAi systems displayed very efficient knockdown of gene expression, even at the single-copy level [18]. Furthermore, miRNA-based approaches do not disrupt cellular processes that are regulated by endogenous miRNAs and may provide safer therapeutic RNAi expression vectors compared with shRNAs [19, 20]. Second-generation shRNA libraries were constructed based on the most characterized and commonly used pri-miRNA backbone, miR-30, comprising more than 140,000 shRNA expression plasmids and covering all predicted genes in both the human and mouse genomes [21].
In this study, we constructed shRNAs and miR30-based shRNAs against HPSE to compare their safety and their effects on HPSE down-modulation in vitro and in vivo, to develop a more ideal therapeutic RNAi vector targeting HPSE.
Materials and Methods
Construction and transduction of lentiviral vectors with specific miRNAs or shRNAs for HPSE
Three RNAi sequences that target HPSE (NM_006665.3, Figure 1A) were designed and screened in our previous study [6]. The lentiviral vectors with miRNAs that were based on the miR-30 framework [18] (Figure 1B and 1C) or HPSE-shRNAs (Figure 1D) were constructed by the GeneChem Company (Shanghai, China). Briefly, the double-stranded oligonucleotide encoding pre-miRNA or shRNA sequences for HPSE and their negative controls were annealed and inserted into the linearized eukaryotic PP-GFP or pGCSIL-GFP vector (GeneChem). All these vectors were transformed into One Shot TOP10 Chemically Competent E. coli, and the desired expression clones were identified by polymerase chain reaction (PCR) amplification and electrophoresis, and then were confirmed by sequencing (TaKaRa Biotechnology Co. Ltd., Dalian, China). The recombinational RNAi expression vectors (PP-GFP/ HPSE-miRNA and pGCSIL-GFP/ HPSE-shRNA) and the packaging vectors (pHelper 1.0 and pHelper 2.0) (GeneChem) were co-transfected into 293FT cells (Invitrogen, Carlsbad, CA, USA) with Lipofectamine2000 (Invitrogen). The culture supernatants were collected at 48 hours after transfection, concentrated, and used as virus stocks named LV PP-GFP/ HPSE-miRNAs and LV pGCSIL-GFP/ HPSE-shRNAs. All the lentiviral vectors expressed enhanced green fluorescent protein (GFP), which allowed for titering and measuring their infection efficiency in infected cells. The viral titer was determined by counting the GFP-positive cells after infection.
Cell culture and lentiviral infection
The human malignant melanoma cell line A375 was purchased from the Shanghai Institute of Cell Biology, routinely cultured in Dulbecco's modified Eagle's medium (DMEM) (Hyclone Laboratories, Inc., Logan, UT, USA) supplemented with 10% heat-inactivated fetal bovine serum and incubated in a humidified (37°C, 5% CO2) incubator. The Neg-miRNA, HPSE-miRNA1, HPSE-miRNA2, HPSE-miRNA3, Neg-shRNA, HPSE-shRNA1, HPSE-shRNA2, and HPSE-shRNA3 constructs were transduced into the A375 cells by lentiviral infection with a multiplicity of infection (MOI) of 1 or 10 in the presence of polybrene (5μg/mL). Cells of different groups were observed under a fluorescence microscope (Olympus, Tokyo, Japan) and harvested at 72 hours after infection.
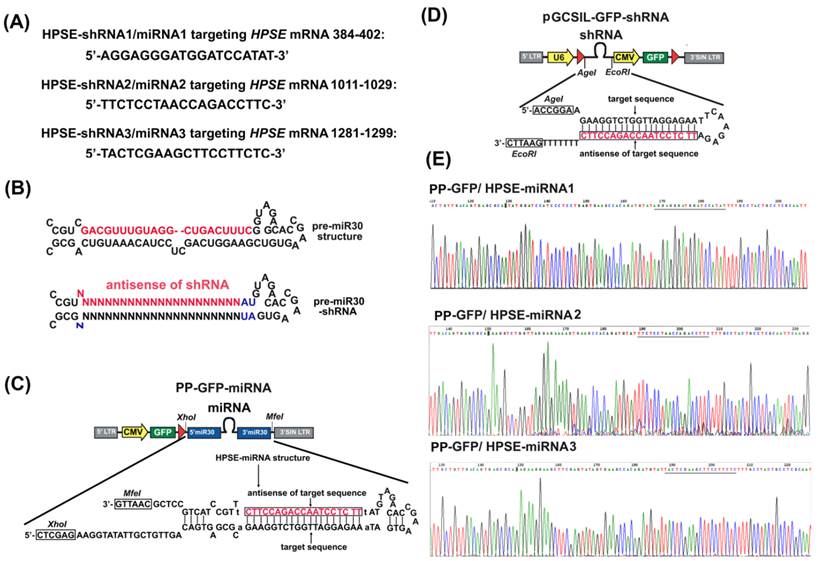
Construction of miR30-based HPSE miRNAs. (A) The sequences of three designed mature miRNAs/shRNAs targeting HPSE and the precise regions of the HPSE mRNA that they targeted. (B) A schematic representation of the pre-miR30 shRNA vector in which the coding region of the mature miR30 miRNA (the upper red indication) was replaced with sequences that encode shRNAs targeting the gene of choice (the lower red indication). (C) A schematic representation of the PP-GFP/CMV-miRNA vector platforms. The synthesized miR30-HPSE-shRNAs (HPSE-miRNAs) were subcloned by site-specific recombination into the PP-GFP vector. (D) A schematic representation of the pGCSIL-GFP/U6-shRNA vector platforms. The synthesized HPSE-shRNAs were subcloned by site-specific recombination into the pGCSIL-GFP vector. (E) The sequences of PP-GFP/HPSE-miRNA1, PP-GFP/HPSE-miRNA2 and PP-GFP/HPSE -miRNA3 were confirmed by sequencing.
Quantitative real-time PCR
Total RNA was isolated from cells of different groups using RNAisoTM PLUS (TaKaRa) following the manufacturer's protocol. Quantitative real-time PCR was performed using the ABI Prism 7900 (Applied Biosystems, Foster City, CA, USA) and SYBR PrimeScript RT-PCR kit II (TaKaRa) according to the manufacturer's instructions. Primers for HPSE and β-actin, the internal control, were synthesized by TaKaRa as follows: HPSE-forward, 5′-GAATGGACGGACTGCTAC-3′; HPSE-reverse, 5′-CCAAAGAATACTTGCCTCA-3′; β-actin-forward, 5′-GGCGGCACCACCATGTACCCT-3′; β-actin-reverse, 5′-AGGGGCCGGACTCGTCATACT-3′. The thermal profile for the real-time PCR was 95°C for 30 seconds followed by 40 cycles of 95°C for 5 seconds and 60°C for 30 seconds. The ΔCt data were collected automatically and -ΔΔCt was calculated using the following formula: -ΔΔCt = average ΔCt of control group -ΔCt of the treated group. The relative expression of the target gene was calculated using 2-ΔΔCt [22]. Each group was performed in triplicate. All the experiments were repeated three times.
