ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2013; 9(8):830-841. doi:10.7150/ijbs.7039 This issue Cite
Research Paper
PTHrP Expression in Human MDA-MB-231 Breast Cancer Cells Is Critical for Tumor Growth and Survival and Osteoblast Inhibition
Lu Zheng1,2, Ke Zhu3, Hongli Jiao2,3, Zhongfang Zhao2, Lixiao Zhang2, Mo Liu2, Weimin Deng1, Di Chen3, Zhi Yao1 ![]() , Guozhi Xiao3
, Guozhi Xiao3 ![]()
1. Department of Immunology, Key Laboratory of Educational Ministry of China, Tianjin Medical University, Tianjin 300070, China
2. College of Life Sciences, Nankai University, Tianjin 300071, China
3. Department of Biochemistry, Rush University, Chicago, IL 60612, USA
Abstract
This study examined the effects of parathyroid hormone-related protein (PTHrP) derived from human MDA-MB-231 breast cancer cells on the tumor growth and osteoblast inhibition. Results revealed that knocking down PTHrP expression in the breast cancer cells strikingly inhibited the formation of subcutaneous tumors in nude mice. PTHrP knockdown dramatically decreased the levels of cyclins D1 and A1 proteins and arrested the cell cycle progression at the G1 stage. PTHrP knockdown led to the cleavage of Caspase 8 and induced apoptosis of the tumor cells. Interestingly, knocking down PTHrP increased the levels of Beclin1 and LC3-II and promoted the formation of autophagosomes. Knocking down PTHrP expression significantly reduced the abilities of the breast cancer cells to inhibit osteoblast differentiation and bone formation in vitro and in vivo. Finally, we found that PTHrP activated its own expression through an autocrine mechanism in MDA-MB-231 cells. Collectively, these studies suggest that targeting PTHrP expression in the tumor cells could be a potential therapeutic strategy for breast cancers, especially those with skeletal metastases.
Keywords: PTHrP, MDA-MB-231 cell, autophagy, osteoblast, osteoclast, osteolysis
INTRODUCTION
Parathyroid hormone-related protein (PTHrP) is produced by many tumor cells. Its 13 N-terminal amino acid sequence is highly homologous to that of parathyroid hormone (PTH). PTHrP binds to and functions through the type 1 PTH receptor (PTH1R) (1). PTHrP has endocrine, autocrine, paracrine, and intracrine actions (2,3). PTHrP was originally identified and characterized from cancers that caused hypercalcemia (3,4). There is strong evidence that PTHrP is associated with the growth and progression of important tumors such as breast cancer (5), lung cancer (6), prostate cancer (7) and renal cancer (8). However, how PTHrP promotes the growth and progression of tumor cells is unclear.
Bone is the most frequent site for breast cancer metastases (9). Breast cancer skeletal metastases promote osteoclast formation and activation and osteolysis and also inhibit osteoblast function and prevent new bone formation, which together cause severe complications such as bone pain, pathological fractures, spinal cord compression, and hypercalcemia of malignancy (9). Current clinical strategies are primarily focused on inhibiting the tumor-induced osteoclastic osteolysis (9,10). While osteoclast inhibitors can improve the quality of life in patients, therapies that restore osteoblast function and bone formation are critical for achieving a more complete repair of osteolytic lesions (9). Therefore, it is crucial to define the mechanisms whereby the tumor cells inhibit osteoblast function and bone formation, which is an underexplored area of this field.
In this study, using a combination of in vitro and in vivo strategies, we demonstrate that PTHrP produced by MDA-MB-231 breast cancer cells plays critical roles in promotion of tumor growth and survival and mediates the tumor-induced inhibition of osteoblast differentiation and bone formation.
MATERIALS AND METHODS
Reagents
Tissue culture media and fetal bovine serum were obtained from Thermo Scientific HyClone (Logan, Utah). Ascorbic acid was purchased from Sigma Aldrich (St. Louis, MO), recombinant human PTHrP (Cat#: 100-09) from Peprotech (Hamburg, Germany), rabbit anti-PTHrP (1-34) neutralizing antibody (T-4512) from Bachem (Torrance, CA), and normal rabbit IgG (SC2027) from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). All other chemicals were of analytical grade.
Ethical statement
All animal research protocols reported in this study were approved by the Institutional Animal Care and Use Committee (IACUC) of the Nankai University.
PTHrP knockdown in MDA-MB-231 breast cancer cells and infection
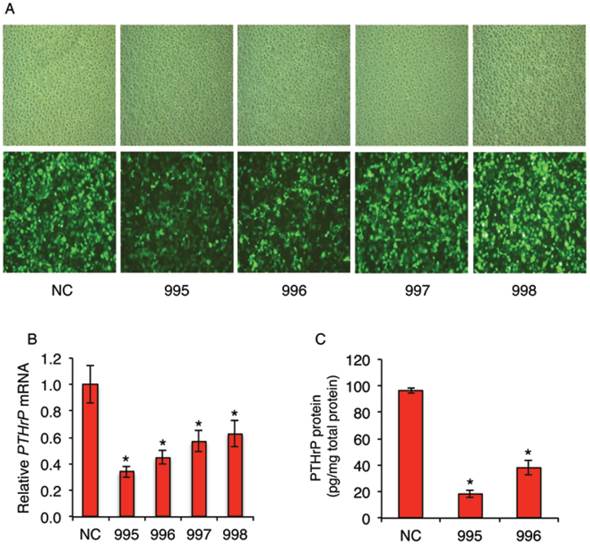
Recombinant lentiviral vectors expressing PTHrP shRNAs were constructed. Four PTHrP-shRNA vectors (PTHLH-homo-465 (995), PTHLH-homo-652 (996), PTHLH-homo-571 (997) and PTHLH-homo-515 (998)) with the following sequences were designed: 995: GGA AGT CCA TCC AAG ATT TAC; 996: GGT GGA GAC GTA CAA AGA GCA; 997: GCC CTC TCC CAA CAC AAA GAA; and 998: GCA GAA ATC CAC ACA GCT GAA. The negative control (NC) vector, which expresses a non-targeting sequence (TTC TCC GAA CGT GTC ACG T), was used as control throughout this study. GFP is expressed in the lentivirus-infected cells to monitor infection efficiency. For infection, the MDA-MB-231 cells were seeded at a density of 3x105 per well in a six-well plate in α-MEM plus 10% fetal bovine serum. LV-PTHrP-shRNAs or LV-NC-shRNA were introduced into cells using polybrene (8 μg/ml) for 24h. Cells were then selected and maintained in the same media containing 0.5 μg/ml puromycin. High infection efficiency was observed under a fluorescence microscopy (Figure 1A, bottom). qPCR analysis revealed that the levels of PTHrP mRNA were significantly decreased by all four PTHrP shRNAs in MDA-MB-231 cells (Figure 1B). ELISA assays confirmed that the levels of PTHrP protein were dramatically decreased in 995 and 996 cells compared to those of the NC cells (Figure 1C).
