ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2014; 10(8):817-824. doi:10.7150/ijbs.9625 This issue Cite
Research Paper
A Small Molecule Agonist THIQ as a Novel Pharmacoperone for Intracellularly Retained Melanocortin-4 Receptor Mutants
Hui Huang, Ya-Xiong Tao ![]()
Department of Anatomy, Physiology and Pharmacology, College of Veterinary Medicine, Auburn University, Auburn, AL 36849, United States
Abstract
Although mutations in the melanocortin-4 receptor (MC4R) gene cause severe early-onset obesity, we still do not have effective approaches to correct the defects of these mutations. Several antagonists have been identified as pharmacoperones of the MC4R whereas no agonist of the MC4R has been reported. In the present study, we investigated the effect of a small molecule agonist of the MC4R, THIQ, on the cell surface expression and signaling of ten intracellularly retained MC4R mutants using different cell lines. We showed that THIQ increased the cell surface expression of three mutants (N62S, C84R, and C271Y) and two of them (N62S and C84R) had increased signaling in HEK293 cells. Interestingly, THIQ increased the signaling of two other mutants (P78L and P260Q) without increasing their cell surface expression in HEK293 cells. In neuronal cells, THIQ exhibited a more potent effect, correcting the cell surface expression and signaling of seven mutants (N62S, I69R, P78L, C84R, W174C, P260Q, and C271Y). Other mutants were not rescued by THIQ. We also showed that THIQ did not rescue MC4R mutants defective in ligand binding or signaling or one intracellularly retained mutant of the melanocortin-3 receptor. In summary, we demonstrated that a small molecule agonist acted as a pharmacoperone of the MC4R rescuing the cell surface expression and signaling of some intracellularly retained MC4R mutants.
Keywords: G protein-coupled receptor, Melanocortin-4 receptor, Obesity, Pharmacological chaperone/Pharmacoperone, THIQ
Introduction
The melanocortin-4 receptor (MC4R), predominantly expressed in the CNS, plays a crucial role in regulating food intake and energy expenditure [1, 2]. The MC4R is activated by α/β-melanocyte-stimulating hormone (MSH) from pro-opiomelanocortin neurons and inhibited by agouti-related peptide from agouti-related peptide neurons in the hypothalamic arcuate nucleus. Activation of the MC4R activates heterotrimeric stimulatory G protein, which subsequently activates adenylyl cyclase to produce cAMP.
Extensive functional studies of MC4R mutants identified from obese patients suggest that most of the dysfunctional mutants are misfolded and retained intracellularly by the cellular quality control system rather than being transported to the cell surface [3-5]. These mutants are dysfunctional mainly due to mislocalization. Several studies have been performed to correct the localization of misfolded MC4Rs through modulating molecular chaperone such as heat shock cognate protein 70 [6], or using chemical chaperones such as 4-phenyl butyric acid [7], or several nonpeptidic MC4R antagonists as pharmacological chaperones (pharmacoperones) [8-11]. A more recent study using endoplasmic reticulum-targeted α-MSH showed that it also promotes the cell surface expression of I316S MC4R [12]. In these studies, most of the misfolded MC4Rs become functional when corrected to the cell surface.
Pharmacoperones have been considered as an alternative to gene therapy and a potential treatment for diseases caused by misfolded G protein-coupled receptors (GPCRs). A number of antagonists, agonists or allosteric modulators have been developed as pharmacoperones for several GPCRs, such as rhodopsin, gonadotropin-releasing hormone receptor, V2 arginine vasopressin receptor (AVPR2), and calcium-sensing receptor (reviewed in [13-15]). Further studies on pharmacoperones have been successfully extended from in vitro using cell lines to in vivo using animals or in a clinical trial [16, 17].
THIQ was the first selective small molecule (MW, 589) agonist of the MC4R reported, with high affinity (IC50, 1.2 nM) and potency (EC50, 2.1 nM) [18]. Several structure-function relationship studies show that THIQ competes with [Nle4,D-Phe7]-α-MSH (NDP-MSH) for binding to the MC4R, sharing common interaction determinants with the endogenous ligands but also possessing several specific interaction determinants [10]. In the present study, we investigated the effects of THIQ on the cell surface expression and signaling of ten misrouted MC4R mutants (N62S, I69R, P78L, C84R, G98R, Y157S, W174C, P260Q, F261S, and C271Y) and on the signaling of four correctly routed MC4R mutants (Δ88-92, D90N, I102S, and N274S).