Western blot analysis
Cells from different groups were collected and lysed using a protein extraction reagent (Applygen, Beijing, China). The amount of protein in each lysate was quantified by the Bradford method [23]. Equivalent amounts of total proteins were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and then were transferred to polyvinylidene difluoride membranes. The membranes were incubated in 5% skim milk for 2 hours at room temperature to block nonspecific interactions before immunoblotting with the HPSE polyclonal antibody (Abcam® Biotechnology plc, San Francisco, CA, USA) overnight at 4°C. After washing three times with TBS/T buffer, the membranes were incubated with horseradish peroxidase-conjugated anti-rabbit IgG for 1 hour at room temperature, followed by EZ-ECL (Biological Industries, Kibbutz Beit-Haemek, Israel) visualization of the bands [22]. The membranes were stripped and probed with the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Kangchen Biotech, Shanghai, China) monoclonal antibody as the internal control [22]. All the experiments were repeated at least three times.
In vitro cell viability assay
A375 cells grown in 96-well plates at a low density of 5×103 cells per well were infected in triplicate with LV HPSE-shRNA2, LV HPSE-miRNA2, LV Neg-shRNA, or LV Neg-miRNA with an MOI of 10. At 24 and 72 hours after infection, microscopic or fluorescent images were captured at 100× magnification. Next, 20 μL of MTT (3-(4, 5-dimethylthiazol-2-yl)-2, 5 -diphenyltetrazolium bromide, 5 mg/mL) (Sigma-Aldrich Co. LLC, St Louis, MD, USA) was added to each well, and the plates were incubated for an additional 4 hours at 37°C. The MTT solution in the medium was then aspirated, and 150 μL of dimethyl sulfoxide (DMSO) (Sigma-Aldrich Co. LLC) was added before measurement of the absorbance at 570 nm. The cellular viability was evaluated using the A570 value [24]. Each group was performed in quadruplicate, and the experiment was repeated three times.
Cellular adhesion assay, transwell migration assay and Matrigel invasion assay in vitro
Cells were collected at 72 hours after infection with LV HPSE-shRNA2, LV HPSE-miRNA2, LV Neg-shRNA, or LV Neg-miRNA. Cells were added into prepared 96-well plates, coated with Matrigel (1:3 dilution ratio; Becton Dickinson, Franklin Lakes, NJ, USA) and 2% bull serum albumin at a concentration of 1×104 per well. The cells that did not adhere to the Matrigel were washed off with phosphate-buffered saline (PBS) after 1 hour incubation at 37°C, and then were subjected to the MTT assay to determine cellular adhesion ability [22]. The migratory and invasive abilities of the tumor cells were determined using 24-well transwell chambers [22] without or with Matrigel coating (1:3 dilution ratio; BD), with upper and lower culture compartments separated by polycarbonate membranes with 8-μm pores (Corning Costar Corp., Cambridge, MA, USA). The transduced cells (2×104) from each group were resuspended in 100 μL of serum-free DMEM and seeded in the top chambers. The bottom chambers were filled with 0.5 mL of DMEM medium containing 10% fetal bovine serum as a chemoattractant. After subculturing for 24 hours, the cells on the upper surface of the membrane were removed using cotton tips. The cells that migrated to or invaded the lower surface were fixed in 10% formalin at room temperature for 30 minutes and stained with hematoxylin-eosin (H&E). The migratory or invasive abilities were determined by counting the H&E-stained cells at 400× magnification. Each treatment was performed in triplicate, and all the experiments were repeated three times.
In vivo assays for tumor proliferation, metastasis and evaluation of toxicity of LV shRNA or LV miRNA
All our animal protocols were approved by the Animal Care and Use Committee of the Zhejiang University of Traditional Chinese Medicine (Hangzhou, China). Five-week-old BALB⁄c-nu mice were bred in laminar flow cabinets and maintained at a constant humidity and temperature (25-28°C). The A375 cells (5×106) were injected into the flank of nude mice (3 in each group) on day 0. The mice were then treated with 100 μL of LV Neg-shRNA, LV Neg-miRNA, LV HPSE-shRNA2, or LV HPSE-miRNA2 by injection into tumors on days 7, 14 and 21. An equal volume of PBS was used as a mock control. Tumor growth was monitored by the tumor volume, which was calculated as described (17): Volume (mm3) = width2 (mm2) × length (mm) /2. The mice were sacrificed on day 35, and the lungs and livers were removed. Consecutive sections were made for every tissue block of the lungs or livers and stained with H&E. The incidences of lung or liver metastasis or inflammation were calculated and evaluated independently by two pathologists. Furthermore, the metastases were classified as grade I-IV according to the number of A375 cells in the metastatic lesion [17]. The in vivo experiment was repeated three times.
Expression of proinflammatory cytokines by ELISA and endogenous miRNAs in the lung by quantitative real-time PCR
Partial fresh lung tissues were prepared for homogenates and further used for enzyme-linked immunosorbent assay (ELISA) [25]. ELISAs for transforming growth factor beta1 (TGF-β1), interleukin-6 (IL-6) and interferon gamma (IFN-γ) were performed according to the manufacturer's instructions using the mouse ELISA kits (R&D Systems® Inc., Minneapolis, MN, USA). Total RNAs, including miRNAs, from formalin-fixed and paraffin-embedded lung sections were purified using the miRNeasy FFPE Kit (Qiagen, Inc., Valencia, CA, USA) according to the manufacturer's specifications. Briefly, 320 μL of Deparaffinization Solution (Qiagen) was added to the 4 pieces of 20-μm thick FFPE sections to remove traces of paraffin. The tissues were digested with protease K at 56°C for 15 minutes followed by heat treatment at 80°C for another 15 minutes. Next, 25 μL of DNase Booster Buffer and 10μL DNase I stock solution were added to the supernatant. After washing, total RNA, including a small miRNA fraction, was eluted with RNase-free water [26]. The primers for mmu-miR-U6, as a reference, and the most highly expressed endogenous miRNAs, including mmu-let-7a-5p, mmu-let-7b-5p, mmu-let-7c-5p, and mmu-miR-21a-5p, as shown on microRNA.org (www.microrna.org/microrna/getExprForTissues.do?tissue=mmu_Lung+), were purchased from Ruibo Company (Guangzhou, China). Each treatment was performed in triplicate, and all the experiments were repeated three times. Furthermore, we performed miRNA target prediction using the two most common algorithms online: Targetscan Human 6.2 (http://www.targetscan.org/) and miRanda (http://www.microrna.org/microrna/home.do).
Statistical Analysis
The results are expressed as means ± SD. All the experiments were performed at least in triplicate. Statistical analysis was performed using one-way analysis of variance (ANOVA) followed by the Dunnett's-t test and the SNK-q test, where a P value less than 0.05 was deemed to be statistically significant.
Results
Development of an improved miR30-based HPSE-RNAi shuttle (HPSE-miRNA) and lentiviral vector
We developed the PP-GFP-miRNA vector, which carried the short hairpins that were embedded within a miRNA transcript from the improved miR-30 and were driven by the CMV-Pol II promoter [12, 15]. The vector had been designed to allow replacement of the miR-30 encoding region with shRNA sequences that target any transcript of choice (Figure 1B). Here, the three HPSE-target sequences that were screened in our previous study were redesigned based on the miR-30 scaffold (HPSE-miRNA1, HPSE-miRNA2 and HPSE-miRNA3) and cloned into the PP-GFP-shRNA vector (Figure 1C). Similarly, the non-targeting control sequence (Neg-miRNA) was also redesigned. In addition to Neg-miRNA and HPSE-miRNAs, three conventional shRNAs targeting HPSE and a negative control shRNA at the same target area were designed (HPSE-shRNA1, HPSE-shRNA2, HPSE-shRNA3 and Neg-shRNA, Figure 1D) to be further tested alongside the HPSE-miRNAs in vitro and in vivo. These recombinant plasmids were purified and confirmed to be correct by sequencing (Figure 1E), and then they were packed into lentivirus and named LV Neg-miRNA, LV HPSE-miRNA1, LV HPSE-miRNA2, LV HPSE-miRNA3, LV Neg-shRNA, LV HPSE-shRNA1, LV HPSE-shRNA2 and LV HPSE-shRNA3. The dilution method was used to titer the viruses, and the dilution that was used was approximately 5×108 TU/mL (data not shown).
