ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2014; 10(9):957-965. doi:10.7150/ijbs.9773 This issue Cite
Research Paper
KRAS and DAXX/ATRX Gene Mutations Are Correlated with the Clinicopathological Features, Advanced Diseases, and Poor Prognosis in Chinese Patients with Pancreatic Neuroendocrine Tumors
Fei Yuan3*, Min Shi1*, Jun Ji1, Hailong Shi1, Chenfei Zhou1, Yingyan Yu1, Bingya Liu1, Zhenggang Zhu1,2, Jun Zhang1,2 ![]()
1. Department of Surgery, Shanghai Institute of Digestive Surgery, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, No. 197 Ruijin Er Road, Shanghai 200025, China;
2. Department of Clinical Oncology, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, No. 197 Ruijin Er Road, Shanghai 200025, China;
3. Department of Pathology, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, No. 197 Ruijin Er Road, Shanghai 200025, China.
*Equal contribution.
Abstract
Background and Aim: Pancreatic neuroendocrine tumor (pNET) is a clinically rare and heterogeneous group of tumors; its pharmacogenetic characteristics are not fully understood. This study was designed to examine the relationship between key gene variations and disease development and prognosis among Chinese patients with pNET.
Methods: Various pNET associated genes such as DAXX/ATRX, KRAS, MEN1, PTEN, TSC2, SMAD4/DPC, TP53 and VHL were analyzed in high-throughput sequencing. The links between the gene mutations and the clinicopathological features and prognosis of the patients were determined.
Results: The somatic mutation frequencies of the DAXX/ATRX, KRAS, MEN1, mTOR pathway genes (PTEN and TSC2), SMAD4/DPC, TP53, and VHL in Chinese pNET patients were 54.05%, 10.81%, 35.14%, 54.05%, 2.70%, 13.51%, and 40.54%, respectively, while the same figures in Caucasians pNET patients were 43%, 0%, 44%, 15%, 0%, 3%, and 0%, respectively. The numbers of mutated genes were from 0 to 6; 4 patients with more than 3 mutated genes had higher proliferation (Ki-67) index or nerve vascular invasion or organ involvement, but only 9 of 27 patients with 3 or few mutated genes had such features. Mutations in KRAS and DAXX/ATRX, but not other genes analyzed, were associated with a shortened survival.
Conclusion: The mutation rates of these genes in Chinese pNET patients are different from those in Caucasians. A higher number of gene mutations and the DAXX/ATRX and KRAS gene mutations are correlated with a poor prognosis of patients with pNET.
Keywords: pancreatic neuroendocrine tumor, gene mutation, prognosis, KRAS, DAXX/ATRX.
Introduction
Pancreatic neuroendocrine tumors (pNETs) include a heterogeneous group of tumors and account for about 3%-5% of all pancreatic malignancies, with an incidence estimated to range from 0.3 to 0.4 per 100,000 in the United States 1-3. The incidence and prevalence of pNETs have increased about five-fold in the last three decades, partly due to the improvement of diagnostic standards and early detection of asymptomatic lesions 3. The pNETs can be divided into functional and nonfunctional tumors. Functional tumors, such as insulinomas and gastrinomas, can secrete hormones and lead to corresponding clinical symptoms, whereas nonfunctional tumors cause non-specific symptoms, such as pain, an abdominal mass, pernicious vomiting, and anemia 4, 5.
The mainstay of treatment for pNETs is surgery. However, most patients are contraindicated for resection, because 65% of patients have unresectable or metastatic disease at the time of diagnosis 6. Unresectable pNETs are associated with a poor prognosis, with a median overall survival (mOS) of 24 months 3. The 10-year survival rate is only 40% and has not changed significantly over the last 30 years 2. Newer chemotherapeutic agents and molecular targeted therapy have recently been developed to treat the unresectable pNETs, but their efficacy has been modest 7.
The genetic background of pNETs has not been fully understood. One study has revealed that there are multiple genetic mutations in nonfamilial pNETs, including mutations in multiple endocrine neoplasias type 1 (MEN1), DAXX (death domain-associated protein)/ATRX (α-thalassemia/mental retardation syndrome X-linked), and genes in the mammalian target of rapamycin (mTOR) pathway 8. Interestingly, the mutations in the MEN1 and DAXX/ATRX genes are associated with a better prognosis of pNET patients in that study. Furthermore, the investigators suggest that the identification of mutations in genes in the mTOR pathway could be used to stratify pNET patients for treatment with mTOR inhibitors 8.
We have been interested in elucidating the genetic mechanisms for the development and progression of pNETs and believe patients' pharmacogenetic characgteristics could be used to guide their diagnosis and treatment. Additionally, it has been well documented that there are significant differences in genetic/genomic variations for many diseases among various ethnic groups. In the present study, we analyzed the gene mutations of Chinese pNET patients. Considering that all of the published studies had focused on Caucasian and Western populations and no studies have been conducted in Chinese populations, we compared the mutations in our patients with those of the previous studies in Westerners and examined the relationship between the key gene mutations and the clinic-pathological features and prognosis of our pNET patients.
Materials and Methods
Ethics Statement
The study was reviewed and approved by the Institutional Review Board (IRB) of Ruijin Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China. Informed written consent to participate in the study was obtained from each of the patients before the entry of the study.
Patient Characteristics, Tissue Samples, and Follow-Up
This was a retrospective study. From 2005 to 2011, 43 consecutive pNET patients underwent surgical resections of their tumors and had a definite pathological diagnosis of pNET in the Department of Surgery, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China. Six cases also had pancreatic adenocarcinoma and were excluded from the present study; therefore, 37 patients were finally enrolled in the study. All tumor specimens were formalin-fixed and paraffin-embedded. The tumors were classified into three groups according to the 2010 World Health Organization (WHO) classification criteria 9. The clinic-pathological data are shown in Table 1. Patients were followed up, and the overall survival time was analyzed.