Materials and plasmids
NDP-MSH was purchased from Peptides International (Louisville, KY). THIQ (N-[(3R)-1,2,3,4-Tetrahydroisoquinolinium-3-ylcarbonyl]-(1R)-1-(4-chlorobenzyl)-2-[4-cyclohexyl-4-(1H-1,2,4-triazol-1-ylmethyl)piperidin-1-yl]-2-oxoethylamine) was purchased from Tocris Bioscience (Ellisville, MO). Dulbecco's Modified Eagle's Medium, newborn calf serum, fetal bovine serum, and antibiotics were purchased from Invitrogen (Carlsbad, CA). Cell culture plastic wares were purchased from Corning (Corning, NY). Radiolabeled cAMP was iodinated with chloramine T method [19]. The N-terminal c-myc-tagged WT and mutant human MC4Rs and 3×HA-tagged human melanocortin-3 receptors (MC3Rs) subcloned into pcDNA3.1 were previously generated and sequenced [8, 20-23].
Cell culture and transfection
Human embryonic kidney (HEK) 293, Neuro2a, and N1E-115 cells (American Type Culture Collection, Manassas, VA) were maintained in DMEM containing 10% newborn calf serum (HEK293 cells) or 10% fetal bovine serum (Neuro2a and N1E-115 cells) in a 5% CO2-humidified atmosphere at 37 oC. Approximately 24 h after seeding into 0.1% gelatin-coated dishes, HEK293 cells were transfected using calcium phosphate precipitation method and were then selected using 0.2 mg/ml G418 for stable transfection [20]. Neuro2a and N1E-115 cells were transiently transfected using jetPRIME transfection reagent (Polyplus-transfection, New York, NY). Approximately 24 h after plating (HEK293 cells) or transfection (Neuro2a and N1E-115 cells), cells were cultured for an additional 24 h in the presence of 0.1% dimethyl sulphoxide (DMSO) or different concentrations of THIQ at 37 oC. Cells were then rinsed and used for confocal microscopy, flow cytometry, or cAMP assays.
Confocal microscopy
HEK293 cells stably expressing WT or mutant receptors were plated into poly-D-lysine-coated eight well slides (Biocoat cellware from Falcon, B&D Systems, Franklin Lakes, NJ) for 24 h and incubated for another 24 h in the presence of 0.1% DMSO or 10-5 M THIQ. On the day of experiment, cells were processed for confocal imaging as previously described [24]. Briefly, cells were rinsed with phosphate buffered saline for immunohistochemistry (PBS-IH, 137 mM NaCl, 2.7 mM KCl, 1.4 mM KH2PO4, 4.3 mM Na2HPO4, pH 7.4) and fixed with 4% paraformaldehyde for 15 min. Nonspecific sites were blocked with 5% bovine serum albumin (BSA) in PBS-IH for 1 h. Cells were incubated with mouse anti-myc 9E10 antibody (Developmental Studies Hybridoma Bank, The University of Iowa, Iowa City, IA) 1:40 diluted in PBS-IH with 0.5% BSA for 1 h. Cells were then extensively rinsed and incubated with goat anti-mouse Alexa Fluor 488-labeled antibody (Invitrogen) 1:2000 diluted in PBS-IH with 0.5% BSA for 1 h in the dark. After extensive rinsing, slides were mounted with coverslips using Vectashield mounting media (Vector Laboratories, Burlingame, CA) and dried overnight at 4 oC. Cells were viewed using a Nikon A1 confocal microscope. All procedures were performed at room temperature unless noted otherwise.
Flow cytometry
HEK293 cells stably or Neuro2A cells transiently expressing WT or mutant receptors were incubated for 24 h in the presence of 0.1% DMSO or different concentrations of THIQ. On the day of experiment, cells were processed for flow cytometry study as previously described [8]. Briefly, cells seeded into six well plates were plated on ice and rinsed once with ice-cold PBS-IH. Cells were then detached and centrifuged at 500 × g for 5 min. Antibodies used and antibody dilutions were the same as described above for confocal microscopy with the exception that for the study of MC3R expression, cells were incubated with the primary antibody mouse anti-HA.11 (Covance, Princeton, NJ) 1:100 diluted in PBS-IH with 0.5% BSA. Cells were rinsed and analyzed using a C6 Accuri Cytometer (Accuri Cytometers, Ann Arbor, MI). Immunostaining of cells expressing empty vector was taken as the background staining. The cell surface expression level of the mutants was calculated using the following formula: [(mutant - empty vector) / (WT - empty vector) × 100%] [25]. All the procedures were performed at room temperature unless noted otherwise.
cAMP assay
HEK293 cells stably or Neuro2A and N1E-115 cells transiently expressing WT or mutant receptors were incubated for 24 h in the presence of 0.1% DMSO or different concentrations of THIQ. On the day of experiment, cells were processed for intracellular cAMP detection as previously described [20]. Briefly, cells were rinsed twice with warm Waymouth's MB752/1 media (Sigma-Aldrich) containing 1 mg/ml BSA (Waymouth/BSA). Cells were pretreated with 0.5 mM isobutylmethylxanthine (Sigma-Aldrich) in Waymouth/BSA for 15 min and then stimulated with 10-6 M NDP-MSH for 1 h at 37 oC in the presence of isobutylmethylxanthine. Intracellular cAMP was extracted by adding 0.5 M percholoric acid containing 180 μg/ml theophylline (Sigma-Aldrich) and measured by RIA. The cAMP concentration of the mutants was calculated using the following formula: [mutant / WT × 100%] (HEK293 cells) or [(mutant - empty vector) / (WT - empty vector) × 100%] (Neuro2a and N1E-115 cells).