HPSE-miRNAs down-regulated HPSE expression of melanoma cells but were not more potent than HPSE-shRNAs
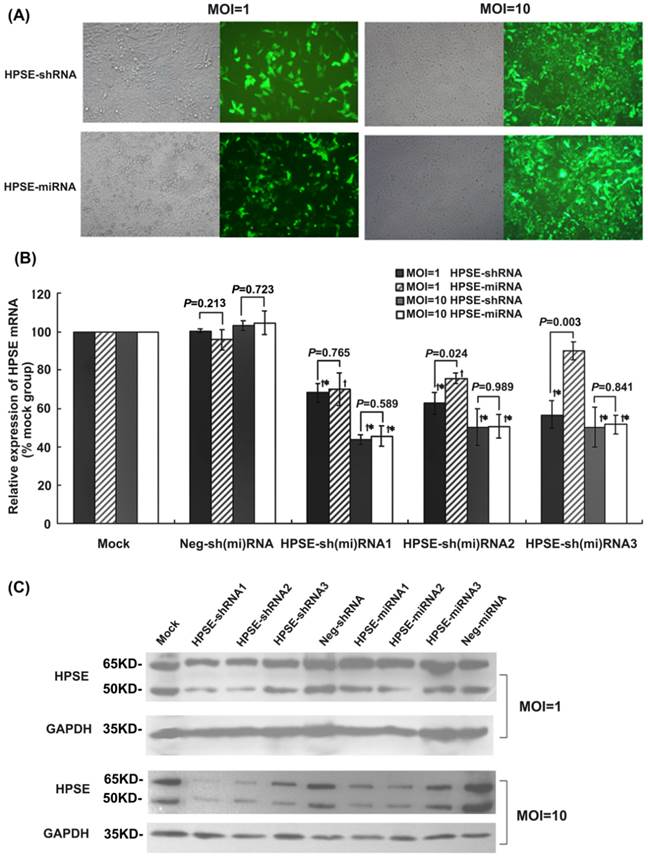
The recombinant lentiviral-mediated shRNAs or miRNAs were transduced into A375 cells in which heparanase was highly expressed [27, 28] with an MOI of 1 or 10. After being transduced for 72 hours, the percentage of GFP-positive cells in both the HPSE-shRNA and HPSE-miRNA groups was approximately 10-20% at an MOI of 1, whereas it was more than 90% at an MOI of 10 (Figure 2A). Compared with the mock control, the mRNA levels of HPSE at an MOI of 1 were down-regulated by 31.83%, 37.23%, 43.14%, respectively, in A375 cells transduced with HPSE-shRNA1, HPSE-shRNA2 or HPSE-shRNA3. Although A375 cells were treated with HPSE-miRNA1, HPSE-miRNA2 or HPSE-miRNA3, the inhibition rate of HPSE expression was about 30.00% (vs. HPSE-shRNA1, P=0.765), 24.07% (vs.HPSE-shRNA2, P=0.024), and 9.97% (vs. HPSE-shRNA3, P=0.003), respectively (Figure 2B). However, the protein levels of HPSE detected by western blot analysis were slightly decreased only in the HPSE-shRNA1 and HPSE-shRNA2 groups. When the MOI was 10, both HPSE-miRNAs and HPSE-shRNAs showed an equal effect regarding HPSE expression, which was approximately 50-60% down-regulated compared with the mock and negative control cells. Furthermore, no significant difference was observed between any pairs of LV HPSE-shRNA/miRNA (P>0.05, Figure 2B and 2C). In addition, HPSE-miRNA2 and HPSE-shRNA2 were selected for further studies.
Both HPSE-miRNA2 and HPSE-shRNA2 attenuated cellular viability without shRNA- mediated toxicity in vitro
At 24 hours after infection, each of the four treatments (Neg-shRNA, Neg-miRNA, HPSE-shRNA2, or HPSE-miRNA2) showed similar levels of cell proliferation and fluorescence. However, at 72 hours, we noted a clear loss of living cells in the HPSE-shRNA2- or HPSE-miRNA2-treated populations compared with the respective negative control groups (Figure 3A). In addition, we performed MTT assays at both 24 hours and 72 hours to measure cellular viability. The A570 values of each group that was subjected to the MTT assay at 24 hours were 1.266 ±0.038, 1.223 ±0.088, 1.210 ±0.017, and 1.202 ±0.132 (P>0.05), indicating that the initial cell density of the four treatments were equal to each other. Upon incubation for an additional 48 hours, the fold increase in cellular proliferation in the HPSE-shRNA2 and HPSE-miRNA2 groups were 0.560±0.036, 0.621 ±0.182, which was significantly lower than those of the respective control groups (1.035±0.152 and 1.172±0.096, respectively; P<0.01) (Figure 3A). However, the shRNA-treated cells presented with a similar proliferative ability relative to miRNA-treated cells (P>0.05, Figure 3A). We inferred that this loss of activity was the result of inhibition of HPSE rather than shRNA-induced toxicity.
Comparison of the effects of HPSE-shRNAs and HPSE-miRNAs on HPSE expression in A375 melanoma cells. A375 cells were infected with the indicated recombinational lentivirus at an MOI of 1 or 10 for 72 hours, and then were harvested for further quantitative RT-PCR and western blot analysis. (A) GFP-positive cells in HPSE-shRNA- and HPSE-miRNA-transduced groups at an MOI of 1 or 10 (magnification of 100×). (B) The inhibitory effects of HPSE-shRNAs or HPSE- miRNAs on HPSE mRNA expression were determined by quantitative real-time PCR. Calculation of the respective HPSE mRNA expression in each group was relative to the mock group (%). (* P <0.05, compared with the mock group; †P <0.05, compared with the respective negative control; n=3) (C) Inhibitory effects of HPSE-shRNAs or HPSE-miRNAs on HPSE protein expression were detected by western blot analysis. Representative blots are shown from three independent experiments with identical results.
