ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2014; 10(3):268-277. doi:10.7150/ijbs.7929 This issue Cite
Research Paper
Metformin Plays a Dual Role in MIN6 Pancreatic β Cell Function through AMPK-dependent Autophagy
Yingling Jiang1, 2*, Wei Huang1*, Jing Wang1, Zhipeng Xu1, Jieyu He1, Xiaohong Lin2, Zhiguang Zhou1, Jingjing Zhang1, ![]()
1. Metabolic Syndrome Research Center and Diabetes Center, Institute of Aging and Geriatric Diseases, Key Laboratory of Diabetes Immunology, Ministry of Education, Institute of Metabolism and Endocrinology, Second Xiangya Hospital, Central South University, Changsha, Hunan, China 410011
2. Endocrinology Department, Zhuzhou Central Hospital, Zhuzhou, Hunan, China 412007
* Y. Jiang and W. Huang contributed equally to this work.
Abstract
Metformin improves insulin sensitivity in insulin sensitive tissues such as liver, muscle and fat. However, the functional roles and the underlying mechanism of metformin action in pancreatic β cells remain elusive. Here we show that, under normal growth condition, metformin suppresses MIN6 β cell proliferation and promotes apoptosis via an AMPK-dependent and autophagy-mediated mechanism. On the other hand, metformin protects MIN6 cells against palmitic acid (PA)-induced apoptosis. Our findings indicate that metformin plays a dual role in β cell survival and overdose of this anti-diabetic drug itself may lead to potential β cell toxicity.
Keywords: metformin, pancreatic β cells, AMPK, autophagy, apoptosis
Introduction
Metformin, the first-line oral therapies for type 2 diabetes, has been used in clinical application since 1950s [1]. Metformin functions mainly in insulin-targeted tissues such as liver, muscle, and adipose tissues, and activation of the AMP-activated protein kinase (AMPK) by metformin suppresses hepatic glucose production and increases glucose utilization [1]. In addition to regulation of metabolism, metformin exerts a protective effect on cardiovascular disease [2] and various cancers [3]. However, metformin induces acute pancreatitis specifically in patients with renal insufficiency [4]. In addition, overactivation of AMPK by metformin impairs the ability of neurons to grow output stalks or axons in animal models [5] thus may exert in Alzheimer's disease [6]. Thus, further research is warranted to understand the complex nature of metformin action and the drug safety.
Despite extensive studies focusing on the functional roles of metformin in regulating insulin sensitivity and glucose homeostasis in insulin-targeted organs, how metformin functions in pancreatic β cells remains controversial. Metformin has been shown to restore insulin secretion under the conditions of chronic free fatty acid (FFA) exposure [7, 8]. In addition, this drug prevents glucose- and fructose-induced permeability transition pore (PTP, a mitochondrial channel) opening and hampers glucose-induced cell death [9]. However, a long-term exposure of MIN6 cells or primary rat β cells to metformin led to increased AMPK activity, reduced protein synthesis, and cell death [10]. Metformin has also been shown to inhibit mitochondrial complex I activity, leading to pancreatic β cell toxicity [11].
Autophagy removes damaged intracellular macromolecules and organelles and plays a protected role for cell survival [12] [13]. However, altered autophagy may trigger cell death in a non-apoptotic manner [14, 15], which is referred as “type 2 programmed cell death” [16]. Therefore, autophagy could either promote cell survival or trigger cells death depending on environmental conditions or cell types [17] [18]. Under specific conditions, autophagy is even interchangeable with apoptosis [19]. However, whether autophagy plays a cytoprotective or a cytotoxic role in pancreatic β cells remains elusive.
In the present study, we sought to explore the functional roles of metformin in regulating pancreatic β cell survival. Our results show that metformin, by inducing AMPK-dependent autophagy, plays a dual role in mouse insulinoma MIN6 cell survival depending on cell culture status.
Materials and Methods
Cell Culture
Mouse insulinoma (MIN6) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 25 mM glucose (GIBCO, 10566016, CA, USA) supplemented with 15% FBS (GIBCO, 10099141, CA, USA), 100 units/ml penicillin, 100 μg/ml streptomycin (GIBCO, 15140122, CA, USA) and 55 uM β-mercaptoethanol (GIBCO, 21985023, CA, USA). The cells were maintained at 37 °C in an atmosphere of 5% CO2/balance air and 100% humidity.
Cell treatment
The following chemicals were used: metformin, palmitic acid (PA), 3-methyladenine (3-MA), and Compound C (all from Sigma-Aldrich Inc.). AICAR was from Toronto Research Chemicals (Toronto, ON, Canada). 3-MA, Compound C and AICAR were diluted in DMSO according to Material Safety Data Sheet in indicated concentrations [20] [21] [22]. Metformin was in ddH2O as described [11]. Cells at 70-80% confluence were washed with phosphate buffered saline (PBS) and preincubated in DMEM containing 25 mM glucose with 0.5% bovine serum albumin (BSA) for 4 hours before PA or metformin treatment. For PA preparation, solution (200 ml) containing 20mmol/L PA and 0.01mol/L NaOH was incubated at 70°C for 30 minutes then mixed with 330 ml 30% BSA and 20ml filter-sterilized DMEM medium [23]. For cell treatment, serum-starved cells were incubated with 200uM PA complexed to 0.5% BSA for 1 hour, followed by metformin, AICAR or Compound C treatment.