Data analysis
Data were calculated as percentage of the DMSO-treated WT receptor and analyzed using Student's t-test performed by GraphPad Prism 4.0 software (San Diego, CA).
Results
The effect of THIQ on the cell surface expression of intracellularly retained MC4R mutants
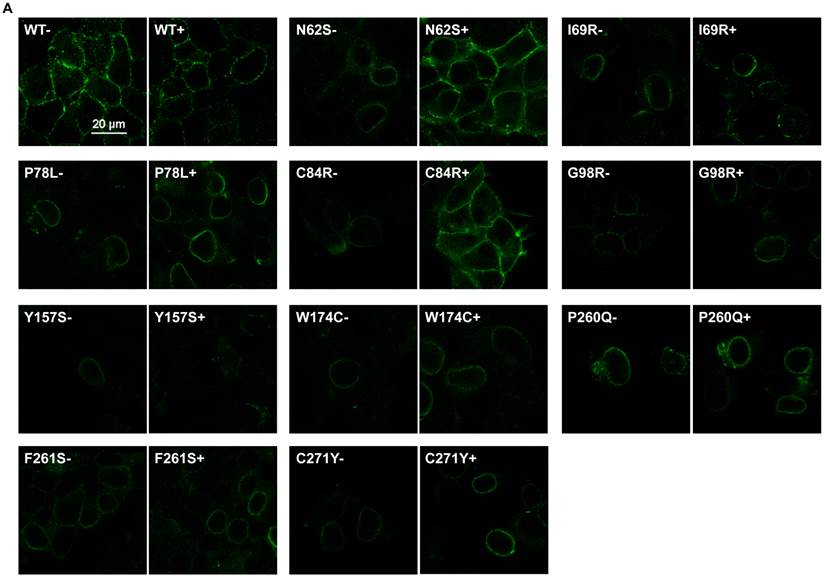
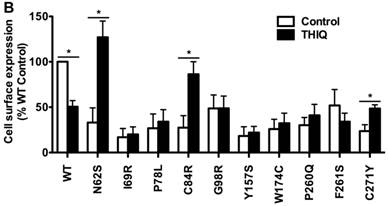
To investigate the effect of THIQ on the cell surface expression of intracellularly retained MC4R mutants, we studied ten misrouted MC4R mutants with decreased cell surface expression (N62S, I69R, P78L, C84R, G98R, Y157S, W174C, P260Q, F261S, and C271Y) [8, 20, 23]. HEK293 cells stably transfected with WT or mutant MC4Rs were treated with 10-5 M THIQ for 24 h and then immunostained at non-permeabilized status for confocal microscopy (Fig. 1A) and flow cytometry study (Fig. 1B). Our results showed that THIQ treatment decreased the signal intensity of WT MC4R by approximately 50% whereas increased that of three mutants (N62S, C84R, and C271Y) in HEK293 cells. THIQ had no obvious effect on the signal intensity of the other mutants.
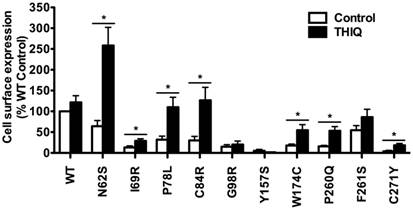
Considering that the MC4R is mainly distributed in the CNS, we further quantitated the effect of THIQ on the cell surface expression of WT and mutant MC4Rs using a neuronal cell line, Neuro2a. Neuro2a cells transiently transfected with the WT or mutant MC4Rs were treated with 10-5 M THIQ for 24 h. Unexpectedly, as shown in Fig. 2, THIQ did not decrease the cell surface expression of WT MC4R in Neuro2a cells. THIQ significantly increased the cell surface expression of seven mutants, including three that were also increased in HEK293 cells (N62S, C84R, and C271Y) and four that were not increased in HEK293 cells (I69R, P78L, W174C, and P260Q).