DNA Extraction and Gene Sequencing
Cellular DNA was extracted from formalin-fixed paraffin-embedded (FFPE) tumor tissues using the QIAamp DNA FFPE Tissue Kit (QIAGEN, China). DNA was extracted only from samples with more than 80% tumor cell content. Target gene amplification was accomplished using Unicorn PCR per-mix kit (Shenzhen, China); the gene sequence and data analyses were applied under the guideline of HYKGENE sequence reagents kit or operation guide (Shenzhen, China). The sequencing flux requirement by a single locus was higher than that of 300×, and the quality requirement of the single locus sequence number was higher than 100× base number accounts for more than 95% of the total length. In somatic mutation selection criteria, the sequencing depth was more than 100, which had on different reads and distribution in different positions. The SNP/somatic mutations of the candidate loci were compared with the SNP database on the HapMap to confirm whether they were reported. Finally, filters were used to remove coding-synon, eventually forming the somatic mutation data of the Chinese pNET patients.
Characteristics of the 37 patients with pNETs from Ruijin Hospital.
| Sample | Age | Gender | Function | WHO classification | TNM(A7) | Ki-67 |
|---|---|---|---|---|---|---|
| pNET1 | 41 | Male | NF | G1 | Ib | ≤2% |
| pNET2 | 37 | Female | NF | G1 | Ib | ≤2% |
| pNET3 | 54 | Male | NF | G2 | Ib | ≤2% |
| pNET4 | 18 | Male | NF | G2 | IV | 3-20% |
| pNET6 | 61 | Male | NF | G1 | Ib | ≤2% |
| pNET7 | 58 | Female | NF | G1 | IIb | ≤2% |
| pNET9 | 32 | Female | Insulinoma | G1 | Ia | ≤2% |
| pNET10 | 66 | Male | NF | G1 | IIa | ≤2% |
| pNET11 | 64 | Female | NF | G1 | Ib | ≤2% |
| pNET13 | 51 | Female | NF | G1 | Ia | ≤2% |
| pNET14 | 48 | Male | NF | G1 | IIb | ≤2% |
| pNET15 | 47 | Male | NF | G1 | Ib | ≤2% |
| pNET16 | 48 | Male | NF | G1 | Ib | ≤2% |
| pNET17 | 61 | Female | NF | G1 | Ib | ≤2% |
| pNET18 | 47 | Male | NF | G3 | IIb | >20% |
| pNET20 | 38 | Female | NF | G2 | Ia | ≤2% |
| pNET21 | 65 | Female | Insulinoma | G2 | IV | 3-20% |
| pNET22 | 67 | Female | NF | G2 | Ia | ≤2% |
| pNET23 | 54 | Male | NF | G2 | Ia | ≤2% |
| pNET24 | 62 | Female | NF | G1 | Ib | ≤2% |
| pNET25 | 47 | Male | NF | G3 | IIa | >20% |
| pNET28 | 57 | Female | NF | G1 | Ib | ≤2% |
| pNET29 | 61 | Female | Insulinoma | G1 | Ia | ≤2% |
| pNET30 | 61 | Female | NF | G1 | Ib | ≤2% |
| pNET31 | 45 | Male | NF | G1 | Ia | ≤2% |
| pNET32 | 55 | Female | NF | G1 | Ia | ≤2% |
| pNET33 | 59 | Female | Insulinoma | G1 | Ia | ≤2% |
| pNET34 | 47 | Male | NF | G2 | IIa | ≤2% |
| pNET35 | 62 | Female | NF | G1 | Ia | ≤2% |
| pNET36 | 67 | Female | NF | G1 | Ib | ≤2% |
| pNET37 | 72 | Male | NF | G1 | Ib | ≤2% |
| pNET38 | 47 | Female | NF | G2 | Ib | 3-20% |
| pNET39 | 57 | Male | NF | G3 | IIa | >20% |
| pNET40 | 37 | Male | NF | G3 | IIb | >20% |
| pNET41 | 64 | Male | NF | G3 | IIa | >20% |
| pNET42 | 61 | Female | NF | G3 | IV | >20% |
| pNET43 | 35 | Female | NF | G2 | Ib | 3-20% |
Literature Review: Inclusion Criteria, Search Strategies, and Data Extraction
In the present study, we also conducted a systemic review on pNET clinical studies. Studies were selected according to the following inclusion criteria: 1) studies must explore the genetic alterations of pNETs; and 2) studies must be published in English in a peer-reviewed journal. We performed a search of the National Center for Biotechnology Information (NCBI) PubMed database using the search terms “Pancreatic endocrine tumors OR Pancreatic neuroendocrine tumors OR pancreatic islet cell tumor OR gastrinoma OR insulinoma OR glucagonoma OR vasoactive intestinal peptide tumor OR somatostatinoma OR nfpets OR growth hormone tumor OR acth tumor OR pancreatic polypeptide tumor OR pets causing carcinoid syndrome OR pets causing hypercalcemia AND mutation”. This search yielded 2278 articles published as of November 1, 2013. After manually screening the studies, we selected 178 articles for data mining, based on the aforementioned criteria. The data were extracted by two investigators independently to avoid any bias due to personal preference. When the two investigators were not in agreement, a third person was consulted to reach a final agreement regarding whether the article was suitable. The following information, if presented, was extracted and tabulated from each article: first author, year of publication, journal, genotype, gene, mutation type, chromosomal location, number of cases, number of mutation cases, population, family history, pNET type, gender, and any other important information.
Statistical Analysis
The chi-squared test was used to analyze the categorical variables. The log-rank test for the Kaplan-Meier method and the Cox regression test were used to assess the patients' outcomes and the prognostic factors. All statistical analyses were performed using the SPSS 17.0 software program. A two-tailed value of P<0.05 was considered statistically significant.