Flow cytometry assay
Apoptotic cells were analyzed by flow cytometer (FACS; Becton Dickinson and Co., San Jose, CA, USA) using the Annexin-V-FLOUS Staining Kit (Roche Diagnostic Corporation, Indianapolis, IN, USA). Annexin-V is a Ca2+-dependent phospholipid-binding protein with a high affinity for phosphotidylserine (PS); hence, this protein can be used as a probe for PS exposure on the outer leaflet of the cell membrane and can be used for the detection of apoptotic cells. The simultaneous application of propidium iodide as a DNA stain allows Annexin-V positively stained cells to be distinguished from necrotic cells. Cells were collected from the culture plates and washed twice with PBS by centrifugation at 200 g for 5 minutes. The cell pellet was suspended in 100 μl of staining solution and incubated for 15 minutes at room temperature. Flow cytometric analysis was performed with a FACScan cytometer (Becton Dickinson and Co., Franklin Lakes, NJ, USA), using LYSIS II analyzer program.
Western Blot
The expression and phosphorylation levels of various proteins in cell lysates were detected by Western blot with specific antibodies. The following primary antibodies were used: The polyclonal anti-Cyclin D2 antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-PCNA, anti-phosphor-Thr172-AMPK, anti-AMPK, anti-LC3B, anti-Cleaved Caspase-3 and anti-p62 antibodies were from Cell Signaling Technology (Danvers, MA). The anti-β-Actin antibody was from AbCam (Cambridge, UK). Quantification of the relative change in protein levels (arbitrary unit; expressed as percentage of control protein levels) was performed by using image J program (ImageJ@list.nih.gov).
TUNEL assay
TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) is a method for detecting DNA fragmentation by labeling the terminal end of nucleic acids. In situ cell apoptosis was examined by detection of fragmented DNA in adherent MIN6 cells, using the DeadEnd Colorimetric TUNEL System (Promega Corporation, Madison, WI, USA).
EdU incorporation assay
EdU (5-ethynyl-2'-deoxyuridine) is a novel superior alternative for BrdU (5-bromo-2'-deoxyuridine) assay to directly measure active DNA synthesis or S-phase synthesis of the cell cycle. For EdU incorporation assay (RiboBio incorporation, Guangdong, China), MIN6 cells were pretreated with or without metformin for 48 hours, and then incubated with EdU (10 µM) for 4 hours and fixed with 4% paraformaldehyde for another 15 minutes. Subsequently, 100 µL 1×Apollo mixture was added for 1 hour and the DNA contents of cells in each well were stained with 100 μL of DAPI (0.5 μg/mL) for 5 minutes and visualized under a fluorescent microscope. The counts of EdU+ cells were calculated by Image-Pro Plus (Version 5.0) system (Media Cybernetics, Inc).
Islet Isolation
Mouse islets were isolated by collagenase digestion of the pancreas according to the procedures described [24]. In brief, male C57/Bl6 mice (4 month old) were anesthetized by I.P. injection of avertin (1 ml/40 g body weight). Mouse pancreas was inflated by injection of 3 ml of a collagenase P solution (Sigma Chemical, St. Louis, MO; 1 mg/ml in Hank's buffered salt solution (HBSS)). Pancreases were removed and incubated at 37℃ for about 12 minutes to allow complete digestion. Digestion was stopped by the addition of 30 mL of HBSS, followed by washing the pancreases with 20 ml RPMI medium three times. Isolated islets were selected with the aid of a pipette under a stereoscopic microscope from the medium, and maintained in RPMI with or without 10% FBS, or HEPES-balanced Krebs-Ringer bicarbonate buffer (KRB; 119 mmol/l NaCl, 4.74 mmol/l KCl, 2.54 mmol/l CaCl2, 1.19 mmol/l MgCl2, 1.19 mmol/l KHPO, 25 mmol/l NaHCO3, and 10 mmol/l HEPES, pH 7.4) containing 0.5% BSA at 37°C.
For signaling studies, the freshly isolated islets (30 islets/experiments) were serum-starved for 2 hours and then treated with 200 uM PA for 1 hour. Islets were then incubated with 10 μl DMSO or 5 mM 3-MA for 1 hour, followed with or without metformin for 24 hours. Islets were lysed and phosphorylation of AMPK at Thr172, as well as the protein levels of AMPK, LC3B-II, and Cleaved Caspase-3 was determined by Western blot using specific antibodies. β-Actin was used as internal control.
Data Analysis
Statistical analyses of the data were performed using analysis of variance (ANOVA). Data are presented as mean±SEM. Statistical significance was set at p values of <0.05 (*), <0.01 (**), <0.001(***), and p<0.05 was considered statistically significant.