The effect of THIQ on the signaling of intracellularly retained MC4R mutants
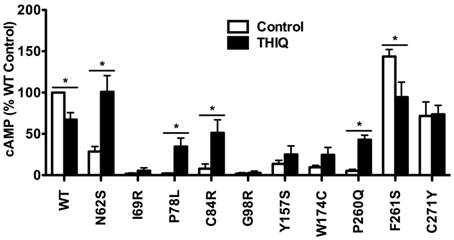
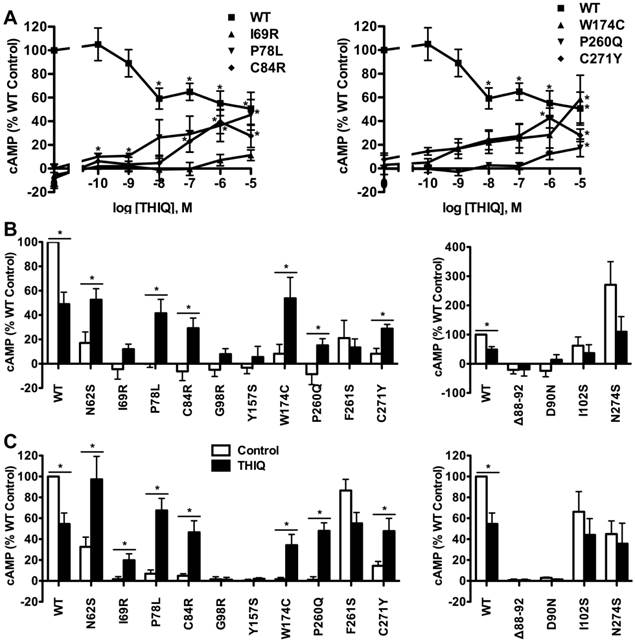
To investigate the effect of THIQ on the signaling of intracellularly retained MC4R mutants, different cell lines (HEK293, Neuro2a, and N1E-115 cells) were used. HEK293 cells stably transfected with the WT or mutant MC4Rs were treated with 10-5 M THIQ for 24 h. Cells were washed twice and stimulated with 10-6 M NDP-MSH for 1 h and then the intracellular cAMP concentration was measured. As shown in Fig. 3, THIQ decreased the cAMP production of WT MC4R by approximately 30%. THIQ significantly increased the signaling of four mutants (N62S, P78L, C84R, and P260Q) and decreased that of one mutant (F261S) in HEK293 cells.
In Neuro2a cells transiently transfected with the WT or mutant MC4Rs, THIQ began to significantly decrease the cAMP production of WT MC4R at 10-8 M and decreased it by approximately 50% at 10-5 M (Fig. 4A). THIQ dose-dependently increased the cAMP production of the mutants with different potencies for different mutants. At 10-5 M, THIQ significantly increased the cAMP production of six mutants (N62S, P78L, C84R, W174C, P260Q, and C271Y) (Fig. 4B). In N1E-115 cells, 10-5 M THIQ significantly increased the signaling of the six mutants that were also rescued in Neuro2a cells and one additional mutant (I69R) that had an increase that did not reach statistical significance in Neuro2a cells (Fig. 4C).
The effect of THIQ on the cell surface expression of MC4R mutants in HEK293 cells. HEK293 cells stably transfected with WT or mutant MC4Rs were incubated with 10-5 M THIQ for 24 h. Cells were then processed for cell surface immunofluorescent detection of myc-MC4R using a Nikon A1 confocal microscope (A) or a C6 Accuri cytometer (B). (A) Experiments were done at least twice. (B) Data are shown as percentage of DMSO-treated WT MC4R after correcting the background staining from cells transfected with the empty vector. Quantification was done from at least three independent experiments and shown as mean ± S.E.M. * Significantly different from the DMSO-treated control group, p < 0.05.
The effect of THIQ on the cell surface expression of MC4R mutants in Neuro2a cells. Neuro2a cells transiently transfected with WT or mutant MC4Rs were incubated with 10-5 M THIQ for 24 h. Quantification was done from at least three independent experiments and shown as mean ± S.E.M. See the legend to Fig. 1 for details.
The effect of THIQ on the signaling of MC4R mutants in HEK293 cells. HEK293 cells stably transfected with WT or mutant MC4Rs were incubated with 10-5 M THIQ for 24 h. Cells were rinsed twice before stimulated with 10-6 M NDP-MSH for 1 h. Intracellular cAMP concentrations were measured by RIA. Data are shown as percentage of DMSO-treated WT MC4R. Quantification was done from at least three independent experiments and shown as mean ± S.E.M. * Significantly different from the DMSO-treated control group, p < 0.05.