Gene expression studies
RNA isolation and reverse transcription (RT) were previously described (11). Quantitative real-time RT-PCR (qPCR) analysis was performed to measure the relative mRNA levels using SYBR Green kit (Bio-Rad Laboratories Inc., Hercules, CA) (12). Melting curve analysis was used to confirm the specificity of the PCR products. Four samples were run for each primer set. The levels of mRNAs were calculated by the ddCt method. Samples were normalized to Gapdh expression. The DNA sequences of human and mouse primers used for qPCR were summarized in Tables 1 and 2, respectively. Western blot analysis was performed as previously described (13,14). Antibodies used were from the following sources: antibodies against cyclins A1 and D3 and anti-mouse antibodies conjugated with horseradish peroxidase from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); antibodies against LC3-I /II (ab58610), Beclin1 (ab137355), cyclin D1 (ab16663) and Bcl-2 (ab7973) from Abcam (Cambridge, MA), anti-P62 antibody (#PM045) from MBL International (Woburn, MA), antibody against caspase-8 (D35G2) (#4790), and mouse monoclonal antibody against β-actin from Sigma Aldrich (St. Louis, MO).
human qPCR primers
| Name | 5' primer | 3' primer |
|---|---|---|
| ACTIN | CGGGAAATCGTGCGTGACAT | ATCTTCATTGTGCTGGGTGCC |
| GAPDH | TCCACCACCCTGTTGCTGTA | ACCACAGTCCATGCCATCA |
| PTHrP | GGAAGCAACCAGCCCACCAG | ACCGCGTAGCTCAGCAGGAA |
mouse qPCR primers
| Name | 5' primer | 3' primer |
|---|---|---|
| Alp | TCCCACGTTTTCACATTCGG | CCCGTTACCATATAGGATGGCC |
| Bsp | AAGAGGAAGAAAATGAGAACGA | GCTTCTTCTCCGTTGTCTCC |
| Gapdh | CAGTGCCAGCCTCGTCCCGTAGA | CTGCAAATGGCAGCCCTGGTGAC |
| Ocn | AGGGAGGATCAAGTCCCG | GAACAGACTCCGGCGCTA |
| Osx | AGAGGTTCACTCGCTCTGACGA | TTGCTCAAGTGGTCGCTTCTG |
| Runx2 | TAAAGTGACAGTGGACGGTCCC | TGCGCCCTAAATCACTGAGG |
Cell growth assay, flow cytometry cell cycle analysis, and in situ apoptosis detection
Tumor cell growth was measured by the CCK assays using the Cell Counting Kit-8 (CK04) from the Dojindo Molecular Technologies (Gaithersburg, MD) as previously described (15). The absorbance was recorded at 490 nm using a 96-well plate reader according to the manufacturer's instruction. For cell cycle analysis, cells were harvested and washed with 1xPBS, fixed with 75% ethanol and labeled with propidium iodide (PI), followed by flow cytometry using a BD FACSCalibur flow cytometer (Becton Dicknson, USA) as described (16). Cell survival was evaluated using the ApopTag Peroxidase In Situ Apoptosis Detection Kit according to the manufacturer's instruction (Millipore, Billerica, MA), which was based on the classical TUNEL assay to examine apoptosis by detecting DNA fragmentation (17). Apoptotic tumor cells were counted and normalized to the total tumor cells from the same area.
ELISA assays
The levels of PTHrP in the MDA-MB-231 cell cultures were measured using a kit from the USCN Life Science Inc. (Cat: E90819Hu) (Wuhan, China) according to the manufacturer's instruction.
Intratibial injection of nude mouse model and Immunohistochemistry (IHC)
For intratibial injection, both left and right tibiae of male nude mice (4 weeks of age, 10 mice or 20 tibiae/group) were injected with 1x106 NC, 995 or 996 cells in 20 μl PBS. Four weeks later, mice were sacrificed and injected tibiae were harvested. For bone histology, all specimens were fixed in 10% formalin at 4 oC for 24 h, decalcified in 10% EDTA (pH 7.4) for 10-14d, and embedded in paraffin. Sections of tibiae were stained with the indicated antibodies using the EnVision+System-HRP (DAB) kit (Dako North America, Inc, Carpinteria, CA) as described previously (18,19). Briefly, slides were baked at 55°C for 45 min, deparaffinized in three washes of xylene, and rehydrated in a decreasing ethanol gradient. Antigen retrieval was performed using 0.1% trypsin for 10 min at 37°C in a humidified chamber. Endogenous peroxidases were deactivated with 3% H2O2 in 1x PBS for 10 min, and sections were blocked in blocking solution for 30 min at room temperature. Sections were incubated with primary antibody (1:200 dilution for Osterix) in blocking solution for 2h at 4°C. Sections were washed in PBS three times and incubated with a donkey-anti-rabbit IgG-HRP secondary antibody solution for 30 min at room temperature. After washing with PBS three times, HRP activity was detected using a DAB substrate solution at room temperature. Sections were counter-stained with a Mayer's hematoxylin solution.
In vitro and in vivo osteoblast differentiation and bone formation assays
Isolation of mouse bone marrow stromal cells (BMSC) was described previously (20). Briefly, mice were sacrificed and tibiae and femurs were isolated and the epiphyses were cut. Marrow was flushed with DMEM containing 20% FBS and 1% penicillin/streptomycin into a 100-mm dish and the cell suspension was aspirated up and down with a 20-gauge needle in order to break clumps of marrow. The cell suspension (marrow from 2 mice/flask) was then cultured in a T75 flask in the same media. After 10d, cells reached confluency and were ready for experiments. MC-4 cells or BMSCs were differentiated as described previously (21). Briefly, cells were cultured in osteoblast differentiation media (complete α-MEM containing 50 µg/ml L-ascorbic acid and 2.0 mM β-glycerophosphate). Media were changed every 2d. At the indicated times, cells were used for RNA isolation and qPCR analysis or Alizarin red staining. ALP assay was performed as previously described (14). Briefly, BMSCs were differentiated for 7d and harvested in 1x Passive Buffer (Promega, Madison, WI). Lysates were clarified by centrifugation (20 min, 13,000 x g, 4°C). Five µl of cell extracts were added to each well (96-well plate) containing 150 µl p-nitrophenyl phosphate at 37 oC for 10-60 min depending on the ALP activity in the extracts. ALP activity was determined by absorbance measurement at 405 nm on a 96-well plate reader. ALP activity was normalized to total protein.