Results
Metformin inhibits MIN6 pancreatic β cell proliferation and promotes apoptosis under normal cell culture conditions
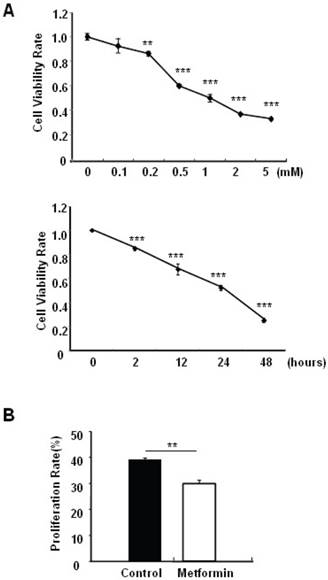
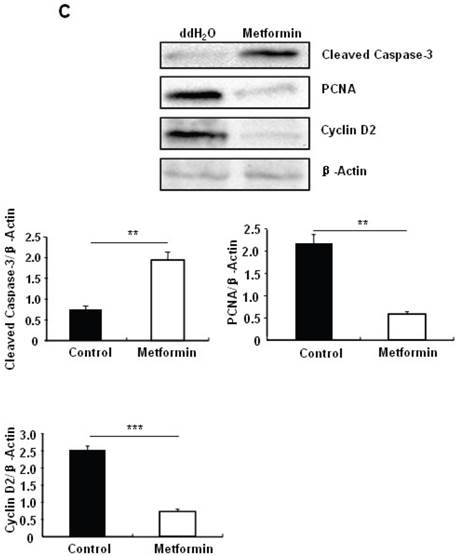
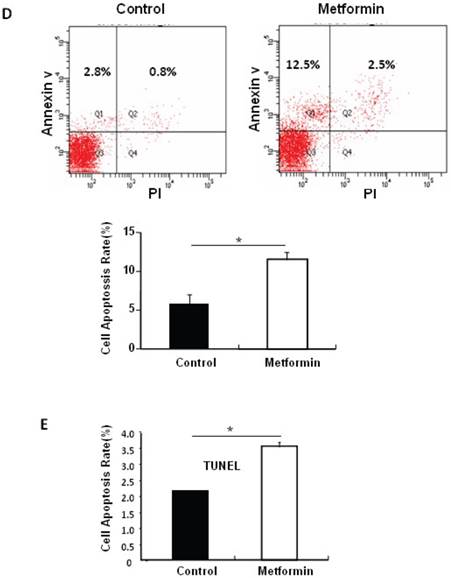
Metformin treatment led to a dose- and time-dependent inhibition of MIN6 cell viability (Fig. 1A). To determine whether the effects of metformin on pancreatic β cell numbers are mediated by increasing apoptosis or decreasing proliferation, MIN6 cells were treated with 2 mM metformin or vehicle for 24 hours. Western blot of cell lysates showed increased cleavage of Caspase-3 to a 17 kDa fragment (Fig.1C), an important biomarker of apoptosis [25]. Consistent with this result, treating MIN6 cells with metformin for 24 hours led to a dramatic increase in cell apoptosis, as demonstrated by both flow cytometry and TUNEL assays (Fig 1D and 1E). After treated with or without metformin for 48 hours, proliferation rate of MIN6 cells was analyzed by EdU incorporation assay [26]. EdU positive cells were significantly decreased in metformin-treated cells compared with control group (Fig. 1B). Metformin treatment also led to a significant decrease in PCNA and cyclin D2 levels (Fig. 1C), confirming reduced cell proliferation. Metformin inhibited insulin secretion at stimulatory glucose concentration (Supplementary Material: Fig. S1), which may partly due to reduced cell viability under standard cell culture condition (Fig. 1 A and 1B and Supplementary Material: Fig. S2).
Metformin inhibits MIN6 pancreatic β cells proliferation and promotes apoptosis under standard cell culture condition. (A) (Upper panel) MIN6 cells were treated with ddH2O as control or metformin at the indicated concentrations for 48 hours. (Lower panel) MIN6 cells were treated with ddH2O or 2 mM metformin for different times as indicated for 24 hours. 10 ul CCK-8 solution was added for 1 hour and cell viability rate was tested by Bio-Rad iMark™ microplate absorbance reader under 450nm wavelength. Cell viability rate= (sample OD-Blank OD)/ (Control OD-Blank OD). (B) MIN6 cells were treated with ddH2O as control or 2mM metformin for 48 hours. Cell proliferation rate were measured by EdU incorporation assay (RiboBio incorporation, Guangdong, China). (C-E) MIN6 cells were incubated with ddH2O or 2mM metformin for 24 hours, and then (C) apoptosis protein marker Cleaved Caspase-3, proliferation protein markers PCNA and Cyclin D2 were tested by Western blot using specific antibodies as indicated; β-Actin was used as internal control. Apoptosis rate was tested by (D) Flow cytometry assay and (E) TUNEL assay Analysis. Data are presented as means ±SEM of three separate experiments. * P<0.05; ** P<0.01; ***P<0.001.