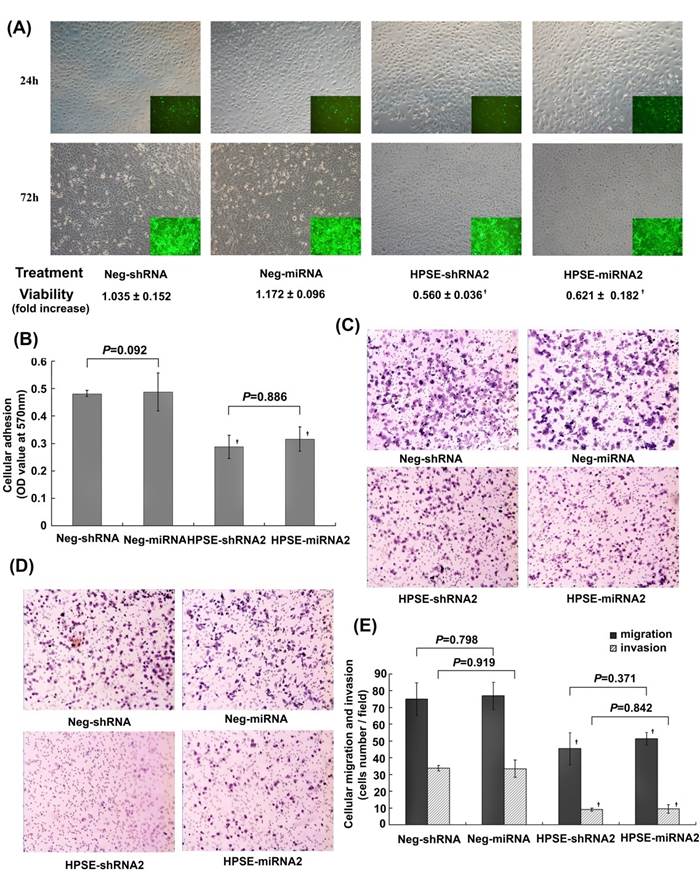
Comparison of the effects of HPSE-shRNA2 and HPSE-miRNA2 on A375 cells in vitro. A375 cells were transduced with HPSE-shRNA2, HPSE-miRNA2 and respective negative controls at an MOI of 10 for 24 hours or 72 hours. (A) The effects of HPSE-shRNA2 and HPSE-miRNA2 on cellular viability. Photomicrographs depicting GFP expression at 24 hours and 72 hours after treatment were shown (magnification of 100×). Cell viability was further measured by the MTT assay (n=4). (B) The cell-Matrigel adhesion assay. A375 cells were transduced with the indicated shRNA or miRNA and seeded in the Matrigel-prepared 96-well plates for 1 hour, washed with PBS, and then were subjected to the MTT assay (n=3). (C) Representative images of migratory cells in HPSE-shRNA2-transduced cells, HPSE-miRNA2-transduced cells and both negative control groups using the transwell migration assay (H&E staining, magnification of 100×, n=3). (D) Representative images of nvasive cells in HPSE-shRNA2 -transduced cells, HPSE-miRNA2-transduced cells and both negative control groups using the Matrigel invasion assay (H&E staining, magnification of 100×, n=3). (E) A diagram of the migratory cells or invasive cells, as determined by counting the cell number under the microscope with a magnification of 400×, in the transwell migration assay or the Matrigel-invasion assay. (†P <0.05, compared with the respective negative control, n=3). All the experiments were performed three times.
Both HPSE-miRNA2 and HPSE-shRNA2 abolished adhesion, migration, and invasion of melanoma cells in vitro
Adhesion of cells to Matrigel was evaluated using the MTT assay at the indicated time points. The A570 value in the HPSE-shRNA2 (0.288±0.043) and HPSE-miRNA2 (0.316 ±0.044) groups were significantly lower than those in the respective negative control groups (0.481 ±0.011 and 0.487 ±0.069; P=0.002 and 0.003) (Figure 3B). In the transwell migration assays, the numbers of migrated A375 cells infected with LV HPSE-shRNA2 (45.333±9.000) and LV HPSE-miRNA2 (51.333±9.333) at a high power of 400× were much lower than those of the respective negative controls (75.000 ±9.849 and 77.000 ±8.000; P=0.020 and 0.018) (Figure 3C and 3E), suggesting that both HPSE-shRNA2 and HPSE-miRNA2 could significantly suppress the migration of A375 cells. Similar effects were also detected in Matrigel invasion assays of A375 cells. Both HPSE-shRNA2 and HPSE-miRNA2 could significantly inhibit the in vitro invasion ability of A375 cells (P=0.0001, compared with Neg-shRNA or Neg-miRNA) (Figure 3D and E). Notably, no significant difference was found between the A375 cells transduced with HPSE-shRNA2 and those with HPSE-miRNA2 in the adhesion, migration, or invasion assays (P>0.05, Figure 3B and E).
Both HPSE-miRNA2 and HPSE-shRNA2 suppressed tumor growth and lung metastasis of melanoma in vivo.
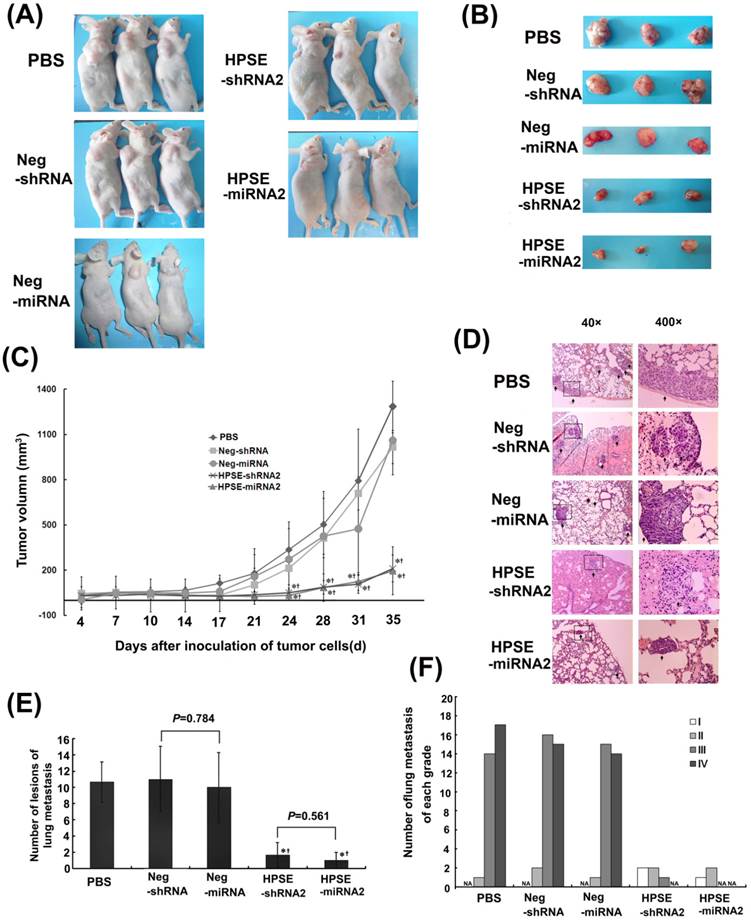
On day 7, the volumes of the tumors among all groups were not different when the recombinational LV and PBS were injected into tumors (P>0.05). In the tumor growth assay, detectable tumors in both RNAi groups were not significantly different (211.847±89.011 vs. 195.842±59.578); however, they did differ from those in the mock and negative control groups on days 24, 28, 31 and 35 (P<0.05; Figure 4A, 4B and 4C). The numbers of lung metastatic lesions in the LV HPSE-shRNA2 and LV HPSE-miRNA2 groups were greatly decreased compared with the respective controls and mock group (P=0.019 and 0.025, Figure 4D and E). Furthermore, most of the lung metastases in both RNAi groups were grade I (≤20 cells) or grade II (20-50 cells), whereas those in the negative control groups or PBS group were grade III (50-100 cells) or grade IV (>100 cells) (Figure 4D and F). However, the difference in the number of lung metastases between the HPSE-shRNA2 and HPSE-miRNA2 groups was not significant (P=0.561, Figure 4D).