The effect of THIQ on the signaling of MC4R mutants in neuronal cells. Neuro2a cells (A and B) and N1E-115 cells (C) transiently transfected with WT or mutant MC4Rs were incubated with different concentrations of THIQ (A) or 10-5 M THIQ (B and C) for 24 h. Data are shown as percentage of DMSO-treated WT MC4R after correcting the cAMP production from cells transfected with the empty vector. Quantification was done from at least three independent experiments and shown as mean ± S.E.M. See the legend to Fig. 3 for details.
The effect of THIQ on the signaling of MC4R mutants with other defects and on one intracellularly retained MC3R mutant
We also studied the effect of THIQ on four correctly routed MC4R mutants with defects in ligand binding or signaling, or with no defect (Δ88-92, D90N, I102S, and N274S) [20, 21, 26, 27]. As shown in Fig. 4B and Fig. 4C in Neuro2a or N1E-115 cells, respectively, 10-5 M THIQ did not increase the signaling of the four mutants.
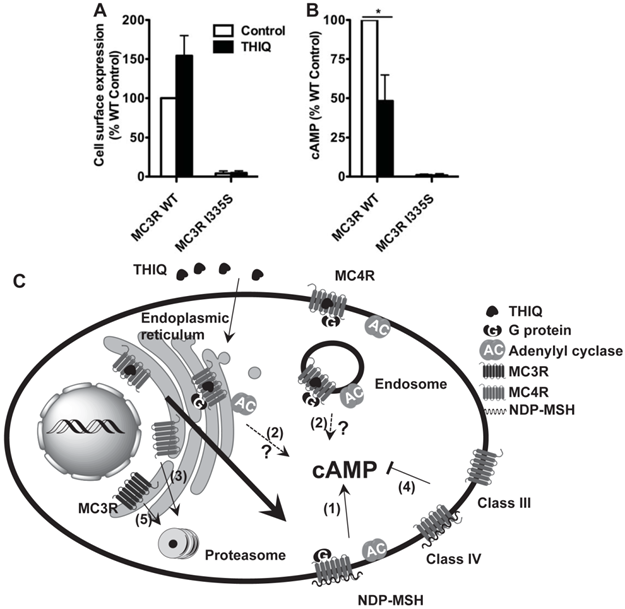
To investigate whether THIQ also rescued misrouted MC3R mutants, we studied one intracellularly retained MC3R mutant, I335S. 10-5 M THIQ did not increase the cell surface expression (Fig. 5A) or signaling (Fig. 5B) of I335S MC3R whereas it decreased the signaling of WT MC3R by approximately 50%.
Discussion
Although mutations in the MC4R are the most common cause of monogenic form of obesity [28], we still have no effective therapeutics specific for obese patients harboring these mutations. One approach is to develop MC4R pharmacoperones. Several antagonists have been identified as pharmacoperones for the MC4R and, to our knowledge, no agonist has been reported acting as a pharmacoperone for the MC4R [8-11]. In the present study, we investigated the effect of a small molecule agonist of the MC4R, THIQ, on the cell surface expression and signaling of ten misrouted MC4R mutants (N62S, I69R, P78L, C84R, G98R, Y157S, W174C, P260Q, F261S, and C271Y) using different cell lines.
The effect of THIQ on the cell surface expression and signaling of the MC3R (A) and model of the effect of THIQ on the MC4R and the MC3R (B). (A) Neuro2a cells transiently transfected with WT or I335S MC3Rs were treated with 10-5 M THIQ for 24 h. The cell surface immunofluorescence of 3×HA-MC3R and intracellular cAMP production were measured. Data are shown as percentage of DMSO-treated WT MC3R after correcting the background staining or the cAMP production from cells transfected with the empty vector. Quantification was done from at least three independent experiments and shown as mean ± S.E.M. * Significantly different from the DMSO-treated control group, p < 0.05. (B) Model of the effect of THIQ on the MC4R and the MC3R. THIQ stabilized the conformation of misfolded MC4R mutants and coaxed them to the cell surface. The rescued mutants were functional in cAMP production (1). THIQ rescued the function of some mutants without increasing their cell surface expression in HEK293 cells possibly by increasing their cell surface expression that was not detected (1) or inducing intracellular signaling by residual THIQ that was not washed away (2). THIQ did not rescue some MC4R mutants (3). THIQ did not rescue the signaling of MC4R mutants defective in ligand binding (Class III) or signaling (Class IV) (4). THIQ did not rescue one MC3R intracellularly retained mutant (5).
THIQ significantly decreased the cell surface expression and signaling of WT MC4R in HEK293 cells (Table 1). Several studies reported that agonists dose-dependently induce MC4R internalization and that nonpeptide agonists induce less internalization than peptide agonists do [29-31]. However, other studies suggested that the MC4R undergoes constitutive internalization and that α-MSH reduces the cell surface expression of the MC4R by blocking the recycling of internalized receptor rather than by increasing its endocytosis rate [32, 33]. THIQ-mediated desensitization, internalization, or inhibition of recycling of the MC4R was probably responsible for the reduced cell surface expression and signaling of WT MC4R after treatment with THIQ.