The measurement of mineral apposition rate (MAR) was conducted as previously described (22). Briefly, mice were double injected with calcein subcutaneously (20 mg/kg) at 6 and 2 d before sacrifice. Undecalcified tibiae were fixed in 70% ethanol, embedded in methylmethacrylate and sectioned at 10 μm. Calcein labeling was visualized using a Nikon E800 fluorescence microscope. The metaphyseal trabecular bones projected into the marrow space were evaluated and the distance between all double-labeled bands was measured at a magnification of 200x. MAR was calculated as mean distance between the double labels divided by the number of the days between the calcein injections. Histomorphometric analysis was performed using an Image Pro Plus 7.0 software (Rockville, MD).
Statistical Analysis
Data were analyzed with a GraphPad Prism software (4.0) (San Diego, CA). A one-way ANOVA analysis was used, followed by the Tukey test. Results were expressed as means ± standard deviation (SD). Differences with a P <0.05 was considered as statistically significant. All in vitro experiments were repeated 2-5 times and similar results were obtained.
RESULTS
Knocking down PTHrP expression inhibits the formation of subcutaneous tumors in nude mice
Although PTHrP is highly expressed in many breast tumor cells, direct effect of PTHrP knockdown in breast cancer cells on tumor growth and progression was not examined. For this reason, we stably knocked down PTHrP expression in MDA-MB-231 cells, a widely used human breast cancer cell line, using lentiviral vectors expressing four shRNAs (995, 996, 997 and 998) that target different regions of the human PTHrP mRNAs or a negative control shRNA (NC) (Figure 1A). Results from qPCR analysis showed that the levels of PTHrP mRNA in the 995-, 996-, 997-, and 998-infected cells were reduced by 66%, 55%, 43% and 37%, respectively, compared to the NC-infected cells (Figure 1B). ELISA assays confirmed that the levels of PTHrP proteins were decreased by 82% and 57% in 995 and 996 cells, respectively, compared to NC cells (Figure 1C). The NC, 995 and 996 cells were used for subsequent experiments in this study.
shRNA knockdown of PTHrP expression in MDA-MB-231 breast cancer cells. (A-C) Lentiviral infection. Human MDA-MB-231 breast cancer cells were infected with lentiviral vectors expressing PTHrP shRNAs that target four different regions of the human PTHrP mRNA (referred to as 995, 996, 997 and 998) or a negative control shRNA (NC). After infection, cells were selected and maintained in culture media containing 0.5 μg/ml puromycin. Representative light (A, top) and GFP fluorescence (A, bottom) microscopic images of each group are shown. The effects of PTHrP knockdown were determined by qPCR (B) and ELISA (C) analyses. mRNA levels were normalized to GAPDH mRNA. PTHrP protein levels were normalized to the total protein in the media. The NC, 995 and 996 cells were used for all subsequent experiments in this study. *P < 0.05, versus NC.
Knocking down PTHrP inhibits subcutaneous tumor growth in nude mice. (A-D) 1x106 tumor cells (NC, 995 or 996) were subcutaneously injected into nude mice (6 mice/group). Pictures of subcutaneous tumor tissues (see arrows) from each group are shown at sacrifice (d44) (A and B). Average tumor weights from each group were measured (C). Tumor volumes were measured at the indicated time points as described (37) (D). Briefly, tumor volumes were determined by measuring the major (L) and minor (W) diameters with an electronic caliper. The tumor volume was calculated according to the following formula: tumor volume = π/6 × L × W2. *P < 0.05, versus NC.
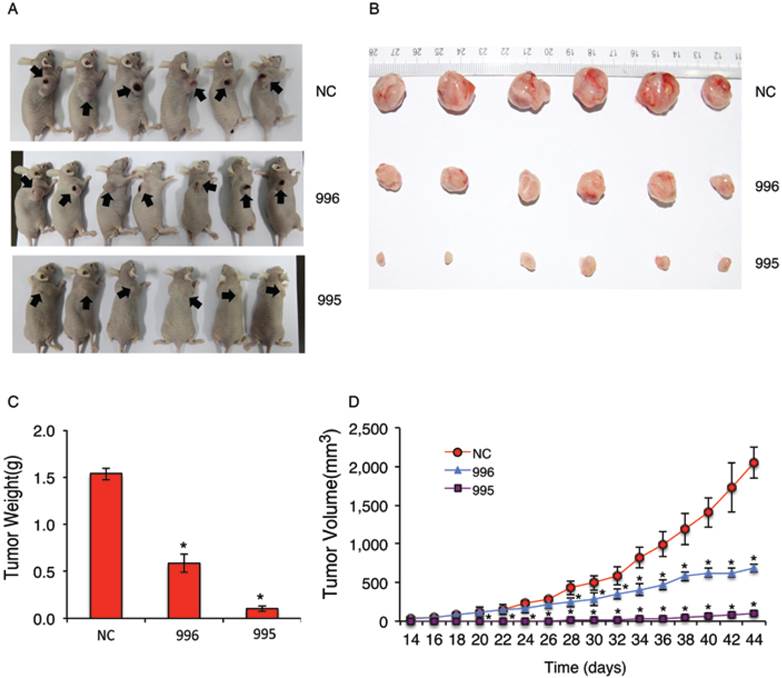
We next examined the effect of PTHrP knockdown on tumor growth in vivo by establishing subcutaneous xenografts in nude mice. As can be seen from Figure 2, A and B, subcutaneous tumors formed by 995 or 996 cells were much smaller than those formed by NC cells. Average tumor weight at sacrifice (44d) was reduced by 94% and 62% in 995 and 996 relative to NC group, respectively (Figure 2C). Knocking down PTHrP expression decreased the growth of subcutaneous tumors over time (Figure 2D).