Results
Literature search results
Based on the literature search, 178 papers were included in the analysis of the mutation frequency after manually screening the full text. These reports described 551 mutations in 11 different genes (SMAD4/DPC4, KRAS, TP53, VHL, PTEN, TSC2, DAXX/ATRX, MEN1, BRAF, BRCA2 and PIK3CA). The mutations and the gene functions were summarized as a table (Supplementary Table 1).
Mutation Frequency in pNET Tissue Samples
By using Sanger sequencing, we identified 133 somatic mutations in eight different genes in our cases (Table 2). The somatic mutation frequency of the DAXX/ATRX, KRAS, MEN1, mTOR pathway genes (PTEN and TSC2), SMAD4/DPC, TP53 and VHL were 54.05%, 10.81%, 35.14%, 54.05%, 2.70%, 13.51%, and 40.54%, respectively.
There were some obvious differences between our findings and the results reported by Jiao et al 8, in which similar genes were investigated. In our study, the KRAS, TP53, mTOR pathway genes (PTEN and TSC2), and VHL genes had significantly higher mutation rates than those in their study (Table 3). The potential reasons for these differences are not clear by may be associated with the following: 1) most of the subjects in their study were Caucasian (85%), while in our study, all of the patients were Chinese; and 2) there were only three patients with Grade 3 (poorly differentiated) tumors in their study (5%), but there were six cases (16.2%) with of poorly differentiated tumors in our study.
The mutations in SMAD4/DPC4. KRAS, TP53, VHL, PTEN, TSC2, DAXX, ATRX, and MEN1 in pNETs.
| Sample | Gene | Nucleotide (genomic) | Amino acid change | Mutation type |
|---|---|---|---|---|
| pNET4 | ATRX | chrx:76778854 | CAG(Q)>CCG(P) | missense |
| ATRX | chrx:76907608 | AGA(R)>ATA(I) | missense | |
| ATRX | chrx:76814262 | GCT(A)>ACT(T) | missense | |
| ATRX | chrx:76814186 | CGC(R)>CAC(H) | missense | |
| pNET9 | ATRX | chrx:76907836-76907837 | indel | |
| pNET15 | ATRX | chrx:76907741 | Gly(GGA)->Arg(AGA) | missense |
| pNET17 | ATRX | chrx:76907744 | Pro(CCT)->Ser(TCT) | missense |
| pNET25 | ATRX | chrx:76874303 | CTC(L)>GTC(V) | missense |
| pNET28 | ATRX | chrx:76907645 | CGT(R)>GGT(G) | missense |
| pNET31 | ATRX | chrx:76778855 | CAG(Q)>TAG(Stop) | nonsense |
| pNET34 | ATRX | chrx:76874303 | CTC(L)>GTC(V) | missense |
| pNET37 | ATRX | chrx:76778844 | AAA(K)>AAT(N) | missense |
| pNET40 | ATRX | chrx:76939403 | CCT(P)>GCT(A) | missense |
| ATRX | chrx:76907612 | TTG(L)>ATG(M) | missense | |
| ATRX | chrx:76907660 | GAA(E)>AAA(K) | missense | |
| pNET41 | ATRX | chrx:76907663 | GAG(E)>CAG(Q) | missense |
| ATRX | chrx:76907840 | AAT(N)>TAT(Y) | missense | |
| pNET42 | ATRX | chrx:76909619 | AAG(K)>AGG(R) | missense |
| ATRX | chrx:76907840 | AAT(N)>TAT(Y) | missense | |
| pNET43 | ATRX | chrx:76907660 | GAA(E)>AAA(K) | missense |
| pNET1 | DAXX | chr6:33287895 | Glu(GAA)->Gly(GGA) | missense |
| pNET3 | DAXX | chr6:33288158 | Glu(GAG)->Val(GTG) | missense |
| pNET4 | DAXX | chr6:33287895 | Glu(GAA)->Gly(GGA) | missense |
| pNET6 | DAXX | chr6:33287869 | Glu(GAA)->Lys(AAA) | missense |
| pNET15 | DAXX | chr6:33289268 | Ala(GCC)->Val(GTC) | missense |
| pNET20 | DAXX | chr6:33289256-33289256 | indel | |
| pNET22 | DAXX | chr6:33289256-33289256 | indel | |
| pNET25 | DAXX | chr6:33286925 | TCC(S)>TTC(F) | missense |
| pNET29 | DAXX | chr6:33288170 | L-Stop 413 | nonsense |
| pNET30 | DAXX | chr6:33287844 | ATG(M)>AGG(R) | missense |
| pNET31 | DAXX | chr6:33287902 | GAG(E)>CAG(Q) | missense |
| DAXX | chr6:33287844 | ATG(M)>AGG(R) | missense | |
| pNET15 | KRAS | chr12:25398299 | Val(GTG)->Ala(GCG) | missense |
| pNET25 | KRAS | chr12:25398284 | rs121913529 | missense |
| Gly(GGT)->Asp(GAT) | ||||
| pNET31 | KRAS | chr12:25398266 | Ala(GCC)->Val(GTC) | missense |
| pNET40 | KRAS | chr12:25398284 | rs121913529 | missense |
| Gly(GGT)->Asp(GAT) | ||||
| pNET1 | MEN1 | chr11:64577464 | Val(GTG)->Leu(TTG) | missense |
| MEN1 | chr11:64577355 | T->I 76 | missense | |
| pNET3 | MEN1 | chr11:64577526 | Val(GTG)->Glu(GAG) | missense |
| pNET4 | MEN1 | chr11:64574502 | Pro(CCA)->Leu(CTA) | missense |
| MEN1 | chr11:64577391 | CAG(Q)>CGG(R) | missense | |
| MEN1 | chr11:64577182 | TTC(F)>CTC(L) | missense | |
| pNET9 | MEN1 | chr11:64577397 | Thr(ACC)->Ile(ATC) | missense |
| pNET13 | MEN1 | chr11:64574514 | Gly(GGC)->Asp(GAC) | missense |
| pNET14 | MEN1 | chr11:64577307 | R->H 92 | missense |