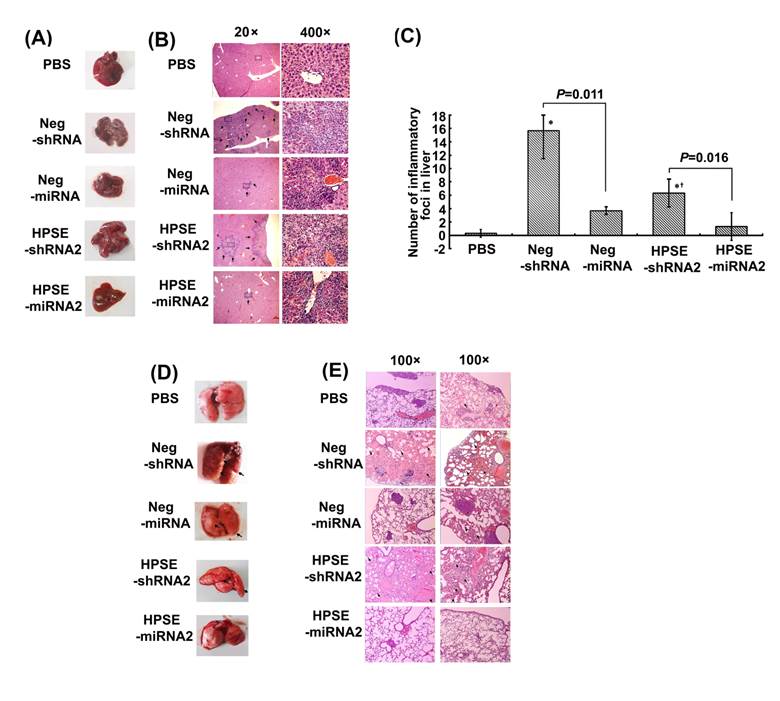
Artificial miRNAs attenuated the shRNA-mediated toxicity of the liver and lung in vivo
Unexpectedly, in our experiments we found no metastasis in the liver, another vulnerable site of tumor metastasis, whereas some inflammatory foci were observed by general or microscopic observation. Upon general observation (Figure 5A), obvious inflammatory or necrotic foci infiltrated by lymphocytes and some swelling hepatocytes were found in both shRNA groups, a finding that was confirmed by histological analysis (Figure 5B). The numbers of inflammatory foci counted at a low power (40×) in the HPSE-shRNA2 or HPSE-miRNA2 group were 6.333±2.082 and 1.333±0.577 (P=0.016), respectively, and those in the respective negative controls were 15.667±4.163 and 3.667±2.082 (P=0.011) (Figure 5C). In the mock group with PBS, inflammatory foci were rarely found (Figure 5B and 5C). Similarly and interestingly, there were lung injuries of inflammatory infiltration or pulmonary edema of differing degrees (Figure 5D and 5E) observed in both Neg-shRNA and HPSE-shRNA2 groups, independent of target sequence and the numbers of metastases. As shown in gross specimens (Figure 5D), obvious edema and consolidation were observed in both shRNAs groups. It was confirmed by histological images that the honeycomb architecture of the lung disappeared, and extensive edema, thickness of alveolar walls or infiltration of inflammatory cells were evident instead (Figure 5E). However, there was little sign of lung injuries, besides metastases, in the PBS or LV miRNAs groups (Figure 5E).
Endogenous miRNAs were predominantly down-regulated in the lungs of LV shRNAs groups, but miR-21 was up-regulated
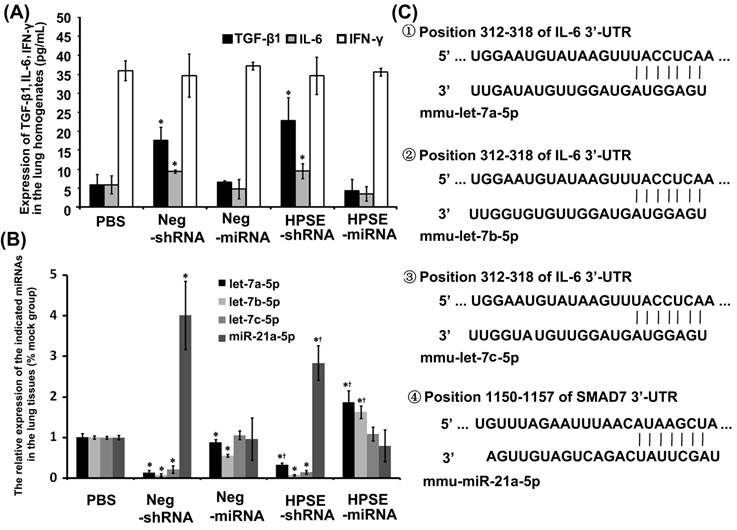
In ELISA assays, the expression levels of TGF-β1 and IL-6 from both shRNAs groups were up-regulated compared with both miRNAs groups and the PBS group (P<0.05), independent of the expression of HPSE, whereas the expression of IFN-γ was not different among the groups (Figure 6A). As shown in Figure 6B, expression of mmu-let-7a-5p, mmu-let-7b-5p and mmu-let-7c-5p were obviously attenuated in the shRNAs groups compared with those in the PBS and miRNAs groups. However, the relative expression levels of mmu-miR-21a-5p in Neg-shRNA and HPSE-shRNA were approximately 4.011±0.838 and 2.832±0.424, respectively, predominantly higher than that of the PBS, Neg-miRNA and HPSE-miRNA groups (1.001±0.047, 0.962±0.518 and 0.795±0.392, respectively). Furthermore, miR-21a-5p in the RNAi group was lower than respective negative control group, although there was no statistical significance between Neg-miRNA and HPSE-miRNA (P=0.452). By prediction of targets of mmu-let-7a-5p, mmu-let-7b-5p, mmu-let-7c-5p and mmu-miR-21a-5p via Targetscan and miRanda, we found that IL-6 was the predicted target gene of mmu-let-7a-5p, mmu-let-7b-5p and mmu-let-7c-5p, whereas the 5′ seed region of mmu-miR-21a-5p targeted the 3′-UTR of the SMAD7 gene, a negative regulator of the TGF-β signaling pathway (Figure 6C).
Comparison of the effects of HPSE-shRNA2 and HPSE-miRNA2 on in vivo tumor proliferation and lung metastasis of A375 cells. A375 cells (5×106) were injected into the flank of nude mice on day 0. The mice were then treated with 100 μL of LV Neg-shRNA, LV Neg-miRNA, LV HPSE-shRNA2, LV HPSE-miRNA2 and PBS by injection into the tumors on days 7, 14 and 21. The mice were sacrificed on day 35, and the lungs and livers were removed. (A-B) Images of the xenograft mice and tumors that were taken at the end of the experiment on day 35 (n=3). (C) Measurements of tumor volumes (mm3) taken twice a week were shown in C. Volume (mm3) = width2 (mm2) × length (mm) /2. (* P <0.05, compared with PBS-treated cells; †P <0.05, compared with the respective negative control; n=3). (D) Representative lung tissue sections from each group (H&E staining, magnification of 40× and 400×, respectively). (E) The number of the melanoma lung metastases in each group. (* P <0.05, compared with PBS-treated cells; †P <0.05, compared with the respective negative control; n=3). (F) Number of lung metastases of melanoma with grade I, II, III or IV in each group.