THIQ significantly increased the cell surface expression of three mutants (N62S, C84R, and C271Y) and increased the signaling of two of them (N62S and C84R) in HEK293 cells (Table 1). The increased amount of the MC4R was a result of the dynamic equilibrium with mutants being stabilized and chaperoned to the plasma membrane and internalized to the cytoplasm. The result suggests that, as illustrated in Fig. 5C, THIQ acted as a pharmacoperone of the MC4R stabilizing the conformation of intracellularly retained mutants and increasing the plasma membrane targeting and/or signaling of these mutants.
Summary of the effect of THIQ on the cell surface expression and signaling of WT and mutant MC4Rs.
| HEK293 cells | Neuronal cells | |||
|---|---|---|---|---|
| Mutants | Expression | Function | Expression | Function |
| WT | ↓ | ↓ | - | ↓ |
| N62S | ↑ | ↑ | ↑ | ↑ |
| I69R | - | - | ↑ | ↑ |
| P78L | - | ↑ | ↑ | ↑ |
| C84R | ↑ | ↑ | ↑ | ↑ |
| G98R | - | - | - | - |
| Y157S | - | - | - | - |
| W174C | - | - | ↑ | ↑ |
| P260Q | - | ↑ | ↑ | ↑ |
| F261S | - | ↓ | - | - |
| C271Y | ↑ | - | ↑ | ↑ |
“↑”, increasing; “↓”, decreasing; “-”, no significant effect.
THIQ did not significantly increase the cell surface expression of the other seven mutants (I69R, P78L, G98R, Y157S, W174C, P260Q, and F261S) but increased the signaling of two of them (P78L and P260Q) in HEK293 cells (Table 1). The increased signaling of P78L and P260Q was probably due to signal amplification so that a small increase in the cell surface expression induced a large increase in the signaling or due to improved signaling efficacy of the mutants at the cell surface as observed in calcium-sensing receptor [34]. It was also possible that residual THIQ induced intracellular signaling. Recently, in addition to the canonical signaling at the plasma membrane, intracellular signaling in the endoplasmic reticulum, Golgi apparatus, or the endosome has also been observed in several GPCRs, such as the β2-adrenoceptor [35]. Similar observation on the AVPR2 was also reported: three nonpeptide AVPR2 agonists activated AVPR2 mutants intracellularly and such activation did not induce degradation [36]. Further studies need to be performed to determine the mechanism of THIQ activating MC4R mutants without increasing their cell surface expression.
Interestingly, THIQ did not decrease the cell surface expression of WT MC4R but decreased its signaling in neuronal cells (Table 1). For all the experiments, cells were treated with THIQ for 24 h; therefore it was not likely that THIQ only induced desensitization but not internalization of the MC4R in neuronal cells. It was more likely that the forward trafficking rate of the MC4R chaperoned with THIQ (pharmacoperone effect) was more efficient than THIQ-induced internalization or THIQ-blocked recycling (agonist effect), resulting in receptor amount at the plasma membrane not changed in neuronal cells. Although the total receptor amount was not decreased, the receptor became desensitized upon prolonged agonist exposure, resulting in reduced signaling. We have not observed such a different rescuing efficiency of antagonists as pharmacoperones of the MC4R in HEK293 and neuronal cells (data unpublished).
THIQ rescued the cell surface expression and signaling of seven mutants (N62S, I69R, P78L, C84R, W174C, P260Q, and C271Y) in neuronal cells (Table 1), which also demonstrated a more efficient pharmacoperone action of THIQ in neuronal cells than in HEK293 cells. The rescuing efficacy of THIQ was varied for different mutants studied herein. Interestingly, G98R, the cell surface expression of which was rescued by two MC4R antagonists ([10] and data unpublished), was not rescued by THIQ. THIQ did not restore the signaling of MC4R mutants with ligand binding or signaling defects.
THIQ has low affinity with the MC3R (IC50, 634-fold over that of the MC4R) [18]. In our study, THIQ treatment decreased the signaling of WT MC3R by approximately 50%. Although THIQ did not correct the cell surface expression or signaling of I335S MC3R, it could not be excluded that THIQ might rescue other MC3R misrouted mutants.
In summary, our results demonstrated that THIQ was a pharmacoperone of the MC4R, rescuing the cell surface expression and signaling of intracellularly retained MC4Rs. THIQ also increased the signaling of two mutants (P78L and P260Q) without increasing their cell surface expression, implying that THIQ might activate the MC4R intracellularly. To our knowledge, THIQ was the first agonist identified as a pharmacoperone of the MC4R.