| MEN1 | chr11:64577397 | Thr(ACC)->Ile(ATC) | missense | |
| pNET15 | MEN1 | chr11:64577185 | Y->H 133 | missense |
| pNET17 | MEN1 | chr11:64574502 | Pro(CCA)->Leu(CTA) | missense |
| pNET30 | MEN1 | chr11:64574565 | CCC(P)>CTC(L) | missense |
| pNET33 | MEN1 | chr11:64577389 | CCC(P)>ACC(T) | missense |
| CCC(P)>TCC(S) | missense | |||
| pNET34 | MEN1 | chr11:64577316 | Leu(CTC)->Arg(CGC) | missense |
| MEN1 | chr11:64577389 | CCC(P)>ACC(T) | missense | |
| CCC(P)>TCC(S) | ||||
| pNET35 | MEN1 | chr11:64574671 | Tyr(TAT)->Stop(TAG) | nonsense |
| pNET38 | MEN1 | chr11:64574502 | Pro(CCA)->Leu(CTA) | missense |
| pNET3 | TP53 | chr17:7579424 | Ala(GCC)->Asp(GAC) | missense |
| pNET25 | TP53 | chr17:7579387 | Q->H 100 | missense |
| pNET36 | TP53 | chr17:7577509-7577509 | indel | |
| pNET37 | TP53 | chr17:7579293-7579294 | indel | |
| pNET41 | TP53 | chr17:7578481 | ACA(T)>AAA(K) | missense |
| pNET3 | PTEN | chr10:89720826 | Asp(GAC)->Ala(GCC) | missense |
| pNET18 | PTEN | chr10:89720870 | Phe(TTT)->Val(GTT) | missense |
| pNET21 | PTEN | chr10:89720870 | Phe(TTT)->Val(GTT) | missense |
| pNET24 | PTEN | chr10:89720826 | Asp(GAC)->Ala(GCC) | missense |
| pNET25 | PTEN | chr10:89720853 | CGA(R)>CAA(Q) | missense |
| pNET32 | PTEN | chr10:89720870 | Phe(TTT)->Val(GTT) | missense |
| pNET34 | PTEN | chr10:89685307 | TAC(Y)>CAC(H) | missense |
| PTEN | chr10:89720816-89720817 | rs121913291 | indel | |
| pNET22 | SMAD4/DPC4 | chr18:48593423 | Lys(AAA)->Gln(CAA) | missense |
| pNET1 | TSC2 | chr16:2134968 | Leu(CTC)->Val(GTC) | missense |
| pNET4 | TSC2 | chr16:2130258 | GCG(A)>ACG(T) | missense |
| pNET7 | TSC2 | chr16:2134973-2134974 | rs137854017 | indel |
| pNET10 | TSC2 | chr16:2134973-2134974 | rs137854017 | indel |
| pNET15 | TSC2 | chr16:2134994 | D->E 151 | missense |
| TSC2 | chr16:2135013 | L->M 1519 | missense | |
| pNET16 | TSC2 | chr16:2134994 | D->E 151 | missense |
| TSC2 | chr16:2135013 | L->M 1519 | missense | |
| TSC2 | chr16:2136843 | G->S 1654 | missense | |
| pNET20 | TSC2 | chr16:2136843 | G->S 1654 | missense |
| pNET24 | TSC2 | chr16:2134992 | D->N 1512 | missense |
| TSC2 | chr16:2136873-2136873 | rs137854005 | indel | |
| pNET25 | TSC2 | chr16:2130168-2130169 | rs137854314 | indel |
| pNET28 | TSC2 | chr16:2134961-2134962 | rs137854160 | indel |
| pNET29 | TSC2 | chr16:2130168-2130169 | rs137854314 | indel |
| pNET30 | TSC2 | chr16:2134329 | CGG(R)>CAG(Q) | missense |
| pNET31 | TSC2 | chr16:2134328 | rs45517328 1369 | missense |
| TSC2 | chr16:2134391 | GAG(E)>CAG(Q) | missense | |
| pNET33 | TSC2 | chr16:2134961-2134962 | rs137854160 | indel |
| TSC2 | chr16:2130259 | rs45448801 | missense | |
| GCG(A)>GAG(E) | ||||
| pNET34 | TSC2 | chr16:2134961-2134962 | rs137854160 | indel |
| pNET35 | TSC2 | chr16:2134961-2134962 | rs137854160 | indel |
| TSC2 | chr16:2130168-2130169 | rs137854314 | indel | |
| TSC2 | chr16:2130266-2130266 | rs137854340 | indel | |
| pNET1 | VHL | chr3:10183775 | Arg(CGC)->Cys(TGC) | missense |
| VHL | chr3:10188212 | F->I 119 | missense | |
| VHL | chr3:10188218 | D->H 121 | missense | |
| VHL | chr3:10183734 | Ser(TCG)->Leu(TTG) | missense | |
| pNET3 | VHL | chr3:10183737 | Arg(CGC)->His(CAC) | missense |
| VHL | chr3:10188304-10188305 | indel | ||
| pNET7 | VHL | chr3:10191563 | Glu(GAA)->Lys(AAA) | missense |
| pNET10 | VHL | chr3:10183841 | Gly(GGC)->Ser(AGC) | missense |
| pNET13 | VHL | chr3:10188307-10188308 | rs5030624 | indel |
| pNET14 | VHL | chr3:10183736 | Arg(CGC)->Cys (TGC) | missense |
| VHL | chr3:10183734 | Ser(TCG)->Leu(TTG) | missense | |
| pNET15 | VHL | chr3:10183736 | Arg(CGC)->Cys (TGC) | missense |
| pNET17 | VHL | chr3:10183826 | Pro(CCA)->Ser(TCA) | missense |
| pNET20 | VHL | chr3:10183734 | Ser(TCG)->Leu(TTG) | missense |
| VHL | chr3:10188304-10188305 | indel | ||
| pNET22 | VHL | chr3:10183734 | Ser(TCG)->Leu(TTG) | missense |
| pNET25 | VHL | chr3:10183736 | Arg(CGC)->Cys (TGC) | missense |
| VHL | chr3:10183682 | Glu(GAG)->Stop(TAG) | nonsense | |
| pNET28 | VHL | chr3:10183704 | CGG(R)>CAG(Q) | missense |
| pNET33 | VHL | chr3:10183802 | Phe(TTC)->Leu(CTC) | missense |
| pNET34 | VHL | chr3:10188304-10188305 | indel | |
| pNET38 | VHL | chr3:10183809 | GGC(G)>GAC(D) | missense |
| VHL | chr3:10188230 | CAC(H)>TAC(Y) | missense |
Comparison of the most commonly mutated genes in pNETs between Chinese and Caucasians.