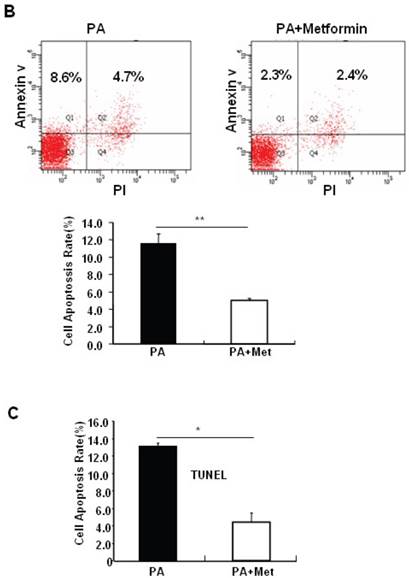
Metformin protects MIN6 pancreatic β cells against PA-induced cell apoptosis
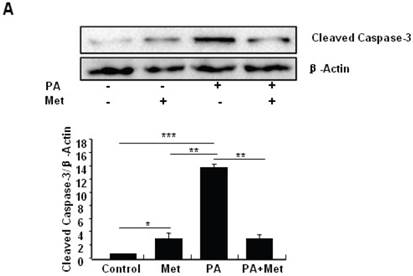
To determine the potential protective effects of metformin on FFA-induced β cell death, serum-starved MIN6 cells were pre-treated with or without PA for 1 hour, followed by metformin or vehicle control. Consistent with previous findings [27], PA treatment greatly induced MIN6 cell apoptosis (Figs. 2A-C). Interestingly, pre-treatment of MIN6 cells with metformin markedly suppressed PA-induced cleaved caspase-3 expression in MIN6 cells (Fig. 2A). Consistent with a suppressive effect of metformin on PA-induced apoptosis, flow cytometry and TUNEL assays revealed a dramatic reduction of PA-induced apoptosis in cells treated with metformin compared to cells treated with PA alone (Figs. 2B and 2C).
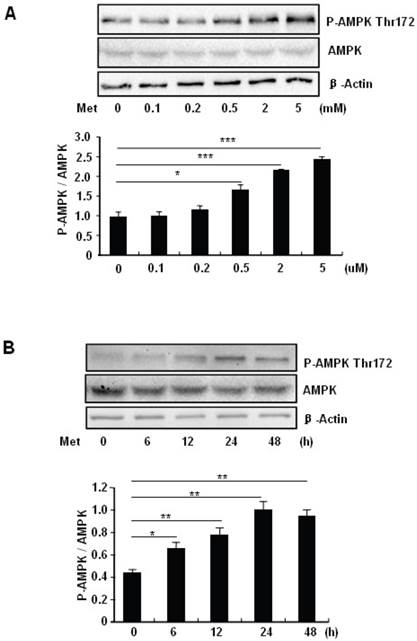
Metformin induces autophagy through AMPK signaling in MIN6 cells
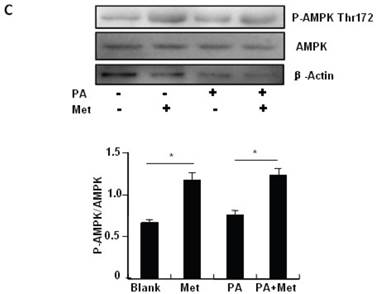
To elucidate the mechanism by which metformin suppresses PA-induced apoptosis in MIN6 cells, we examined the potential roles of AMPK, a well-characterized downstream target of metformin [1]. Metformin treatment led to a dose- (Fig 3A) and time-dependent (Fig. 3B) increase in AMPK phosphorylation at Thr172 in MIN6 cells and the metformin-induced AMPK phosphorylation was not affected by PA treatment (Fig. 3C).
Metformin protects MIN6 cells against PA-induced cell apoptosis. (A) MIN6 cells were co-treated with or without 200 uM PA/NaOH and 2 mM metformin (Met) for 24 hours as indicated, ddH2O was used as control, and then apoptosis marker Cleaved Caspase-3 was tested by Western blot using specific antibodies as indicated; β-Actin was used as internal control. Apoptosis rate was tested by (B) Flow cytometry assay and (C) TUNEL assay Analysis. Data are presented as means ±SEM of three separate experiments. * P<0.05; ** P<0.01; *** P<0.001.
Metformin activates AMPK signaling in MIN6 cells. (A) MIN6 cells were treated with metformin (Met) at the indicated concentrations for 24 hours. (B) MIN6 cells were treated with 2 mM metformin for different times as indicated. (C) MIN6 cells were co-treated with or without 200 uM PA and 2 mM metformin for 24 hours as indicated. ddH2O was used as negative control, and then phosphorylation and protein levels of AMPK Thr172 were determined by Western blot using specific antibodies as indicated. β-Actin was used as internal control. Data are presented as means ±SEM of three separate experiments. * P<0.05; ** P<0.01; *** P<0.001.