Abbreviations
AVPR2, V2 arginine vasopressin receptor; GPCR, G protein-coupled receptor; HEK293, human embryonic kidney 293; MC3R, melanocortin-3 receptor; MC4R, melanocortin-4 receptor; MSH, melanocyte stimulating hormone; WT, wild type.
Acknowledgements
This study was funded by American Diabetes Association Grant 1-12-BS212, Auburn University Intramural Grant Program and Interdisciplinary Grant of College of Veterinary Medicine at Auburn University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gantz I, Konda Y, Tashiro T, Shimoto Y, Miwa H, Munzert G. et al. Molecular cloning of a novel melanocortin receptor. J Biol Chem. 1993;268:8246-50
2. Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR. et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131-41
3. Tao YX. Molecular mechanisms of the neural melanocortin receptor dysfunction in severe early onset obesity. Mol Cell Endocrinol. 2005;239:1-14
4. Tao YX. Mutations in melanocortin-4 receptor and human obesity. Prog Mol Biol Transl Sci. 2009;88:173-204
5. Hinney A, Volckmar AL, Knoll N. Melanocortin-4 receptor in energy homeostasis and obesity pathogenesis. Prog Mol Biol Transl Sci. 2013;114:147-91
6. Meimaridou E, Gooljar SB, Ramnarace N, Anthonypillai L, Clark AJ, Chapple JP. The cytosolic chaperone Hsc70 promotes traffic to the cell surface of intracellular retained melanocortin-4 receptor mutants. Mol Endocrinol. 2011;25:1650-60
7. Granell S, Mohammad S, Ramanagoudr-Bhojappa R, Baldini G. Obesity-linked variants of melanocortin-4 receptor are misfolded in the endoplasmic reticulum and can be rescued to the cell surface by a chemical chaperone. Mol Endocrinol. 2010;24:1805-21
8. Fan ZC, Tao YX. Functional characterization and pharmacological rescue of melanocortin-4 receptor mutations identified from obese patients. J Cell Mol Med. 2009;13:3268-82
9. Rene P, Le Gouill C, Pogozheva ID, Lee G, Mosberg HI, Farooqi IS. et al. Pharmacological chaperones restore function to MC4R mutants responsible for severe early-onset obesity. J Pharmacol Exp Ther. 2010;335:520-32
10. Tao YX. The melanocortin-4 receptor: Physiology, pharmacology, and pathophysiology. Endocr Rev. 2010;31:506-43
11. Ward NA, Hirst S, Williams J, Findlay JB. Pharmacological chaperones increase the cell-surface expression of intracellularly retained mutants of the melanocortin 4 receptor with unique rescuing efficacy profiles. Biochem Soc Trans. 2012;40:717-20
12. Granell S, Molden BM, Baldini G. Exposure of MC4R to agonist in the endoplasmic reticulum stabilizes an active conformation of the receptor that does not desensitize. Proc Natl Acad Sci U S A. 2013;110:E4733-E42
13. Conn PM, Ulloa-Aguirre A, Ito J, Janovick JA. G protein-coupled receptor trafficking in health and disease: lessons learned to prepare for therapeutic mutant rescue in vivo. Pharmacol Rev. 2007;59:225-50
14. Conn PM, Ulloa-Aguirre A. Pharmacological chaperones for misfolded gonadotropin-releasing hormone receptors. Adv Pharmacol. 2011;62:109-41
15. Tao YX, Conn PM. Chaperoning G protein-coupled receptors: From cell biology to therapeutics. Endocr Rev. 2014;35:602-47
16. Bernier V, Morello JP, Zarruk A, Debrand N, Salahpour A, Lonergan M. et al. Pharmacologic chaperones as a potential treatment for X-linked nephrogenic diabetes insipidus. J Am Soc Nephrol. 2006;17:232-43
17. Janovick JA, Stewart MD, Jacob D, Martin LD, Deng JM, Stewart CA. et al. Restoration of testis function in hypogonadotropic hypogonadal mice harboring a misfolded GnRHR mutant by pharmacoperone drug therapy. Proc Natl Acad Sci U S A. 2013;110:21030-5
18. Sebhat IK, Martin WJ, Ye Z, Barakat K, Mosley RT, Johnston DB. et al. Design and pharmacology of N-[(3R)-1,2,3,4-tetrahydroisoquinolinium- 3-ylcarbonyl]-(1R)-1-(4-chlorobenzyl)- 2-[4-cyclohexyl-4-(1H-1,2,4-triazol- 1-ylmethyl)piperidin-1-yl]-2-oxoethylamine (1), a potent, selective, melanocortin subtype-4 receptor agonist. J Med Chem. 2002;45:4589-93