| Genes | pNETs (Chinese) | pNETs (Caucasian) a |
|---|---|---|
| KRAS | 10.81% | 0% |
| TP53 | 13.51% | 3% |
| mTOR (PTEN and TSC2) | 54.05% | 15% |
| VHL | 40.54% | 0% |
| SMAD4/DPC4 | 2.70% | 0% |
| DAXX/ATRX | 54.05% | 43% |
| MEN1 | 35.14% | 44% |
a The data are from Science 2011;331:1199-203.
Gene Mutations in KRAS, TP53, TSC2, and VHL in pNET Tissue Samples
The mutation frequencies of the KRAS, TP53, TSC2, and VHL genes in our study were different from those in Caucasians, so we subjected these genes to a more detailed mutation analysis (Table 2). We detected four cases with KRAS mutations, all of which were missense mutations. There were two G-to-A transitions and one T-to-C transition and one C-to-T transition. All of the mutations were in the GTPase domain. Fifteen of the 30 cases examined for VHL gene mutations were positive, including 23 different mutant sites, among which were 18 missense mutations, four indels, and one nonsense mutations. C-to-T transitions and C-to-A transitions were responsible for 55.6% and 27.8%, respectively, of the missense mutations. The mutations occurred mainly in the β domain and its nearby regions.
Fifteen cases were identified that had TSC2 gene mutations, among which 25 different mutant sites containing 14 missense mutations and 11 indels were found. There were two G-to-A transitions, one G-to-C, one C-to-G, and one C-to-A transition. The TSC2 mutations mainly occurred in or near the RAP-GAP domain. We detected five cases with TP53 mutations, three of which were missense mutations and two of which were indels. Two C-to-A transitions were detected in the missense mutations. One patient had an indel mutation at the 258 region, which is located at the center of the TP53 DNA binding domain (102-292 region). This patient had a higher degree of differentiation and an earlier clinical stage compared to the other patients. His overall survival time was also longer than the average of the remaining patients (45.8 vs. 26.7 months, P<0.05). This suggested that patients with the TP53 indel mutation in the 258 region may have a lower level of malignancy and a better prognosis.
Relationships between Gene Mutations and the Clinico-pathological Characteristics of pNETs
The three most frequently mutated genes found in our study were DAXX/ATRX (54.1%), TSC2 (43.2%) and VHL (40.5%). Missense mutations were the most common type of mutation in DAXX/ATRX and VHL (comprising 82.1% and 78.3% of mutations, respectively), while in TSC2, missense and indel mutations comprised 54.2% and 45.8% of the mutations, respectively (Table 4). The number of mutations among these well-differentiated samples are from 0 to 6 and we cutoff at the median 3, there were 27 patients have 3 or less genes occur mutations and only 9 patients in them have higher Ki-67 index or associated with the nerve vascular invasion or organ involvement. Otherwise in other 4 patients with more than 3 mutated genes all have these features (Table 5). This difference may indicates that well-differentiated patients with mutations of multiple genes tend tumor invasion. Specifically, one patient (No. 25) had six gene mutations, with a Ki-67 index of 80%, with neurological vascular invasion and spleen metastasis. These findings suggested that the number of mutated genes may be correlated with the pNETs' growth and progression.
All gene mutations identified in the 37 Chinese pNETs patients.
| Sample | SMAD4/DPC4 | KRAS | TP53 | VHL | PTEN | TSC2 | DAXX/ATRX | MEN1 |
|---|---|---|---|---|---|---|---|---|
| pNET1 | missense | missense | missense | missense | ||||
| pNET2 | ||||||||
| pNET3 | missense | missense/ indel | indel | missense | missense | |||
| pNET4 | missense | missense | missense | |||||
| pNET6 | missense | |||||||
| pNET7 | missense | Indel | ||||||
| pNET9 | indel | missense | ||||||
| pNET10 | missense | Indel | ||||||
| pNET11 | ||||||||
| pNET13 | indel | missense | ||||||
| pNET14 | missense | missense | ||||||
| pNET15 | missense | missense | missense | missense | missense | |||
| pNET16 | missense | |||||||
| pNET17 | missense | missense | missense | |||||
| pNET18 | missense | |||||||
| pNET20 | missense/ indel | missense | indel | |||||
| pNET21 | missense | |||||||
| pNET22 | Missense | missense | indel | |||||
| pNET23 | ||||||||
| pNET24 | missense | missense/ indel | ||||||
| pNET25 | missense | missense | missense/ nonsense | missense | indel | missense | ||
| pNET28 | missense | indel | missense | |||||
| pNET29 | indel | nonsense | ||||||
| pNET30 | missense | missense | missense | |||||
| pNET31 | missense | missense | missense/ nonsense | |||||
| pNET32 | missense | |||||||
| pNET33 | missense | indel | missense | |||||
| pNET34 | indel | missense/ indel | indel | missense | missense | |||
| pNET35 | indel | nonsense | ||||||
| pNET36 | indel | |||||||
| pNET37 | indel | missense | ||||||
| pNET38 | missense | missense | ||||||
| pNET39 | ||||||||
| pNET40 | missense | missense | ||||||
| pNET41 | missense | missense | ||||||
| pNET42 | missense | |||||||
| pNET43 | missense |
The relationship between the number of mutated genes and the Ki-67 index or TNM stage of well-differentiated pNETs patients.