Knocking down PTHrP arrests the cell cycle progression
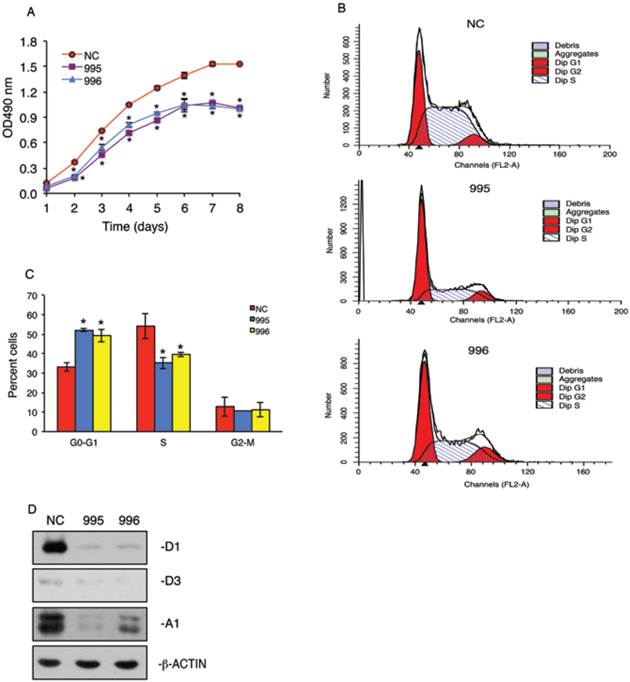
In vitro growth of 995 and 996 cells was significantly reduced compared to that of NC cells in a time-dependent manner (Figure 3A). To determine whether the tumor-derived PTHrP is required for the cell cycle progression, we performed flow cytometric analysis to compare the cell distribution across different phases of the cell cycle among NC, 995 and 996 cells. As shown in Figure 3, B and C, 995 and 996 cells showed significant decreases in the cell distribution into the S phase when compared with values of NC cells. This decrease in the S-phase cell population was accompanied by a concomitant increase in the G0/G1 phase cell populations. In contrast, the fraction of the cells in the G2-M phase was not altered by PTHrP knockdown. Western blot analysis showed that cyclin D1 protein expression, a major driver of the cell cycle progression, was essentially abolished in both 995 and 996 cells (Figure 3D). The expression of cyclin A1 was also markedly decreased in 995 and, to a less extent, 996 cells compared to that of NC cells (Figure 3D). The expression level of cyclin D3 was low and not significantly altered by the PTHrP down-regulation (Figure 3D).
Knocking down PTHrP up-regulates Beclin1 and LC-3-II expression and promotes formation of autophagosome-like vesicles in MDA-MB-231 cells
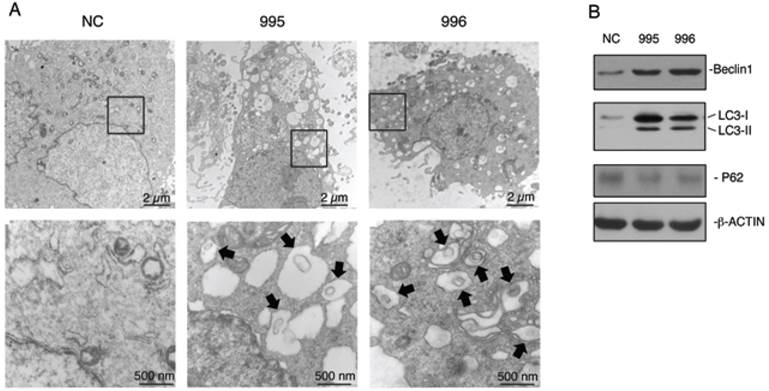
Autophagy is an important survival mechanism, which is activated in cells in response to stress signals such as starvation, growth factor deprivation, ER stress, and pathogen infection (23,24). We examined whether PTHrP knockdown activates autophagy. The formation of double-membrane autophagosomes is central to autophagy, which is responsible for delivering cytoplasmic materials to lysosomes and eventually degrading damaged organelles and proteins (24). Analysis of NC, 995 and 996 cells using a transmission electron microscope (TEM) revealed that PTHrP knockdown resulted in the formation of a number of autophagosome-like double-membrane vesicles, which contained damaged organelles such as mitochondria (see arrows) (Figure 4A). In contrast, the autophagosome-like vesicles were hardly observed in NC cells. We further found that Beclin1, a key autophagy protein, was up-regulated in both 995 and 996 cells (Figure 4B). Furthermore, LC3-II protein, another critical autophagy factor, was undetectable on Western blots in NC cells, which was dramatically up-regulated in both 995 and 996 cells. Although P62 is known to be involved in autophagy activation, its protein level was not altered by the PTHrP knockdown (Figure 4B).
PTHrP knockdown arrests cell cycle progression. (A) Tumor cell growth in vitro. 1x103 tumor cells (NC, 995 or 996)/well were seeded on a 96-well plate, followed by the CCK-8 assay at the indicated times. *P < 0.05, versus NC. (B and C) 5x105 NC, 995 or 996 cells were cultured in proliferation media for 24h and labeled with propidium iodide. Cell population of different cell cycle phases was then measured by flow cytometry using 488 nm excitation and 630 nm emission. Quantitative data from 3 independent experiments were presented in C. *P <0.01, versus NC. (D) Whole cell extracts from NC, 995 and 996 cells were isolated for Western blot analysis for expression of cyclins D1, D3 and A1. β-ACTIN was used as a loading control.
Knocking down PTHrP expression activates autophagy. (A) Analysis of autophagy using transmission electron microscope (TEM). NC, 995 or 996 cells were rinsed with PBS, fixed and processed for TEM analysis as described (38). Representative electron microscopic images with lower magnification images (12000X, top) and higher magnification images of the boxed areas (60000X, bottom) are shown. Arrows indicate autophagosome-like double-membrane vesicles, which contain damaged organelles. (C) Western blot analysis. Whole cell extracts from NC, 995 or 996 cells were used for expression of Beclin1, LC3 and P62. β-ACTIN was used as a loading control.
Knocking down PTHrP accelerates the cleavage of Caspase-8 protein and dramatically induces apoptosis in MDA-MB-231 cells
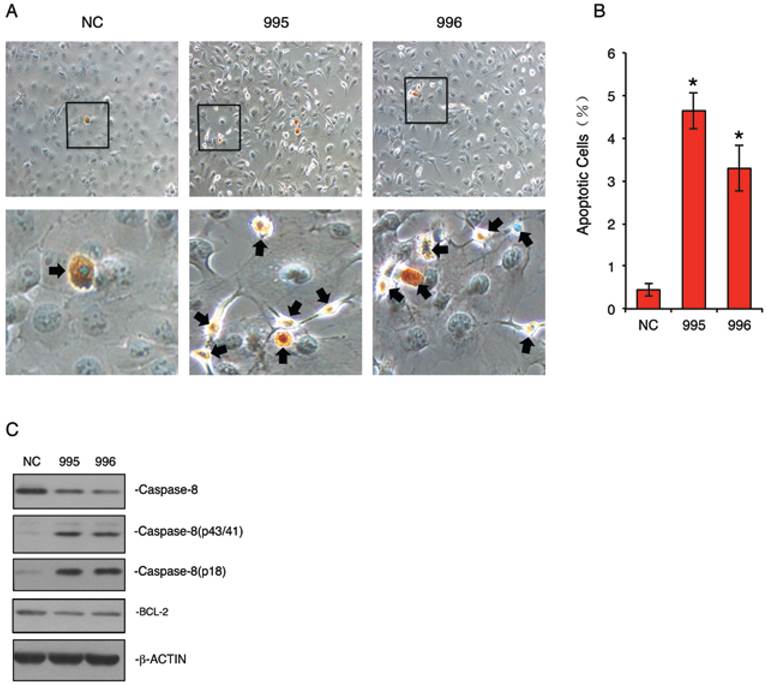
Autophagy can be switched to apoptosis under certain conditions (24-27). We next examined whether knocking down PTHrP expression induces apoptosis in MDA-MB-231 cells. NC, 995 and 996 cells were stained using the ApopTag Peroxidase In Situ Apoptosis Detection Kit, a modified TUNEL staining that measures DNA fragmentation in situ (19). As shown in Figure 5, A and B, the percent apoptotic cells in 995 and 996 cells were increased by 11.7- and 6.4-fold, respectively, when compared to NC cells (P<0.01, NC vs. 995 or 996). Western blot analysis showed that levels of the cleaved forms of Caspase 8 were markedly increased in 995 and 996 cells, which was accompanied by a concomitant decrease in the uncleaved form (Figure 5C). In contrast, the level of BCL-2 protein was not markedly altered by the PTHrP knockdown (Figure 5C).