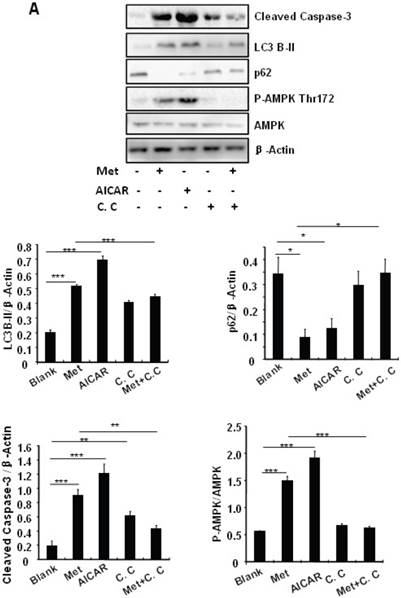
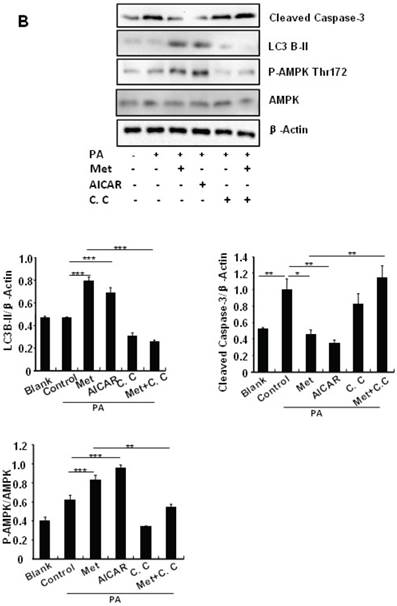
Treating MIN6 cells with metformin increased the cellular levels of cleaved caspase-3 and LC3B-II (Fig. 4A); the latter is an indicator of increased autophagy [28]. Metformin treatment also significantly decreased the cellular levels of p62 (Fig. 4A), another autophagic marker that has been shown to promote cell survival [29]. To determine the mechanism underlying metformin-induced autophagy, we treated MIN6 cells with metformin in the presence or absence of the AMPK activator AICAR (2mM) or inhibitor Compound C (10 uM). Both metformin and AICAR stimulated the cellular levels of LC3B-II and cleaved caspase-3, which was suppressed by Compound C treatment (Fig. 4A). These results indicate that metformin may activate autophagy and induce apoptosis through an AMPK-dependent signaling pathway.
To determine whether metformin has a protective effect on PA-induced β cell survival, MIN6 cells were treated with PA in the presence or absence of metformin, AICAR or Compound C. Treating MIN6 cells with PA enhanced cleaved caspase-3, suggesting enhanced cell apoptosis (Fig. 4B). Both metformin and AICAR enhanced LC3B-II expression and suppressed PA-induced increase in the cellular levels of cleaved caspase-3 (Fig.4B). In addition, the stimulatory effect of metformin on LC3B-II expression was greatly inhibited by Compound C treatment; in the meantime, the inhibitory effect of metformin on caspase-3 was counteracted by Compound C (Fig. 4B), demonstrating a potential involvement of the AMPK signaling pathway in the up-regulation of autophagy and down-regulation of apoptosis.
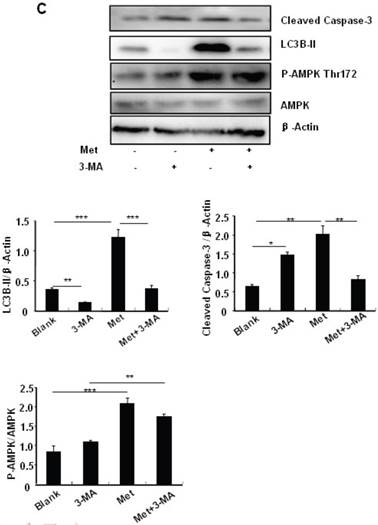
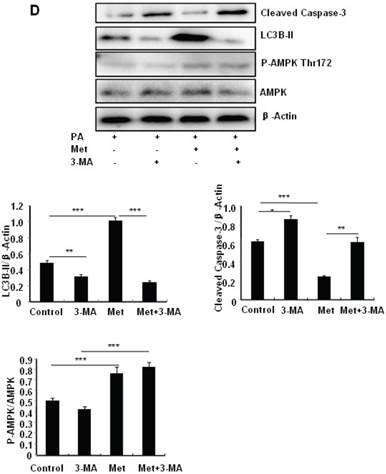
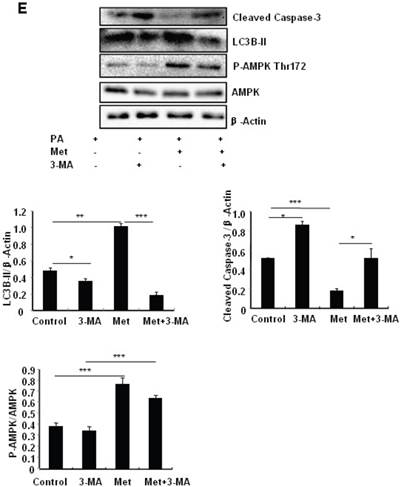
To determine the role of autophagy in the regulation of pancreatic β cell survival, MIN6 cells were co-treated with 2 mM metformin or 5 mM 3-MA, with or without 200 uM PA. 3-MA is a class III PI3K inhibitor that blocks autophagosome formation. Treating cells with 3-MA or metformin alone induced caspase-3 cleavage (Fig. 4C). Interestingly, the promoting effect of metformin on caspase-3 cleavage was protected by 3-MA (Fig. 4C). To determine whether metformin has a protective effect on PA-induced lipotoxicity, MIN6 cells or primary mouse islets were pre-treated with PA followed with metformin or metformin plus 3-MA. Treating cells with 3-MA markedly increased capase-3 cleavage (Fig. 4D). Treating cells with metformin alone increased LC3B-II expression, which was markedly suppressed by 3-MA in both MIN6 cells and primary mice islets (Fig. 4D and 4E). However, metformin suppressed PA-induced capase-3 cleavage, and the protective effect of metformin was diminished in the presence of 3-MA (Fig. 4D and 4E).