| Ki-67 index or TNM stage | ||||
|---|---|---|---|---|
| Number of mutated genes | Cases (n=31) | High | Low | P value |
| ≤ 3 | 27 | 9 | 18 | 0.012 |
| > 3 | 4 | 4 | 0 | |
The KRAS and DAXX/ATRX Gene Mutations Predict a Poor Prognosis in Patients With pNETs
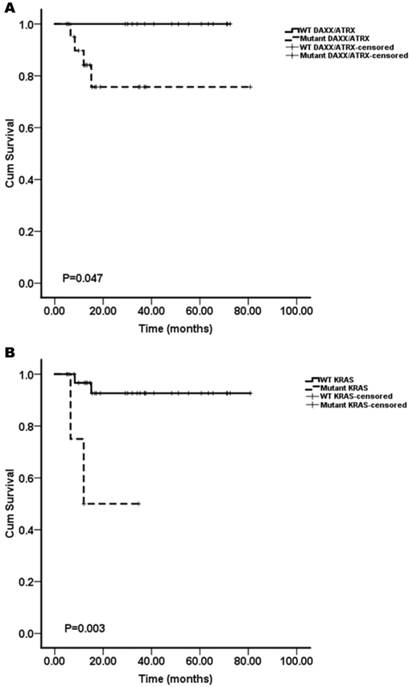
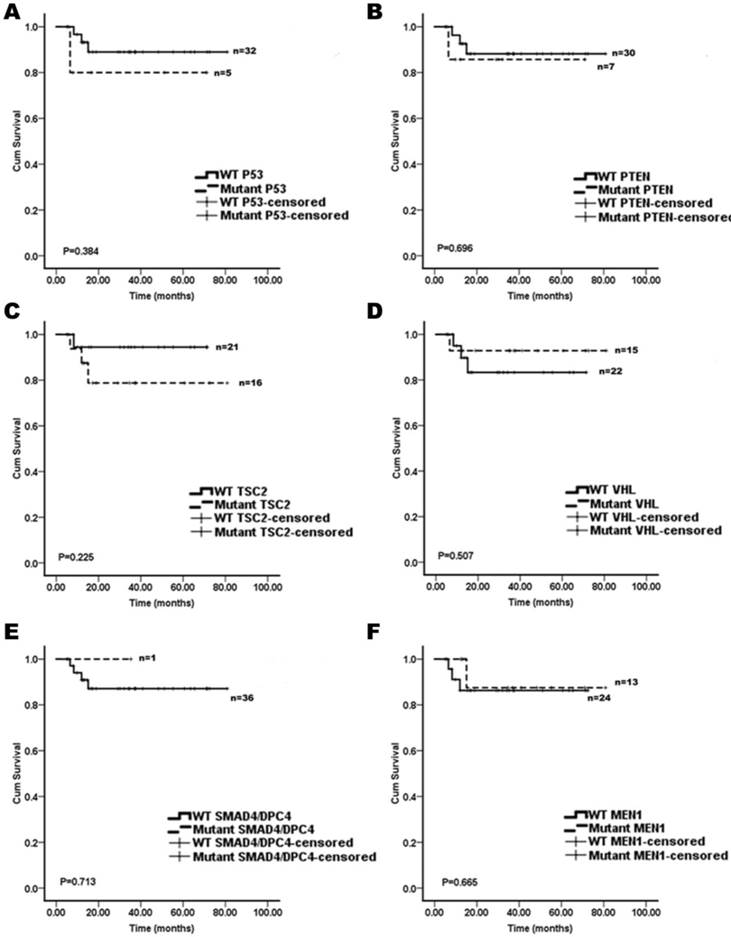
We assessed whether the mutations in KRAS, TP53, mTOR pathway genes (PTEN and TSC2), VHL, SMAD4/DPC4, DAXX/ATRX, and MEN1 were associated with the patient survival. A Kaplan-Meier survival analysis was performed for the 37 pNET patients. Mutations in KRAS and DAXX/ATRX were found to be associated with a shortened survival (Figure 1), while mutations in TP53, mTOR pathway genes (PTEN and TSC2), VHL, SMAD4/DPC4, and MEN1 did not predict the pNET patients' survival (Figure 2). To our knowledge, this is the first report that the presence of KRAS gene mutations was confirmed in pNETs, and furthermore, provides the first evidence that KRAS gene mutations may predict a poor survival.
Interestingly, the result that mutations of DAXX/ATRX were associated with a shortened survival was inconsistent with the report by Jiao et al.8 Their results showed that mutations in MEN1, DAXX/ATRX, or the combination of both MEN1 and DAXX/ATRX were associated with prolonged survival relative to that of the patients whose tumors lacked these mutations. The results reported by Marinoni et al. 10 are in agreement with our results. In their report, the mutations found in the DAXX/ATRX genes correlated with the loss of the corresponding protein expression in pNETs, and loss of DAXX/ATRX expression was significantly correlated with both a shortened relapse-free survival and tumor-specific survival.
The Kaplan-Meier curves of the overall survival of pNET patients, stratified by the status of gene mutations. A: DAXX/ATRX; B: KRAS. Mutations in DAXX/ATRX and KRAS were found to be associated with a shortened survival (n=37; all p<0.05).
The Kaplan-Meier curves of the overall survival of pNET patients, stratified by the status of gene mutations. A: TP53; B: PTEN; C: TSC2; D: VHL; E: SMAD4/DPC4; and F: MEN1. These mutations analyzed were not associated with survivals of the pNET patients (n=37; all p>0.05).