Knocking down PTHrP reduces the tumor inhibition of osteoblast differentiation in vitro
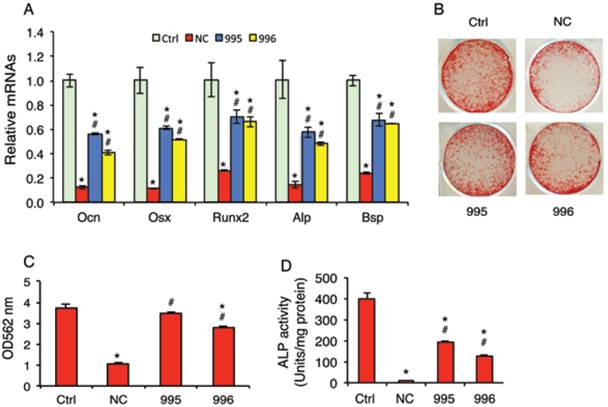
Although breast cancer skeletal metastases can inhibit osteoblast function and prevent new bone formation, the underlying mechanisms are poorly understood. We investigated whether the tumor-derived PTHrP plays a role in the osteoblast inhibition. In vitro studies using the mouse MC-4 preosteoblastic cells treated with or without conditioned media (CM) from NC (NC-CM), 995 (995-CM) or 996 cells (996-CM) showed that the mRNA levels of osteoblast differentiation marker genes including those encoding osteocalcin (Ocn), Osterix (Osx), Runx2, alkaline phosphatase (Alp) and bone sialoprotein (Bsp) were all significantly decreased by NC-CM (Figure 6A). Although 995-CM and 996-CM still inhibited osteoblast gene expression, the magnitude of inhibition was significantly decreased by the PTHrP knockdown. Likewise, the tumor-induced inhibition of osteoblast-mediated mineralization was significantly reduced in 995-CM- or 996-CM- versus NC-CM-treated MC-4 cultures (Figure 6, B and C). Down-regulation of PTHrP expression reduced the tumor-induced inhibition of Alp activity in primary bone marrow stromal cells (BMSC) (Figure 6D).
Knocking down PTHrP decreases the tumor inhibition of osteoblast differentiation and bone formation in vivo
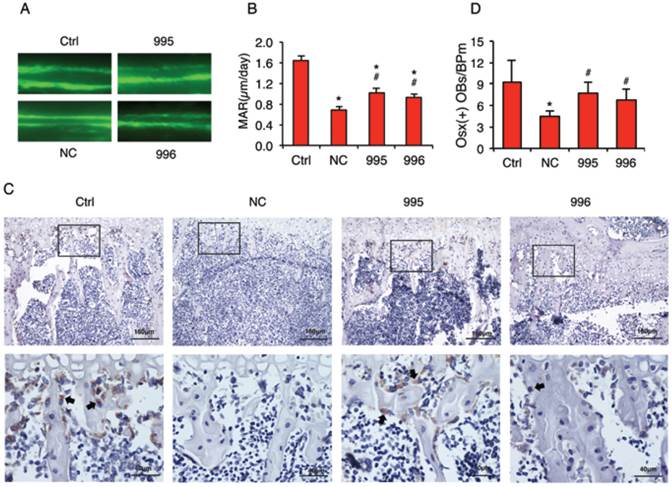
In vivo calcein double labeling studies showed that intratibial injection of NC cells decreased the mineral apposition rate (MAR), an in vivo indicator of the osteoblast-mediated bone formation, which was attenuated by PTHrP knockdown (Figure 7, A and B). To investigate whether the tumor-derived PTHrP plays a role in the tumor-induced inhibition of osteoblast differentiation in bone, we analyzed the expression of Osterix (Osx) protein, a critical transcription factor for osteoblast formation and maturation, by performing immunohistochemical (IHC) staining of tibial sections from each group. The numbers of Osx-positive cells (see arrows) (i.e., differentiating and differentiated osteoblasts) located on trabecular bone surfaces were counted and normalized to trabecular bone perimeter (Osx (+) OB.Nb/BPm). As can be seen from Figure 7C, Osx-positive osteoblasts were identified on all surfaces throughout the trabeculae of tibiae injected with PBS (Ctrl), which was decreased in tibiae injected with NC cells compared to that in tibiae injected with PBS (PBS: 9.2±3.2; NC: 4.7±0.7, P<0.05, PBS versus NC). Importantly, Osx (+) OB.Nb/BPm was significantly increased in tibiae in 995- or 996-injected tibiae compared to that in tibiae injected with NC cells (995: 7.7±1.6; 996: 6.8±1.6, P<0.05, 995 or 996 versus NC) (Figure 7D). In addition, the relative intensity of Osx signal per osteoblast was dramatically reduced in NC versus PBS-injected tibiae, which was markedly increased in the 995- or 996- versus NC-injected tibiae.
Knocking down PTHrP expression induces apoptosis. (A and B) TUNEL staining. NC, 995 or 996 cells were seeded at 105 cells/well in 8-well chamber, cultured in proliferation media for 2d and stained using the ApopTag Peroxidase In Situ Apoptosis Detection Kit according the manufacturer's instruction. Representative lower magnification images and higher magnification images of the boxed areas are shown. Arrows indicate apoptotic cells. Quantitative data from 3 independent experiments were shown in A. *P<0.01 (vs. NC) (C) Western blot analysis. Whole cell extracts from NC, 995 and 996 cells were subject to Western blotting for expression of Caspase-8 and BCL-2. β-ACTIN was used as a loading control.
PTHrP knockdown decreases the tumor-induced inhibition of osteoblast differentiation in vitro. (A-C) MC-4 preosteoblastic cells were cultured in osteoblast differentiation media for 7d (qPCR analysis) (A) or 12d (Alizarin red staining) (B). Cells were treated with and without NC-CM, 995-CM or 996-CM for the last 48h. mRNAs were normalized to Gapdh mRNA. Alizarin red was extracted with 10% cetylpyridinium chloride and the dye concentration in the extracts was determined at OD 562 nm. (C). Bars represent means ± S.D. from three independent experiments. (D) Primary BMSCs were cultured in osteoblast differentiation media for 7d and treated with and without conditioned media (CM) from NC (NC-CM), 995 (995-CM) or 996 cells (996-CM) for the last 48h, followed by ALP (alkaline phosphatase) activity assay. ALP activity was normalized to total protein.