Discussion
In the current study we investigated the molecular mechanisms by which metformin modulates β cell survival and function. We demonstrate that while metformin by itself promotes MIN6 cell apoptosis under normal cell culture conditions, this compound alleviates PA-induced MIN6 cell apoptosis. In addition, our results suggest that the protective effect of metformin on PA-induced MIN6 cell adipotosis is most likely via an AMPK-dependent and autophagy-mediated mechanism. These findings provide new insight into the molecular mechanism of metformin and new inspiration for the prevention and cure of diabetes mellitus.
Metformin, a widely used antidiabetic drug for the treatment of type 2 diabetes, alleviates hyperglycemia by inhibiting hepatic gluconeogenesis, augmenting muscle glucose uptake, suppressing lipogenesis, and inhibiting absorption in gastrointestinal [30]. However, how metformin functions in pancreatic β cells is still uncertain. Chronic exposure to metformin has been shown to suppress both mTOR (mammalian target of rapamycin) and EEF2 (Eukaryotic elongation factor 2), resulting in β cells death [10]. Under normal cell culture condition, metformin has no stimulatory effect on insulin secretion [31] or inhibits glucose-stimulated insulin secretion [32] (Supplementary Material: Fig. S1). However, metformin restores insulin secretion impaired by chronic free fatty acid exposure [7] [8]. In the present study, we demonstrate metformin suppresses MIN6 pancreatic β cell proliferation and promotes cell death under normal cell culture condition. On the other hand, metformin protects MIN6 cells against PA-induced cell apoptosis. Thus, metformin plays a dual role in regulating MIN6 pancreatic β cell survival.
Metformin induces autophagy through AMPK signaling in MIN6 cells. (A) MIN6 cells were pre-treated with 10ul DMSO as control or 10 uM Compound C (C.C) for 1 hour, followed with or without 2 mM metformin (Met) or 2 mM AICAR for 24 hours. (B) After serum starved for 4 hours, MIN6 cells were pre-treated with or without 200 uM PA/NaOH for 1 hour, then with 10ul DMSO or 10uM Compound C for 1 hour, followed with 2 mM metformin or 2 mM AICAR for 24 hours as indicated. (C) MIN6 cells were pre-treated with 10ul DMSO or 5 mM 3-MA for 1 hour, followed with or without 2 mM metformin for 24 hours. (D) Serum-starved MIN6 cells were pre-treated with 200 uM PA for 1 hour. Cells were then incubated with 10ul DMSO or 5 mM 3-MA for 1 hour, followed with or without 2 mM metformin for 24 hours. (E) Serum-starved mice islets were pre-treated with 200 uM PA for 1 hour. Islets were then incubated with 10ul DMSO or 5 mM 3-MA for 1 hour, followed with or without 2 mM metformin for 24 h. For (B)-(E), the expression levels of Cleaved Caspase-3, LC3B-II, p62, AMPK, phosphorylation of AMPK Thr172 were determined by Western blot using specific antibodies as indicated. β-Actin was used as internal control. Data are presented as means ±SEM of three separate experiments. * P<0.05; ** P<0.01; ***P<0.001.