19. Tao YX, Huang H, Wang ZQ, Yang F, Williams JN, Nikiforovich GV. Constitutive activity of neural melanocortin receptors. Methods Enzymol. 2010;484:267-79
20. Tao YX, Segaloff DL. Functional characterization of melanocortin-4 receptor mutations associated with childhood obesity. Endocrinology. 2003;144:4544-51
21. Tao YX, Segaloff DL. Functional analyses of melanocortin-4 receptor mutations identified from patients with binge eating disorder and nonobese or obese subjects. J Clin Endocrinol Metab. 2005;90:5632-8
22. Tao YX. Functional characterization of novel melanocortin-3 receptor mutations identified from obese subjects. Biochim Biophys Acta. 2007;1772:1167-74
23. Wang ZQ, Tao YX. Functional studies on twenty novel naturally occurring melanocortin-4 receptor mutations. Biochim Biophys Acta. 2011;1812:1190-9
24. Huang H, Tao YX. Pleiotropic functions of the transmembrane domain 6 of human melanocortin-4 receptor. J Mol Endocrinol. 2012;49:237-48
25. Wang SX, Fan ZC, Tao YX. Functions of acidic transmembrane residues in human melanocortin-3 receptor binding and activation. Biochem Pharmacol. 2008;76:520-30
26. Biebermann H, Krude H, Elsner A, Chubanov V, Gudermann T, Gruters A. Autosomal-dominant mode of inheritance of a melanocortin-4 receptor mutation in a patient with severe early-onset obesity is due to a dominant-negative effect caused by receptor dimerization. Diabetes. 2003;52:2984-8
27. Donohoue PA, Tao YX, Collins M, Yeo GSH, O'Rahilly S, Segaloff DL. Deletion of codons 88-92 of the melanocortin-4 receptor gene: a novel deleterious mutation in an obese female. J Clin Endocrinol Metab. 2003;88:5841-5
28. Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O'Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085-95
29. Gao Z, Lei D, Welch J, Le K, Lin J, Leng S. et al. Agonist-dependent internalization of the human melanocortin-4 receptors in human embryonic kidney 293 cells. J Pharmacol Exp Ther. 2003;307:870-7
30. Shinyama H, Masuzaki H, Fang H, Flier JS. Regulation of melanocortin-4 receptor signaling: agonist-mediated desensitization and internalization. Endocrinology. 2003;144:1301-14
31. Nickolls SA, Fleck B, Hoare SR, Maki RA. Functional selectivity of melanocortin 4 receptor peptide and nonpeptide agonists: evidence for ligand-specific conformational states. J Pharmacol Exp Ther. 2005;313:1281-8
32. Mohammad S, Baldini G, Granell S, Narducci P, Martelli AM, Baldini G. Constitutive traffic of melanocortin-4 receptor in neuro2A cells and immortalized hypothalamic neurons. J Biol Chem. 2007;282:4963-74
33. McDaniel FK, Molden BM, Mohammad S, Baldini G, McPike L, Narducci P. et al. Constitutive cholesterol-dependent endocytosis of melanocortin-4 receptor (MC4R) is essential to maintain receptor responsiveness to α-melanocyte-stimulating hormone (α-MSH). J Biol Chem. 2012;287:21873-90
34. Rus R, Haag C, Bumke-Vogt C, Bahr V, Mayr B, Mohlig M. et al. Novel inactivating mutations of the calcium-sensing receptor: the calcimimetic NPS R-568 improves signal transduction of mutant receptors. J Clin Endocrinol Metab. 2008;93:4797-803
35. Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J. et al. Conformational biosensors reveal GPCR signalling from endosomes. Nature. 2013;495:534-8
36. Robben JH, Kortenoeven ML, Sze M, Yae C, Milligan G, Oorschot VM. et al. Intracellular activation of vasopressin V2 receptor mutants in nephrogenic diabetes insipidus by nonpeptide agonists. Proc Natl Acad Sci U S A. 2009;106:12195-200
Author contact
![]() Corresponding author: Ya-Xiong Tao, PhD, Department of Anatomy, Physiology and Pharmacology, 212 Greene Hall, College of Veterinary Medicine, Auburn University, Auburn, AL 36849, United States. Tel: 01-334-844-5396; FAX: 01-334-844-5388; Email: taoyaxiedu
Corresponding author: Ya-Xiong Tao, PhD, Department of Anatomy, Physiology and Pharmacology, 212 Greene Hall, College of Veterinary Medicine, Auburn University, Auburn, AL 36849, United States. Tel: 01-334-844-5396; FAX: 01-334-844-5388; Email: taoyaxiedu
Received 2014-5-11
Accepted 2014-7-1
Published 2014-7-20