PTHrP knockdown decreases the tumor-induced inhibition of osteoblast differentiation and bone formation in vivo. (A and B) Calcein double labeling of metaphyseal trabecular bone. This assay measures the osteoblast-mediated bone formation. 1x106 NC, 995 or 996 cells in 20 μl PBS or 20 μl PBS (Ctrl) alone were injected into both left and right tibiae of 4-weeks-old male nude mice (10 mice or 20 tibiae per group). Four weeks later, mice were sacrificed. Sections of undecalcified tibiae (6 tibiae/group) were used for the MAR assay. Magnification: x200, *P<0.05 (vs. Ctrl), #P<0.05 (versus NC). (C and D) IHC staining. Five-μm sections of decalcified tibiae (6 tibiae/group) were immunohistochemically stained with an anti-Osx antibody. Representative lower magnification images (100x, top) and higher magnification images of the boxed areas (400x, bottom) are shown. Arrows in C indicate the Osx-positive osteoblasts that were stained brown. The numbers of Osx-positive cells located on trabecular bone surfaces were counted and normalized to trabecular bone perimeter (Osx (+) OBs/BPm) using an Image Pro Plus 7.0 software (G). *P <0.05 (vs. Ctrl), #P <0.05 (vs. NC).
PTHrP activates its own expression in MDA-MB-231 cells
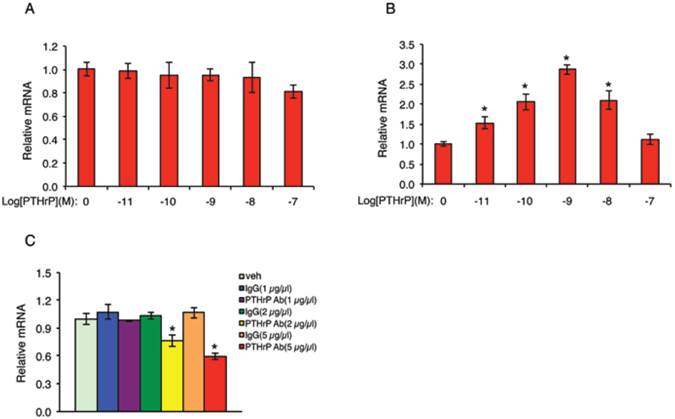
The fact that PTHrP knockdown inhibits the growth of MDA-MB-231 cells suggests that PTHrP may auto-regulate its own expression in these cells. To test this interesting possibility, NC cells were treated with increasing concentrations of PTHrP for 6h, followed by qPCR for PTHrP mRNA. Results showed that addition of human recombinant PTHrP protein in the cultures did not increase endogenous PTHrP mRNA level in NC cells (Figure 8A). We reasoned that this could be due to a high endogenous PTHrP expression in NC cells (Figure 1, B and C), which may mask the stimulatory effect of the exogenous PTHrP. To test if this is the case, we next performed the same experiment in 995 cells, in which endogenous PTHrP expression was knocked down (Figure 1, B and C). Indeed, exogenous PTHrP dramatically increased PTHrP mRNA expression in these cells in a dose-dependent manner with a maximal stimulation at 10-9 molar concentration of PTHrP (Figure 8B). To further confirm whether endogenous PTHrP expression is required for its own expression, NC cells were treated with increasing concentrations of an anti-PTHrP neutralizing antibody or equal amounts of a control IgG. Results showed that the levels of PTHrP mRNA were dose-dependently reduced by the anti-PTHrP neutralizing antibody but not control IgG (Figure 8C).
PTHrP auto-activates its own expression in MDA-MB-231 cells. (A and B) NC (A) and 995 (B) cells were treated with the indicated concentrations of PTHrP for 6h. Total RNA was isolated for qPCR analysis for PTHrP mRNA. Samples were normalized to β-ACTIN mRNA. *P<0.01 (vs. 0 µm PTHrP). (C) NC cells were treated with the indicated concentrations of an anti-PTHrP neutralizing antibody or control IgG for 6h. Total RNA was isolated for qPCR analysis for PTHrP mRNA, which was normalized to β-ACTIN mRNA. *P<0.01 (vs. veh).
DISCUSSION
PTHrP is a secreted factor that is produced by a number of normal and tumor cells. PTHrP is critical for skeletal development, placental calcium transport, smooth muscle relaxation, mammary gland, teeth and skin, and pancreas development as well as the growth and progression of multiple tumors through endocrine, autocrine, paracrine, and intracrine mechanisms (2,3,7,8,28-30). In the present study, we use a combination of in vitro and in vivo strategies and demonstrate that PTHrP expression in MDA-MB-231 breast cancer cells plays a direct role in driving the tumor growth and survival as well as the tumor inhibition of osteoblast differentiation and bone formation.
Autophagy is a cellular process that is used by cells to degrade damaged organelles, cell membranes and proteins. Autophagy is critical for cell survival in response to starvation, growth factor deprivation, ER stress, and pathogen infection (23,24). In the present study we found that down-regulation of PTHrP in MDA-MB-231 cells activated autophagy as demonstrated by the formation of autophagosomes, which is central for autophagy, and increased expression of Beclin1 and LC3-II. It should be noted that although P62 is known to be involved in autophagy, its expression level was not altered by the PTHrP knockdown. This would suggest that distinct components of autophagy pathway are activated by the PTHrP knockdown. Increased autophagy could represent a survival mechanism for the tumor cells in response to the PTHrP down-regulation. In addition, there is an accumulation of evidence that autophagy can be switched to cell apoptosis under certain condictions (31-34). Interestingly, we found that knocking down PTHrP expression significantly induced apoptosis in MDA-MB-231 cells as demonstrated by a dramatic increase of TUNEL-positive cells and accelerated cleavage of Caspase-8.
The cell cycle includes a series of molecular events, which leads to cell division and duplication. The cell cycle has four distinct phases: G1, S (synthesis), G2 (interphase) and M (mitosis) phases. In addition, there is a G0 phase in which cells are in a quiescent state. We found in this study that PTHrP down-regulation decreased the cell distribution into the S phase but increased the cell distribution in G1 phase. This suggests a G1 arrest. PTHrP modulates the cell cycle progression through, at least in part, control of cyclins D1 and A1 expression, both major drivers of cell cycle progression. Accelerated cell apoptosis and decelerated cell cycle progression could together contribute to the reduction of tumor growth and progression of the subcutaneous tumors observed in this study.