Comparison of the toxicity induced by shRNAs or miRNAs. The xenograft mice were treated with 100 μL of LV Neg-shRNA, LV Neg-miRNA, LV HPSE-shRNA2, LV HPSE-miRNA2 and PBS by injection into the tumors on days 7, 14 and 21. The mice were sacrificed on day 35, and the lungs and livers were removed. (A) The representative liver morphology of each group. (B) Representative liver tissue sections from each group (H&E staining, magnification of 20× and 400×, respectively). (C) The number of liver inflammatory foci in each group (* P <0.05, compared with the PBS-treated cells; †P <0.05, compared with the respective negative control). (D) The representative lung morphology of each group. (E) Representative manifestation of lung tissue sections from each group (H&E staining, magnification of 100×). Obvious pulmonary edema or inflammatory infiltration was observed in shRNA-treated groups but not in PBS- or miRNA-treated groups.
Expression of some cytokines and endogenous miRNAs in the lung tissues of each group. (A) The expression of TGF-β1, IL-6 and IFN-γ in the lung homogenates of each group (*P <0.05, compared with the PBS- and miRNAs- treated groups). (B) The relative expression of let-7a-5p, let-7b-5p, let-7c-5p and miR-21a-5p in the mouse lung FFPE tissues of each group (*P <0.05, compared with the PBS- treated groups; †P <0.05, compared with the respective negative control). (C) Murine IL-6 might be the molecular target of let-7a-5p, let-7b-5p and let-7c-5p, and SMAD7 might be the molecular target of miR-21a-5p. Shown was the sequence alignment of let-7a-5p, let-7b-5p, let-7c-5p and miR-21a-5p, and their target sites in the 3′-UTR of IL-6 or SMAD7.
Discussion
RNAi is an evolutionarily conserved mechanism that triggers sequence-specific inhibition of complementary mRNAs in eukaryotes and has been widely utilized as a powerful tool to knockdown particular genes of interest for basic research or therapeutic purposes. Previous studies from several laboratories, including ours, demonstrated that siRNA or shRNA targeting HPSE could lead to slower growth, reduced clonogenic capacity, and invasive potential of aggressive tumor cell lines [28, 29]. However, the use of synthetic siRNA for RNAi in mammalian cells is limited by their transient nature and lack of an efficient delivery system [7, 8]. Although ~19- to 29-bp shRNAs delivered as plasmids or viral vectors are developed to allow the stable expression of RNAi, and shRNA constructs are generally driven by RNA polymerase III promoters, including the H1, U6 and tRNA promoters [30, 31], they are constitutively expressed in all cell types, a finding that became the major handicap of the application of shRNA [31-33]. Recently, the original shRNA design has been further optimized by embedding the hairpins in a miRNA-based context via the inclusion of structural motifs of pri-miRNAs, with the characteristic features of larger loops, internal mismatches, bulges, and flanking sequences [34, 35]. Compared with shRNAs, artificial miRNAs more naturally resemble endogenous RNAi substrates and are more amenable to Pol-II transcription [36], allowing for tissue-specific expression of RNAi and even polycistronic or reversible strategies [14, 37, 38]. Furthermore, Pol II-transcribed, miRNA-based shRNA displayed very efficient knockdown of gene expression, even the single-copy level [18]. However, contradictory results have been reported by other groups [39, 40]. When minimizing the variables between shRNA and miRNA-based vectors, taking into consideration the transcribed product, processing sites, and strand bias, shRNAs demonstrated improved silencing potency relative to their miRNA-based counterparts [40].
In the current study, we designed three HPSE-miRNA sequences that were embedded in the precursor miRNA context derived from miR-30 and then cloned the sequences into the Pol II promoter CMV-miRNA expression cassette of a lentiviral vector. Concurrently, we also constructed shRNA-based lentiviral vectors, which, after processing, yield similar RNAi sequences. At a low MOI of 1, HPSE-miRNAs did not display effective knockdown, whereas the respective HPSE-shRNAs showed a slight interference effect. It was ever shown that shRNAs yielded abundant levels of precursor and fully processed antisense RNA, even at a 10-fold lower dose compared with artificial miRNAs [41], thus providing mechanistic insight to explain the increased potency of shRNAs at a low dose in our study. When the infection efficiency was greater than 90% at a high MOI of 10, both HPSE-miRNAs and HPSE-shRNAs exhibited a 50-60% down-regulation of HPSE transcriptionally and translationally compared with the parental cells and negative controls, without a difference between HPSE-miRNAs and respective HPSE-shRNAs. Upon further observation, both HPSE-shRNA and HPSE-miRNA could similarly suppress proliferation, adhesion, migration and invasion of A375 melanoma cells in vitro, a result that was confirmed by xenograft experiments in mice. Our data demonstrated that miRNAs targeting HPSE could create effective RNAi mediated by Pol II promoters in vitro and in vivo, although these miRNAs were not more potent than HPSE-shRNAs, as expected.
The safety profile of these treatments is another important issue that should be considered. There are increasing reports that reveal siRNAs and shRNAs can induce immunological responses or off-target effects, thus causing toxicity [26, 42-44]. It is worth noting that many commonly used tumor cell lines [45, 46], including resistant A375 cells [47, 48], have a defective interferon response or cause down-regulation of components of the interferon pathway, findings that may explain why shRNA- treated cells in the current study presented with similar proliferative ability relative to the miRNA- treated cells in vitro. However, in vivo hepatotoxicity, characterized by lobular collapse, multifocal hepatocyte necrosis, and even cancer or death in mice, was ever reported in short- or long- term observations [49, 50]. Several controls proved that the toxicity was both shRNA-specific and dose-dependent and was not restricted to a particular shRNA or target [43, 49, 51]. In 2006, Grim and colleagues found that highly expressed 'toxic' shRNAs competed with endogenous miRNAs for intracellular processing. That study was supported by the results of further RNA analyses that toxicity and morbidity were correlated with decreased levels of the natural miRNAs miR-122 or let-7a and disruption of their biogenesis and function [49]. Interestingly, Grim and colleagues induced liver regeneration by surgery or chemical injury, but this technique did not result in miRNA inhibition. It was also observed by Beer that shRNAs globally dysregulated expressions of hepatic miRNAs and even at marginal doses, shRNAs could trigger histologically detectable hepatoxicity that could facilitate the ability of the MYC oncogene to induce liver tumorigenesis [50]. It has been demonstrated that the introduction of bulges, which are invariably present in miRNA stems, are instrumental to prevent activation of dsRNA-dependent IFN-stimulated genes (ISGs) as well as oligo adenylate synthetase1 (OAS1) pathway-associated off-target effects [20, 43, 52]. Our findings here corroborated data from work by others [19-21], revealing that embedding an shRNA into a miRNA scaffold could alleviate shRNA-specific hepatotoxicity in vivo independent of the target.