How metformin promotes MIN6 β cell death under normal culture condition but protects cells from PA-induced apoptosis? One possible mechanism is that metformin treatment leads to altered autophagy. Autophagy is a well-conserved cellular self-eating process that involves the degradation of cellular components through the lysosomal machinery, which plays an important role in cell growth, development, and homeostasis [33]. When autophagy is induced, a cytosolic form of LC3 (LC3-I) is conjugated to phosphatidylethanolamine to form LC3-phosphatidylethanolamine conjugate (LC3-II), which is recruited to autophagosomal membranes. p62, initially isolated as an interacting partner of atypical protein kinase C (aPKC), binds directly to LC3 and degraded by lysosomal hydrolases after the fusion of autophagosomes with lysosomes. Therefore, LC3-II as lysosomal turnover and p62 degradation reflect starvation-induced autophagic activity. While moderating autophagy has been shown to protect high fat diet feeding-induced stress and cell death [13, 34], overactivated autophagy may lead to excessive digestion of essential cellular components, leading to cell death [19]. Consistent with this, altered autophagy has been shown to trigger apoptosis [17-19] or result in non-apoptotic cell death in a type 2 programmed cell death manner [35]. Thus, enhanced autophagy could either boost or protect cell death, depending on the cell type and environmental context [18]. Genes involved in autophagy and apoptosis were both upregulated in dying salivary gland cells [36]. The autophagy marker p62 regulates cell survival by triggering the TRAF6-NF-κB pathway or by promoting the aggregation of caspase-8 [37]. Multiple additional signals, including p53, Death associated protein kinase (DAPK), BH3 (BCL-2 homology 3)-only proteins and JUN N-terminal kinase (JNK), have been shown to induce both apoptosis and autophagy [38]. So far, how cell fate is determined after induction of autophagy and the transformation mechanism between in apoptosis and autophagy remain to be further elucidated. Our data demonstrate for the first time that metformin, by enhancing autophagy, affects MIN6 β cell survival under different conditions. In addition to promoting autophagy, we also show that metformin plays an important role in MIN6 cell proliferation. Further investigations will be needed to elucidate the underlying mechanisms.
In conclusion, our study shows for the first time a dual role of metformin in MIN6 cell survival, depending on cell growth conditions. We also demonstrate that AMPK-dependent activation of autophagy may play a critical role in the protection of MIN6 cells from PA-induced apoptosis. It should be pointed out that while our study suggests that metformin by itself may be detrimental for MIN6 cell survival, this negative effect could be compensated by the positive effects of metformin in other insulin targeted organ in vivo. Analysis of clinical studies indicate that metformin prevents impaired glucose tolerance progression to diabetes [39]. Our study underscores the merits and caveats of metformin used to date, which should be informative for appropriate use of metformin as an anti-diabetic treatment in the clinical setting.
Abbreviations
AMPK, AMP-activated protein kinase; FFA, free fatty acid; PA, palmitic acid; PTP, permeability transition pore; DMEM, Dulbecco's modified Eagle's medium; MIN6 cells, mouse insulinoma cells; 3-MA, 3-methyladenine; AICAR, 5-Aminoimidazole-4-carboxyamide ribonucleoside; DMSO, dimethyl sulfoxide; PS, phosphotidylserine; mTOR, mammalian target of rapamycin; EEF2, eukaryotic elongation factor 2; aPKC, atypical protein kinase C; DAPK, Death-Associated Protein kinase; JNK, JUN N-terminal kinase; BH3, BCL-2 homology 3; PI3K, phosphatidyl inositol 3-kinase; KRB, Krebs-Ringer bicarbonate buffer; HBSS, Hank's Balanced Salt Solution; FBS, fetal bovine serum; HEPES, hydroxyethyl piperazineethanesulfonic acid; BSA, bovine serum albumin; PCNA, proliferating cell nuclear antigen; TUNEL, Terminal deoxynucleotidyl transferase dUTP nick end labeling; EdU, 5-ethynyl-2'-deoxyuridine.
Supplementary Material
Supplementary figures (fig. S1 & S2) and methods.