In addition to the osteoclast-mediated osteolysis, breast cancers also inhibit osteoblast function and prevent new bone formation. Previous studies have been primarily focused on the mechanisms whereby the breast tumors activate osteoclast formation and activation and cause osteolytic lesions. In contrast, much less attention was paid to how the tumors impact osteoblast function and bone formation. In the present study we found that PTHrP expression in the breast cancer cells plays a critical role in tumor inhibition of osteoblast differentiation and bone formation in vitro and in bone. It should be noted that down-regulation of PTHrP did not completely restore the defects in osteoblast differentiation and bone formation, indicating that other factor(s) are also involved in this regulation. Dickkopf-related protein 1 (DKK1) and sclerostin, both Wnt antagonists, were reported to inhibit osteoblast function and bone formation in breast cancer bone metastases (35,36).
Our finding that PTHrP auto-regulates its own expression is of particular interest. It suggests that the breast cancer cells can promote their own growth by expressing PTHrP via a positive feedback mechanism involving autocrine. It is interesting to examine if this mechanism also exists in other PTHrP-producing tumors. It should be noted that PTHrP at higher concentration (e.g., 10-7 M) failed to activate its own expression in the MDA-MB-231 cells under our experimental conditions. This could be because PTHrP at higher concentration desensitizes its receptor (PTH1R). This finding also suggests that the tumor-derived PTHrP promotes the growth of tumors in vivo only when the protein is produced at a proper level.
In summary, we demonstrate that the tumor-derived PTHrP drives the breast cancer growth and progression and plays a major role in the tumor inhibition of osteoblast differentiation and bone formation. Li and coworkers recently showed that ablation of the Pthrp gene in the mammary epithelium of the PyMT-MMTV breast cancer mouse model caused a delay in primary tumor initiation and progression and reduced metastasis to distal sites such as lung (6). Collectively, these results along with our finding from the present study suggest that targeting PTHrP in breast cancer and probably other PTHrP-expressing tumors could be a potential therapeutic strategy for blocking tumor growth and progression in cancer patients, especially those with skeletal metastases.
Acknowledgements
We wish to thank Qiang Zhao, Xi Zhao and Shuai Li of Nankai University for technical support. This work was supported by a Chinese Ministry of Science and Technology grant (2009CB918902) and by a US National Institutes of Health grant (AR059647).
Author Contributions
Study design: GX and LZ. Study conduct and data collection: LZ, GX, KZ, HJ, ZZ, LZ, and ML. Data interpretation: GX, LZ, WD, ZY and DC. Drafting manuscript: GX. GX takes the responsibility for the integrity of the data analysis.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gensure RC, Gardella TJ, Juppner H. Parathyroid hormone and parathyroid hormone-related peptide, and their receptors. Biochem Biophys Res Commun. 2005;328:666-78 doi:10.1016/j.bbrc.2004.11.069
2. Tovar Sepulveda VA, Shen X, Falzon M. Intracrine PTHrP protects against serum starvation-induced apoptosis and regulates the cell cycle in MCF-7 breast cancer cells. Endocrinology. 2002;143:596-606
3. McCauley LK, Martin TJ. Twenty-five years of PTHrP progress: from cancer hormone to multifunctional cytokine. J Bone Miner Res. 2012;27:1231-9 doi:10.1002/jbmr.1617
4. Broadus AE, Mangin M, Ikeda K, Insogna KL, Weir EC, Burtis WJ. et al. Humoral hypercalcemia of cancer. Identification of a novel parathyroid hormone-like peptide. The New England journal of medicine. 1988;319:556-63 doi:10.1056/NEJM198809013190906
5. Kozlow W, Guise TA. Breast cancer metastasis to bone: mechanisms of osteolysis and implications for therapy. Journal of mammary gland biology and neoplasia. 2005;10:169-80 doi:10.1007/s10911-005-5399-8
6. Li J, Karaplis AC, Huang DC, Siegel PM, Camirand A, Yang XF. et al. PTHrP drives breast tumor initiation, progression, and metastasis in mice and is a potential therapy target. J Clin Invest. 2011;121:4655-69 doi:10.1172/JCI46134
7. Kremer R, Goltzman D, Amizuka N, Webber MM, Rhim JS. ras Activation of human prostate epithelial cells induces overexpression of parathyroid hormone-related peptide. Clin Cancer Res. 1997;3:855-9
8. Agouni A, Sourbier C, Danilin S, Rothhut S, Lindner V, Jacqmin D. et al. Parathyroid hormone-related protein induces cell survival in human renal cell carcinoma through the PI3K Akt pathway: evidence for a critical role for integrin-linked kinase and nuclear factor kappa B. Carcinogenesis. 2007;28:1893-901 doi:10.1093/carcin/bgm106
9. Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2:584-93 doi:10.1038/nrc867
10. Chirgwin JM, Guise TA. Skeletal metastases: decreasing tumor burden by targeting the bone microenvironment. J Cell Biochem. 2007;102:1333-42 doi:10.1002/jcb.21556
11. Yu S, Jiang Y, Galson DL, Luo M, Lai Y, Lu Y. et al. General transcription factor IIA-gamma increases osteoblast-specific osteocalcin gene expression via activating transcription factor 4 and runt-related transcription factor 2. J Biol Chem. 2008;283:5542-53
12. Yang S, Xu H, Yu S, Cao H, Fan J, Ge C. et al. Foxo1 mediates insulin-like growth factor 1 (IGF1)/insulin regulation of osteocalcin expression by antagonizing Runx2 in osteoblasts. J Biol Chem. 2011;286:19149-58 doi:M110.197905 [pii]10.1074/jbc.M110.197905
13. Yu S, Franceschi RT, Luo M, Zhang X, Jiang D, Lai Y. et al. Parathyroid hormone increases activating transcription factor 4 expression and activity in osteoblasts: requirement for osteocalcin gene expression. Endocrinology. 2008;149:1960-8
14. Yu S, Zhu K, Lai Y, Zhao Z, Fan J, Im HJ. et al. ATF4 Promotes beta-Catenin Expression and Osteoblastic Differentiation of Bone Marrow Mesenchymal Stem Cells. International journal of biological sciences. 2013;9:256-66 doi:10.7150/ijbs.5898