In addition to hepatotoxicity, it was reported that artificial miRNAs mitigated the shRNA-mediated toxicity in brain tissue [19, 41]. Some, but not all, shRNA expression vectors targeting the mouse Huntington's disease homolog (HDh) gene could cause neurotoxicity in the mouse brain, which would be alleviated by moving those toxic shRNAs into an artificial miRNA scaffold without HDh silencing efficiency [19]. However, shRNA-mediated acute lung injury, such as pulmonary edema, was seldom revealed. In our in vivo study, HPSE-miRNA and HPSE-shRNA similarly suppressed the tumor growth and lung metastasis of melanoma. Strikingly, obvious lung edema or inflammatory infiltration was observed in both shRNA groups but not in miRNA groups. We also collected mouse lung homogenates to detect the expression of cytokines of TGF-β1, IL-6 and IFN-γ, which are critical mediators of lung injury [53-55]. It was found that, in both shRNA groups, the expression levels of TGF-β1 and IL-6 were up-regulated compared with both the miRNA and PBS-treatment groups, independent of HPSE expression, whereas the expression of IFN-γ was not different among each group. Based on the known published mechanism on hepatotoxicity, we further detected the expression of let-7a-5p, let-7b-5p, let-7c-5p and miR-21a-5p, which highly expressed in mouse lung tissues. The expressions of let-7a-5p, let-7b-5p and let-7c-5p were down-regulated obviously in the shRNA groups as expected, whereas miR-21a-5p presented with opposite results regarding a significant up-regulation. More interestingly, IL-6 was the predicted target gene for all of the former three. Thus, we speculated that up-regulation of IL-6 may partly result from attenuation of endogenous miRNAs let-7a-5p, let-7b-5p and let-7c-5p that were disrupted by shRNA of high doses. However, it is necessary to validate our speculation by further loss- and gain-of-function miRNA experiments and luciferase reporter assays. It was further reported that miR-21a-5p displayed profibrotic miRNA, notably in the heart or kidney [56, 57]. miR-21a-5p was also up-regulated in either experimental or idiopathic pulmonary fibrosis [58]. On the one hand, miR-21a-5p could be induced directly by TGF-β mediated signaling [59]. Conversely, miR-21a-5p promoted TGF-β1-induced fibrogenic activation by inhibition of the inhibitory SMAD-SMAD7, resulting in TGF-β1 signal amplification that eventually promoted fibrosis [58, 60]. However, in murine models of acute or chronic asthma sensitized by allergen ovalbumin, miR-21a-5p was attenuated in the long-term treatment group [61]. It was suggested that miRNAs function in a time- and model-dependent manner. In our results, obvious lung injuries in the shRNA groups may be associated with up-regulation of TGF-β1 and miR-21a-5p. However, there are still many unresolved problems, including i) identification of the mechanism of enhancement of TGF-β1 and miR-21a-5p caused by shRNAs and the causal relationship between TGF-β1 and miR-21a-5p in our experimental models; ii) whether shRNA-mediated lung injuries are dose-, target sequence- or vector- dependent; iii) whether there are other inflammatory cytokines and miRNAs involved in lung injuries caused by shRNAs, and what the regulatory network amongst them would be. All these issues need to be further addressed by much stricter controlled experiments in our future series study.
In conclusion, in our present study, the artificial miRNAs displayed an improved safety profile and lowered lung injury or hepatotoxicity relative to shRNAs in vivo, which in turn increased the flexibility of this system for functional genomic and gene therapy applications.
Acknowledgements
This study was financially supported by National Natural Science Foundation of China Grants (No.81000761) and Natural Science Foundation of Zhejiang Province (No. LY12H16010). This study was performed in the Institute of Infectious Diseases, the First Affiliated Hospital, College of Medicine, Zhejiang University. Animal experiments were performed in Animal Care and Use Committee of the Zhejiang University of Traditional Chinese Medicine.
Abbreviations
DMEM: Dulbecco's modified Eagle's medium; DMSO: dimethyl sulfoxide; ECM: extracellular matrix; ELISA: enzyme-linked immunosorbent assay; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GFP: green fluorescent protein; H&E: hematoxylin-eosin; HPSE: heparanase; IFN-γ: interferon gamma; IL-6: interleukin-6; LV: lentivirus; miRNA/miR: microRNA; MOI: multiplicity of infection; MTT: (3-(4,5-dimethylthiazol-2-yl)-2,5 -diphenyltetrazolium bromide; PBS: phosphate-buffered saline; PCR: polymerase chain reaction; Pol II/III: polymerase II/III; RNAi: Ribonucleic acid interference; shRNA: short-hairpin RNA; TGF-β1: Transforming growth factor, beta1.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Abboud-Jarrous G, Atzmon R, Peretz T. et al. Cathepsin L is responsible for processing and activation of proheparanase through multiple cleavages of a linker segment. J Biol Chem. 2008;283:18167-76
2. Barash U, Cohen-Kaplan V, Dowek I. et al. Proteoglycans in health and disease: new concepts for heparanase function in tumor progression and metastasis. FEBS J. 2010;277:3890-903
3. Kudchadkar R, Gonzalez R, Lewis KD. PI-88: a novel inhibitor of angiogenesis. Expert Opin Investig Drugs. 2008;17:1769-76
4. Khasraw M, Pavlakis N, McCowatt S. et al. Multicentre phase I/II study of PI-88, a heparanase inhibitor in combination with docetaxel in patients with metastatic castrate-resistant prostate cancer. Ann Oncol. 2010;21:1302-7
5. Tan FL, Yin JQ. RNAi, a new therapeutic strategy against viral infection. Cell Res. 2004;14:460-6
6. Liu XY, Fang H, Yang ZG. et al. Construction of small interfering RNA targeting heparanase gene and its inhibitory effect on the invasiveness of human malignant melanoma cell line A375 in vitro. Chin J Dermatol. 2007;11:676-9
7. Leung RK, Whittaker PA. RNA interference: from gene silencing to gene-specific therapeutics. Pharmacol Ther. 2005;107:222-39
8. Akhtar S, Benter IF. Nonviral delivery of synthetic siRNAs in vivo. J Clin Invest. 2007;117:3623-32
9. Yuan J, Wang X, Zhang Y. et al. shRNA transcribed by RNA Pol II promoter induce RNA interference in mammalian cell. Mol Biol Rep. 2006;33:43-9
10. Scherr M, Eder M. Gene silencing by small regulatory RNAs in mammalian cells. Cell Cycle. 2007;6:444-9
11. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-97
12. Lee Y, Kim M, Han J. et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051-60
13. Dickins RA, McJunkin K, Hernando E. et al. Tissue-specific and reversible RNA interference in transgenic mice. Nat Genet. 2007;39:914-21
14. Snyder LL, Esser JM, Pachuk CJ. et al. Vector design for liver-specific expression of multiple interfering RNAs that target hepatitis B virus transcripts. Antiviral Res. 2008;80:36-44
15. Shin KJ, Wall EA, Zavzavadjian JR. et al. A single lentiviral vector platform for microRNA-based conditional RNA interference and coordinated transgene expression. Proc Natl Acad Sci U S A. 2006;103:13759-64
16. Wu J, Bonsra AN, Du G. pSM155 and pSM30 vectors for miRNA and shRNA expression. Methods Mol Biol. 2009;487:205-19
17. Sun BS, Dong QZ, Ye QH. et al. Lentiviral-mediated miRNA against osteopontin suppresses tumor growth and metastasis of human hepatocellular carcinoma. Hepatology. 2008;48:1834-42