Acknowledgements
This study was supported by grants from National Nature Science Foundation of China (81000316 and 81370017 to JZ) and Natural Science Foundation of Hunan Province, China (14JJ3034 to JZ).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Viollet B. et al. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond). 2012;122(6):253-70
2. El Messaoudi S. et al. The cardioprotective effects of metformin. Curr Opin Lipidol. 2011;22(6):445-53
3. Bost F. et al. Metformin and cancer therapy. Curr Opin Oncol. 2012;24(1):103-8
4. Fimognari F.L. et al. Metformin-induced pancreatitis: A possible adverse drug effect during acute renal failure. Diabetes Care. 2006;29(5):1183
5. Williams T. et al. AMP-activated protein kinase (AMPK) activity is not required for neuronal development but regulates axogenesis during metabolic stress. Proc Natl Acad Sci U S A. 2011;108(14):5849-54
6. Mairet-Coello G. et al. The CAMKK2-AMPK Kinase Pathway Mediates the Synaptotoxic Effects of Abeta Oligomers through Tau Phosphorylation. Neuron. 2013;78(1):94-108
7. Patane G. et al. Metformin restores insulin secretion altered by chronic exposure to free fatty acids or high glucose: a direct metformin effect on pancreatic beta-cells. Diabetes. 2000;49(5):735-40
8. Piro S. et al. Effects of Metformin on oxidative stress, adenine nucleotides balance and glucose-induced insulin release impaired by chronic FFA exposure in rat pancreatic islets. J Endocrinol Invest. 2012
9. Lablanche S. et al. Protection of pancreatic INS-1 beta-cells from glucose- and fructose-induced cell death by inhibiting mitochondrial permeability transition with cyclosporin A or metformin. Cell Death Dis. 2011;2:e134
10. Wang Q. et al. Interaction of glibenclamide and metformin at the level of translation in pancreatic beta cells. J Endocrinol. 2011;208(2):161-9
11. Hinke S.A. et al. Methyl succinate antagonises biguanide-induced AMPK-activation and death of pancreatic beta-cells through restoration of mitochondrial electron transfer. Br J Pharmacol. 2007;150(8):1031-43
12. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27-42
13. Ebato C. et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008;8(4):325-32
14. Kim E.H. et al. Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res. 2007;67(13):6314-24
15. Pyo J.O. et al. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem. 2005;280(21):20722-9
16. Boya P. et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25(3):1025-40
17. Chen Z.F. et al. The double-edged effect of autophagy in pancreatic beta cells and diabetes. Autophagy. 2011;7(1):12-6
18. Levine B, Yuan J. Autophagy in cell death: an innocent convict?. J Clin Invest. 2005;115(10):2679-88
19. Clarke M, Bennett M, Littlewood T. Cell death in the cardiovascular system. Heart. 2007;93(6):659-64
20. Zhou L. et al. Berberine acutely inhibits insulin secretion from beta-cells through 3',5'-cyclic adenosine 5'-monophosphate signaling pathway. Endocrinology. 2008;149(9):4510-8
21. Chen Z.F. et al. Liraglutide prevents high glucose level induced insulinoma cells apoptosis by targeting autophagy. Chin Med J (Engl). 2013;126(5):937-41
22. Kefas B.A. et al. AMP-activated protein kinase can induce apoptosis of insulin-producing MIN6 cells through stimulation of c-Jun-N-terminal kinase. J Mol Endocrinol. 2003;30(2):151-61
23. Tian D. et al. Overexpression of steroidogenic acute regulatory protein in rat aortic endothelial cells attenuates palmitic acid-induced inflammation and reduction in nitric oxide bioavailability. Cardiovasc Diabetol. 2012;11:144
24. Zhang J. et al. Disruption of growth factor receptor-binding protein 10 in the pancreas enhances beta-cell proliferation and protects mice from streptozotocin-induced beta-cell apoptosis. Diabetes. 2012;61(12):3189-98
25. Cohen G.M. Caspases: the executioners of apoptosis. Biochem J. 1997;326( Pt 1):1-16
26. Salic A, Mitchison T.J. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A. 2008;105(7):2415-20
27. Maedler K. et al. Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes. 2003;52(3):726-33
28. Yang W. et al. Protein kinase B/Akt1 inhibits autophagy by down-regulating UVRAG expression. Exp Cell Res. 2013;319(3):122-33
29. Mathew R. et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137(6):1062-75
30. Joshi S.R. Metformin: old wine in new bottle--evolving technology and therapy in diabetes. J Assoc Physicians India. 2005;53:963-72
31. Langelueddecke C. et al. Effect of the AMP-Kinase Modulators AICAR, Metformin and Compound C on Insulin Secretion of INS-1E Rat Insulinoma Cells under Standard Cell Culture Conditions. Cell Physiol Biochem. 2012;29(1-2):75-86
32. Leclerc I. et al. Metformin, but not leptin, regulates AMP-activated protein kinase in pancreatic islets: impact on glucose-stimulated insulin secretion. Am J Physiol Endocrinol Metab. 2004;286(6):E1023-31
33. Levine B and KlionskyD.J. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6(4):463-77
34. Jung H.S. et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008;8(4):318-24
35. Tanemura M. et al. Rapamycin causes upregulation of autophagy and impairs islets function both in vitro and in vivo. Am J Transplant. 2012;12(1):102-14
36. Gorski S.M. et al. A SAGE approach to discovery of genes involved in autophagic cell death. Curr Biol. 2003;13(4):358-63
37. Moscat J, Diaz-Meco M.T. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137(6):1001-4
38. Marino G. et al. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014
39. DeFronzo R.A, Abdul-Ghani M.A. Preservation of beta-cell function: the key to diabetes prevention. J Clin Endocrinol Metab. 2011;96(8):2354-66
Author contact
![]() Corresponding author: J.Z.: Metabolic Syndrome Research Center, Second Xiangya Hospital, Central South University, No. 139, Middle Renmin Road, Changsha, Hunan 410011, China. Fax: +86 0731 85292148; E-mail address: Doctorzhangjjcom
Corresponding author: J.Z.: Metabolic Syndrome Research Center, Second Xiangya Hospital, Central South University, No. 139, Middle Renmin Road, Changsha, Hunan 410011, China. Fax: +86 0731 85292148; E-mail address: Doctorzhangjjcom
Received 2013-10-18
Accepted 2014-2-7
Published 2014-2-20