15. Tanaka H, Kono E, Tran CP, Miyazaki H, Yamashiro J, Shimomura T. et al. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nature medicine. 2010;16:1414-20 doi:10.1038/nm.2236
16. Shi X, Zhao Y, Jiao Y, Shi T, Yang X. ROS-Dependent Mitochondria Molecular Mechanisms Underlying Antitumor Activity of Pleurotus abalonus Acidic Polysaccharides in Human Breast Cancer MCF-7 Cells. PLoS One. 2013;8:e64266. doi:10.1371/journal.pone.0064266
17. Zhang X, Yu S, Galson DL, Luo M, Fan J, Zhang J. et al. Activating transcription factor 4 is critical for proliferation and survival in primary bone marrow stromal cells and calvarial osteoblasts. J Cell Biochem. 2008;105:885-95
18. Yu S, Sharma R, Nie D, Jiao H, Im HJ, Lai Y. et al. ADAR1 ablation decreases bone mass by impairing osteoblast function in mice. Gene. 2013;513:101-10 doi:10.1016/j.gene.2012.10.068
19. Cao H, Yu S, Yao Z, Galson DL, Jiang Y, Zhang X. et al. Activating transcription factor 4 regulates osteoclast differentiation in mice. J Clin Invest. 2010;120:2755-66 doi:42106 [pii]10.1172/JCI42106
20. Jiang D, Franceschi RT, Boules H, Xiao G. Parathyroid Hormone Induction of the Osteocalcin Gene: REQUIREMENT FOR AN OSTEOBLAST-SPECIFIC ELEMENT 1 SEQUENCE IN THE PROMOTER AND INVOLVEMENT OF MULTIPLE SIGNALING PATHWAYS. J Biol Chem. 2004;279:5329-37
21. Xiao G, Cheng H, Cao H, Chen K, Tu Y, Yu S. et al. Critical role of filamin-binding LIM protein 1 (FBLP-1)/migfilin in regulation of bone remodeling. J Biol Chem. 2012;287:21450-60 doi:10.1074/jbc.M111.331249
22. Yu S, Franceschi RT, Luo M, Fan J, Jiang D, Cao H. et al. Critical role of activating transcription factor 4 in the anabolic actions of parathyroid hormone in bone. PLoS One. 2009;4:e7583
23. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annual review of genetics. 2009;43:67-93 doi:10.1146/annurev-genet-102808-114910
24. Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nature cell biology. 2007;9:1102-9 doi:10.1038/ncb1007-1102
25. Gump JM, Thorburn A. Autophagy and apoptosis: what is the connection?. Trends in cell biology. 2011;21:387-92 doi:10.1016/j.tcb.2011.03.007
26. Bhutia SK, Das SK, Azab B, Dash R, Su ZZ, Lee SG. et al. Autophagy switches to apoptosis in prostate cancer cells infected with melanoma differentiation associated gene-7/interleukin-24 (mda-7/IL-24). Autophagy. 2011;7:1076-7
27. Rikiishi H. Novel Insights into the Interplay between Apoptosis and Autophagy. International journal of cell biology. 2012;2012:317645. doi:10.1155/2012/317645
28. Vargas SJ, Gillespie MT, Powell GJ, Southby J, Danks JA, Moseley JM. et al. Localization of parathyroid hormone-related protein mRNA expression in breast cancer and metastatic lesions by in situ hybridization. J Bone Miner Res. 1992;7:971-9 doi:10.1002/jbmr.5650070814
29. Iwamura M, di Sant'Agnese PA, Wu G, Benning CM, Cockett AT, Deftos LJ. et al. Immunohistochemical localization of parathyroid hormone-related protein in human prostate cancer. Cancer Res. 1993;53:1724-6
30. Lu Y, Xiao G, Galson DL, Nishio Y, Mizokami A, Keller ET. et al. PTHrP-induced MCP-1 production by human bone marrow endothelial cells and osteoblasts promotes osteoclast differentiation and prostate cancer cell proliferation and invasion in vitro. Int J Cancer. 2007;121:724-33 doi:10.1002/ijc.22704
31. Luo S, Rubinsztein DC. Atg5 and Bcl-2 provide novel insights into the interplay between apoptosis and autophagy. Cell Death Differ. 2007;14:1247-50 doi:10.1038/sj.cdd.4402149
32. Bhutia SK, Dash R, Das SK, Azab B, Su ZZ, Lee SG. et al. Mechanism of autophagy to apoptosis switch triggered in prostate cancer cells by antitumor cytokine melanoma differentiation-associated gene 7/interleukin-24. Cancer Res. 2010;70:3667-76 doi:10.1158/0008-5472.CAN-09-3647
33. Jiang Q, Wang Y, Li T, Shi K, Li Z, Ma Y. et al. Heat shock protein 90-mediated inactivation of nuclear factor-kappaB switches autophagy to apoptosis through becn1 transcriptional inhibition in selenite-induced NB4 cells. Molecular biology of the cell. 2011;22:1167-80 doi:10.1091/mbc.E10-10-0860
34. Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571-80 doi:10.1038/cdd.2010.191
35. Bu G, Lu W, Liu CC, Selander K, Yoneda T, Hall C. et al. Breast cancer-derived Dickkopf1 inhibits osteoblast differentiation and osteoprotegerin expression: implication for breast cancer osteolytic bone metastases. Int J Cancer. 2008;123:1034-42 doi:10.1002/ijc.23625
36. Mendoza-Villanueva D, Zeef L, Shore P. Metastatic breast cancer cells inhibit osteoblast differentiation through the Runx2/CBFbeta-dependent expression of the Wnt antagonist, sclerostin. Breast cancer research: BCR. 2011;13:R106. doi:10.1186/bcr3048
37. Lin W, Zheng L, Zhuang Q, Zhao J, Cao Z, Zeng J. et al. Spica prunellae promotes cancer cell apoptosis, inhibits cell proliferation and tumor angiogenesis in a mouse model of colorectal cancer via suppression of stat3 pathway. BMC complementary and alternative medicine. 2013;13:144. doi:10.1186/1472-6882-13-144
38. Fong MY, Jin S, Rane M, Singh RK, Gupta R, Kakar SS. Withaferin A synergizes the therapeutic effect of doxorubicin through ROS-mediated autophagy in ovarian cancer. PLoS One. 2012;7:e42265. doi:10.1371/journal.pone.0042265
Author contact
![]() Corresponding author: Zhi Yao, Department of Immunology, Key Laboratory of Educational Ministry of China, Tianjin Medical University, Tianjin 300070, China, Phone: 0118613920771116; Email: yaozhi23com; Guozhi Xiao, Rm 518, Cohn Research Building, 1735 West Harrison Street, Chicago, IL 60612. Phone: 312-942-4879 (O); Fax: 312-942-3053; Email: Guozhi_Xiaoedu
Corresponding author: Zhi Yao, Department of Immunology, Key Laboratory of Educational Ministry of China, Tianjin Medical University, Tianjin 300070, China, Phone: 0118613920771116; Email: yaozhi23com; Guozhi Xiao, Rm 518, Cohn Research Building, 1735 West Harrison Street, Chicago, IL 60612. Phone: 312-942-4879 (O); Fax: 312-942-3053; Email: Guozhi_Xiaoedu
Received 2013-6-28
Accepted 2013-8-6
Published 2013-8-21