Discussion
In the present study with 37 Chinese pNET patients, we identified 133 somatic mutations in eight different genes, including KRAS, TP53, PTEN, TSC2, VHL, SMAD4/DPC4, DAXX/ATRX, and MEN1. Compared to the results from Caucasians, there were several remarkable differences: 1) the KRAS, TP53, mTOR pathway genes (PTEN and TSC2), and VHL genes had significantly higher mutation rates in Chinese patients than in Caucasian patients; 2) the number of gene mutations was correlated with the growth and progression of well-differentiated PNETs patients; and 3) KRAS and DAXX/ATRX gene mutations were associated with a shortened survival in Chinese pNET patients.
The mutations of such genes may be involved in the development and progression of pNETs. The VHL protein contains two structural domains, called α and β. The α domain can bind to Elongin B or C, while the β domain can interact with hypoxia-inducible factor-1α (HIF-1α). Fifteen of the 30 cases that were evaluated were identified to have VHL gene mutations, and the mutations occurred mainly in or near the β domain. This may have caused VHL to fail to interact with HIF-1α, which would lead to a decrease in HIF-1α ubiquitination and degradation. As a result, the subsequent downstream genes of HIF-1α, such as VEGF, TGF-α and various cytokines, would exhibit sustained activation, which could promote tumor development and progression 11, 12.
It has been demonstrated that pNETs exhibit overexpression of VEGF, its receptor, VEGFR, and platelet-derived growth factor receptor (PDGFR) 13, 14. Sunitinib malate, an oral tyrosine kinase inhibitor, has activity against VEGFRs, PDGFRs, stem-cell factor (KIT) receptor, glial cell line-derived neurotrophic factor, and FMS-like tyrosine kinase-3 15. Recently, several phase II and phase III trials have demonstrated that the administration of sunitinib can significantly improve the progression-free survival and overall survival, and that it shows an acceptable safety profile in patients with advanced pNETs 16-18.
During the process of pNET development, the receptor tyrosine kinase (RTK)-PI3K/PTEN-AKT-TSC1/2-mTOR signaling pathway plays an important role in the control of cell growth, proliferation, survival, and differentiation 19, 20. TSC2 and TSC1 can form a dimer and inhibit the GTPase activity of Rheb, a critical activator of mTOR signaling 21. TSC2 contains two structural domains: one is necessary for the binding to TSC1, but the deletion of this domain has no effect on its GAP activity; another is the RAP-GAP domain. When this domain is mutated, the GTPase activity of Rheb is enhanced, resulting in the activation of mTOR 22. In our study, TSC2 mutations mainly occurred in or near the RAP-GAP domain. Everolimus is an mTOR inhibitor, and many studies have shown the efficacy and safety of everolimus in the treatment of advanced pNETs 23-25. Based on these findings, the results of our study are consistent with the previous reports, and may help predict the response of pNETs to molecular targeted therapies 8.
Mutations in DAXX or ATRX have been detected in about 40% of pNETs. DAXX is an H3.3-specific histone chaperone, while ATRX encodes a protein that has an ADD (ATRX-DNMTT3-DNMT3L) domain at the amino-terminus and a carboxy-terminal helicase domain 10, 26. The interaction between DAXX and ATRX is required for H3.3 incorporation at the telomeres, and ATRX, but not DAXX, could suppress the telomeric repeat-containing RNA expression. Furthermore, ATRX was also found to target CpG islands and G-rich tandem repeats, which exist close to telomeric regions 27-30. In our study, the relationship between DAXX/ATRX mutations and the poor prognosis of pNET patients was inconsistent with the findings of a previous report. In that report, Jiao et al. showed that DAXX/ATRX mutations were associated with prolonged survival in pNET patients. In contrast, Marinoni et al. showed that the loss of DAXX/ATRX expression caused by a DAXX/ATRX gene mutation was significantly correlated with both a shortened relapse-free and tumor-specific survival. The reasons for these differences may include the following: 1) The DAXX/ATRX mutation rate in our study was higher than that in the other studies; 2) The patients in our study were all Chinese, while those in the other studies were mostly Caucasian; and 3) The mutated regions of DAXX/ATRX in our study were different from those in the other studies.
KRAS, a member of the rat sarcoma viral oncogene homolog (RAS) proto-oncogene family, is a key protein in the EGFR pathway, and plays an important role in the activation of various downstream proteins, including Raf, MEK and ERK 31. The mutant status of KRAS may inhibit GTP enzymatic activity, resulting in sustained activation of the EGRF signaling pathway. Codons 12, 13, and 61 are the most common mutation sites in KRAS 32, 33. The mutation rate of KRAS in pancreatic cancer is about 90%, and there is a major region of mutation in codon 12. However, the KRAS mutation rate in pNETs has rarely been reported. In our study, the mutation rate of KRAS was 10.81%, while there were no KRAS mutations reported by Jiao et al. The KARS mutations in our study were found in codons 7, 12, and 18. Interestingly, our study suggests that the presence of a KRAS gene mutation may predict a poorer survival. This was a novel finding, but needs to be further analyzed in a larger number of patients.
In conclusion, the significance of our study is the first to report the gene mutations of pNETs in Chinese patients. Our data indicated that DAXX/ATRX and KRAS gene mutations were correlated with a poor prognosis of pNET patients, and the patients bearing a larger number of gene mutations had a worse prognosis. The limitation of this study may be associated with the small sample size and the sequence methods; thus, future studies enrolling a larger number of pNET patients should be performed, and whole exome sequencing should be conducted to further explore the role of newly identified gene mutations in pNET development and progression.