18. Stegmeier F, Hu G, Rickles RJ. et al. A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc Natl Acad Sci U S A. 2005;102:13212-7
19. McBride JL, Boudreau RL, Harper SQ. et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc Natl Acad Sci U S A. 2008;105:5868-73
20. Bauer M, Kinkl N, Meixner A. et al. Prevention of interferon-stimulated gene expression using microRNA-designed hairpins. Gene Therapy. 2009;16:142-7
21. Jose MS, Mamie ZL, Ken Chang. et al. Second-generation shRNA libraries covering the mouse and human genomes. Nature genetics. 2005;37:1281-7
22. Liu X, Fang H, Chen H. et al. An Artificial miRNA against HPSE Suppresses Melanoma Invasion Properties, Correlating with a Down-Regulation of Chemokines and MAPK Phosphorylation. PLoS One. 2012;7:e38659
23. Lu TS, Yiao SY, Lim K. et al. Interpretation of biological and mechanical variations between the Lowry versus Bradford method for protein quantification. N Am J Med Sci. 2010;2:325-8
24. Sui M, Xiong X, Kraft AS. et al. Combination of gemcitabine antagonizes antitumor activity of paclitaxel through prevention of mitotic arrest and apoptosis. Cancer Biol Ther. 2006;5:1015-21
25. Barada KA, Mourad FH, Sawah SI. et al. Up-regulation of nerve growth factor and interleukin-10 in inflamed and non-inflamed intestinal segments in rats with experimental colitis. Cytokine. 2007;37:236-45
26. Chen L, Li Y, Fu Y. et al. Role of deregulated microRNAs in breast cancer progression using FFPE tissue. PLoS One. 2013;8:e54213
27. Roy M, Marchetti D. Cell surface heparan sulfate released by heparanase promotes melanoma cell migration and angiogenesis. J Cell Biochem. 2009;106:200-9
28. Zhang Y, Li L, Wang Y. et al. Downregulating the expression of heparanase inhibits the invasion, angiogenesis and metastasis of human hepatocellular carcinoma. Biochem Biophys Res Commun. 2007;358:124-9
29. Zheng L, Jiang G, Mei H. et al. Small RNA interference-mediated gene silencing of heparanase abolishes the invasion, metastasis and angiogenesis of gastric cancer cells. BMC Cancer. 2010;10:33
30. Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550-3
31. H. Kawasaki, K. Taira. Short hairpin type of dsRNAs that are controlled by tRNA (Val) promoter significantly induce RNAi-mediated gene silencing in the cytoplasm of human cells. Nucleic Acids Res. 2003;31:700-7
32. Bridge AJ, Pebernard S, Ducraux A. et al. Induction of an interferon response by RNAi vectors in mammalian cells. Nature Genet. 2003;34:263-4
33. Jackson AL, Bartz SR, Schelter J. et al. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. 2003;21:635-7
34. Zeng Y, Wagner EJ, Cullen BR. Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol Cell. 2002;9:1327-33
35. Chang K, Elledge SJ, Hannon GJ. Lessons from nature: microRNA-based shRNA libraries. Nat Methods. 2006;3:707-14
36. Zhou H, Xia XG, Xu Z. An RNA polymerase II construct synthesizes short-hairpin RNA with a quantitative indicator and mediates highly efficient RNAi. Nucleic Acids Res. 2005;33:e62
37. Chung KH, Hart CC, Al-Bassam S. et al. Polycistronic RNA polymerase II expression vectors for RNA interference based on BIC/miR-155. Nucleic Acids Res. 2006;34:e53
38. Dickins RA, McJunkin K, Hernando E. et al. Tissue-specific and reversible RNA interference in transgenic mice. Nat Genet. 2007;39:914-21
39. Li L, Lin X, Khvorova A. et al. Defining the optimal parameters for hairpin-based knockdown constructs. RNA. 2007;13:1765-74
40. Boudreau RL, Monteys AM, Davidson BL. Minimizing variables among hairpin-based RNAi vectors reveals the potency of shRNAs. RNA. 2008;14:1834-44
41. Boudreau RL, Martins I, Davidson BL. Artificial microRNAs as siRNA shuttles: improved safety as compared to shRNAs in vitro and in vivo. Mol Ther. 2009;17:169-75
42. Sledz CA, Holko M, de Veer MJ. et al. Activation of the interferon system by short-interfering RNAs. Nat Cell Biol. 2003:5834-9
43. Kleinman ME, Yamada K, Takeda A. et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452:591-7
44. Bergé M, Bonnin P, Sulpice E. et al. Small interfering RNAs induce target-independent inhibition of tumor growth and vasculature remodeling in a mouse model of hepatocellular carcinoma. Am J Pathol. 2010;177:3192-201
45. Stojdl DF, Lichty B, Knowles S. et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6:821-5
46. Wollmann G, Robek MD, van den Pol AN. Variable deficiencies in the interferon response enhance susceptibility to vesicular stomatitis virus oncolytic actions in glioblastoma cells but not in normal human glial cells. J Virol. 2007;81:1479-91
47. Chawla-Sarkar M, Leaman DW, Jacobs BS. et al. Resistance to interferons in melanoma cells does not correlate with the expression or activation of signal transducer and activator of transcription 1 (Stat1). J Interferon Cytokine Res. 2002;22:603-13
48. Bae SI, Cheriyath V, Jacobs BS. et al. Reversal of methylation silencing of Apo2L/TRAIL receptor 1 (DR4) expression overcomes resistance of SK-MEL-3 and SK-MEL-28 melanoma cells to interferons (IFNs) or Apo2L/TRAIL. Oncogene. 2008;27:490-8
49. Grimm D, Streetz KL, Jopling CL. et al. Fatality in mice due to oversaturation of cellular microRNA/ short hairpin RNA pathways. Nature. 2006;441:537-41
50. Beer S, Bellovin DI, Lee JS. et al. Low-level shRNA cytotoxicity can contribute to MYC-induced hepatocellular carcinoma in adult mice. Mol Ther. 2010;18:161-70
51. Bridge AJ, Pebernard S, Ducraux A. et al. Induction of an interferon response by RNAi vectors in mammalian cells. Nat Genet. 2003;34:263-4
52. Pebernard S, Iggo RD. Determinants of interferon-stimulated gene induction by RNAi vectors. Differentiation. 2004;72:103-11
53. Anscher MS. Targeting the TGF-beta1 pathway to prevent normal tissue injury after cancer therapy. Oncologist. 2010;15:350-9
54. Pedroza M, Schneider DJ, Karmouty-Quintana H. et al. Interleukin-6 contributes to inflammation and remodeling in a model of adenosine mediated lung injury. PLoS One. 2011;6(7):e22667
55. Yamada M, Kubo H, Kobayashi S. et al. Interferon-gamma: a key contributor to hyperoxia-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1042-7
56. Thum T, Gross C, Fiedler J. et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980-4
57. Denby L, Ramdas V, McBride MW. et al. miR-21 and miR-214 are consistently modulated during renal injury in rodent models. Am J Pathol. 2011;179:661-72
58. Liu G, Friggeri A, Yang Y. et al. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med. 2010;207:1589-97
59. Davis BN, Hilyard AC, Lagna G. et al. SMAD proteins control DROSHA mediated microRNA maturation. Nature. 2008;454:56e61
60. Chung AC, Dong Y, Yang W. et al. Smad7 suppresses renal fibrosis via altering expression of TGF-β/Smad3-regulated microRNAs. Mol Ther. 2013;21:388-98
61. Garbacki N, Di Valentin E, Huynh-Thu VA. et al. MicroRNAs profiling in murine models of acute and chronic asthma: a relationship with mRNAs targets. PLoS One. 2011;6:e16509
Author contact
![]() Corresponding author: Hong Fang, Department of Dermatology, the First Affiliated Hospital, College of Medicine, Zhejiang University79# Qingchun Road, Hangzhou 310003, China. Tel: 86-571-87236340. Fax: 86-571-87236628. Email: fanghongzycom
Corresponding author: Hong Fang, Department of Dermatology, the First Affiliated Hospital, College of Medicine, Zhejiang University79# Qingchun Road, Hangzhou 310003, China. Tel: 86-571-87236340. Fax: 86-571-87236628. Email: fanghongzycom
Received 2012-10-21
Accepted 2013-5-18
Published 2013-6-12