Supplementary Material
Supplementary Table 1Acknowledgements
The study was supported by National Science Foundation of China (81372645) and Shanghai Natural Science Foundation from Municipal Government (13ZR1425900) and Shanghai Jiao Tong University School of Medicine Science and Technology Foundation (13XJ10035) and FONG SHU FOOK TONG Foundation.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rindi G, Wiedenmann B. Neuroendocrine neoplasms of the gut and pancreas: new insights. Nat Rev Endocrinol. 2012;8:54-64
2. Halfdanarson TR, Rabe KG, Rubin J. et al. Pancreatic neuroendocrine tumors (PNETs): incidence, prognosis and recent trend toward improved survival. Ann Oncol. 2008;19:1727-33
3. Yao JC, Hassan M, Phan A. et al. One hundred years after "carcinoid": epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063-72
4. Ramage JK, Ahmed A, Ardill J. et al. Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumours (NETs). Gut. 2012;61:6-32
5. Turaga KK, Kvols LK. Recent progress in the understanding, diagnosis, and treatment of gastroenteropancreatic neuroendocrine tumors. CA Cancer J Clin. 2011;61:113-32
6. Valle JW, Eatock M, Clueit B. et al. A systematic review of non-surgical treatments for pancreatic neuroendocrine tumours. Cancer Treat Rev. 2013
7. Sharma J, Duque M, Saif MW. Emerging therapies and latest development in the treatment of unresectable pancreatic neuroendocrine tumors: an update for clinicians. Therap Adv Gastroenterol. 2013;6:474-90
8. Jiao Y, Shi C, Edil BH. et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199-203
9. Klimstra DS, Modlin IR, Coppola D. et al. The pathologic classification of neuroendocrine tumors: a review of nomenclature, grading, and staging systems. Pancreas. 2010;39:707-12
10. Marinoni I, Kurrer AS, Vassella E. et al. Loss of DAXX and ATRX Are Associated With Chromosome Instability and Reduced Survival of Patients With Pancreatic Neuroendocrine Tumors. Gastroenterology. 2013
11. Jung YS, Lee SJ, Yoon MH. et al. Estrogen receptor alpha is a novel target of the Von Hippel-Lindau protein and is responsible for the proliferation of VHL-deficient cells under hypoxic conditions. Cell Cycle. 2012;11:4462-73
12. Sourbier C, Srivastava G, Ghosh MC. et al. Targeting HIF2alpha translation with Tempol in VHL-deficient clear cell renal cell carcinoma. Oncotarget. 2012;3:1472-82
13. Wiedenmann B, Pavel M, Kos-Kudla B. From targets to treatments: a review of molecular targets in pancreatic neuroendocrine tumors. Neuroendocrinology. 2011;94:177-90
14. Zhang J, Jia Z, Li Q. et al. Elevated expression of vascular endothelial growth factor correlates with increased angiogenesis and decreased progression-free survival among patients with low-grade neuroendocrine tumors. Cancer. 2007;109:1478-86
15. Mena AC, Pulido EG, Guillen-Ponce C. Understanding the molecular-based mechanism of action of the tyrosine kinase inhibitor: sunitinib. Anticancer Drugs. 2010;21(Suppl 1):S3-11
16. Kulke MH, Lenz HJ, Meropol NJ. et al. Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol. 2008;26:3403-10
17. Bajetta E, Guadalupi V, Procopio G. Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol. 2009;27:319-20 author reply 20
18. Raymond E, Dahan L, Raoul JL. et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:501-13
19. Populo H, Lopes JM, Soares P. The mTOR Signalling Pathway in Human Cancer. Int J Mol Sci. 2012;13:1886-918
20. Zhou CF, Ji J, Yuan F. et al. mTOR activation in well differentiated pancreatic neuroendocrine tumors: a retrospective study on 34 cases. Hepatogastroenterology. 2011;58:2140-3
21. Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179-90
22. Hoogeveen-Westerveld M, van Unen L, van den Ouweland A. et al. The TSC1-TSC2 complex consists of multiple TSC1 and TSC2 subunits. BMC Biochem. 2012;13:18
23. Yao JC, Phan AT, Chang DZ. et al. Efficacy of RAD001 (everolimus) and octreotide LAR in advanced low- to intermediate-grade neuroendocrine tumors: results of a phase II study. J Clin Oncol. 2008;26:4311-8
24. Yao JC, Shah MH, Ito T. et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514-23
25. Ito T, Okusaka T, Ikeda M. et al. Everolimus for advanced pancreatic neuroendocrine tumours: a subgroup analysis evaluating Japanese patients in the RADIANT-3 trial. Jpn J Clin Oncol. 2012;42:903-11
26. Lewis PW, Elsaesser SJ, Noh KM. et al. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci U S A. 2010;107:14075-80
27. Drane P, Ouararhni K, Depaux A. et al. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010;24:1253-65
28. Goldberg AD, Banaszynski LA, Noh KM. et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678-91
29. Wong LH, McGhie JD, Sim M. et al. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res. 2010;20:351-60
30. Law MJ, Lower KM, Voon HP. et al. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell. 2010;143:367-78
31. Yan J, Roy S, Apolloni A. et al. Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J Biol Chem. 1998;273:24052-6
32. Guerrero S, Casanova I, Farre L. et al. K-ras codon 12 mutation induces higher level of resistance to apoptosis and predisposition to anchorage-independent growth than codon 13 mutation or proto-oncogene overexpression. Cancer Res. 2000;60:6750-6
33. Mao C, Huang YF, Yang ZY. et al. KRAS p.G13D mutation and codon 12 mutations are not created equal in predicting clinical outcomes of cetuximab in metastatic colorectal cancer: a systematic review and meta-analysis. Cancer. 2013;119:714-21
Author contact
![]() Corresponding author: Departments of Surgery and Clinical Oncology, Shanghai Institute of Digestive Surgery, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, No. 197 Ruijin Er Road, Shanghai 200025, China; Tel: +86-21-64741635; Fax: +86-21-64741635; E-mail: jun_zj10977com.
Corresponding author: Departments of Surgery and Clinical Oncology, Shanghai Institute of Digestive Surgery, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, No. 197 Ruijin Er Road, Shanghai 200025, China; Tel: +86-21-64741635; Fax: +86-21-64741635; E-mail: jun_zj10977com.
Received 2014-5-30
Accepted 2014-8-12
Published 2014-8-30