ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2014; 10(10):1108-1115. doi:10.7150/ijbs.9797 This issue Cite
Research Paper
Dexmedetomidine-induced Contraction Involves Phosphorylation of Caldesmon by JNK in Endothelium-denuded Rat Aortas
Jiseok Baik1*, Seong-Ho Ok2*, Hyunhoo Cho3, Jongsun Yu3, Woochan Kim3, In-Koo Nam3, Mun-Jeoung Choi4, Heon-Keun Lee2, Ju-Tae Sohn2 ![]()
1. Department of Anesthesiology and Pain Medicine, Pusan National University Hospital, Biomed Research Institute, Pusan National University School of Medicine, Busan, Republic of Korea
2. Department of Anesthesiology and Pain Medicine, Institute of Health Sciences, Gyeongsang National University School of Medicine, Gyeongsang National University Hospital, Jinju, Republic of Korea
3. Department of Anesthesiology and Pain Medicine, Gyeongsang National University Hospital, Jinju, Republic of Korea
4. Department of Oral and Maxillofacial Surgery, Gyeongsang National University Hospital, Jinju, Republic of Korea
* These authors contributed equally to this study as co-first authors.
Abstract
Caldesmon, an inhibitory actin binding protein, binds to actin and inhibits actin-myosin interactions, whereas caldesmon phosphorylation reverses the inhibitory effect of caldesmon on actin-myosin interactions, potentially leading to enhanced contraction. The goal of this study was to investigate the cellular signaling pathway responsible for caldesmon phosphorylation, which is involved in the regulation of the contraction induced by dexmedetomidine (DMT), an alpha-2 adrenoceptor agonist, in endothelium-denuded rat aortas. SP600125 (a c-Jun NH2-terminal kinase [JNK] inhibitor) dose-response curves were generated in aortas that were pre-contracted with DMT or phorbol 12,13-dibutyrate (PDBu), a protein kinase C (PKC) activator. Dose-response curves to the PKC inhibitor chelerythrine were generated in rat aortas pre-contracted with DMT. The effects of SP600125 and rauwolscine (an alpha-2 adrenoceptor inhibitor) on DMT-induced caldesmon phosphorylation in rat aortic vascular smooth muscle cells (VSMCs) were investigated by western blot analysis. PDBu-induced caldesmon and DMT-induced PKC phosphorylation in rat aortic VSMCs was investigated by western blot analysis. The effects of GF109203X (a PKC inhibitor) on DMT- or PDBu-induced JNK phosphorylation in VSMCs were assessed. SP600125 resulted in the relaxation of aortas that were pre-contracted with DMT or PDBu, whereas rauwolscine attenuated DMT-induced contraction. Chelerythrine resulted in the vasodilation of aortas pre-contracted with DMT. SP600125 and rauwolscine inhibited DMT-induced caldesmon phosphorylation. Additionally, PDBu induced caldesmon phosphorylation, and GF109203X attenuated the JNK phosphorylation induced by DMT or PDBu. DMT induced PKC phosphorylation in rat aortic VSMCs. These results suggest that alpha-2 adrenoceptor-mediated, DMT-induced contraction involves caldesmon phosphorylation that is mediated by JNK phosphorylation by PKC.
Keywords: dexmedetomidine, contraction, alpha-2 adrenoceptor, caldesmon, c-Jun NH2-terminal kinase, protein kinase C, rat aorta.
Introduction
Dexmedetomidine (DMT), a highly selective alpha-2 adrenoceptor agonist, is widely used during the perioperative period because DMT decreases the neuroendocrine response and analgesic drug and opioid requirements as well as produces sedation without respiratory depression [1]. As a full alpha-2B adrenoceptor agonist, DMT causes vasoconstriction by stimulating the alpha-2B adrenoceptor, which is primarily involved in vascular smooth muscle contraction, thereby leading to transiently increased blood pressure [2-5]. High-dose DMT produces hypertension, which appears to be associated with the DMT-induced alpha-2B adrenoceptor stimulation of vascular smooth muscles [3,4,6-9]. In addition, in isolated rat aortic smooth muscle, DMT-induced contraction primarily involves the phosphorylation of c-Jun NH2-terminal kinase (JNK) among the mitogen-activated protein kinase isoforms [10]. DMT-induced contraction, which involves increased myofilament calcium sensitization, is mediated by protein kinase C (PKC) via the alpha-2 adrenoceptor [11]. The PKC-dependent pathway contributes to mitogen-activated protein kinase activation, thereby leading to smooth muscle contraction [12,13]. Caldesmon is an inhibitory actin-binding protein that inhibits actomyosin ATPase activity and actin-myosin interactions in smooth muscle [14]. Caldesmon phosphorylation induced by an extracellular signal-regulated kinase reverses the inhibitory effect of caldesmon on actin-myosin interactions, which may lead to enhanced contraction [14,15]. However, the cellular signaling pathway that is associated with caldesmon phosphorylation, which is potentially involved in regulating DMT-induced contraction, remains unknown. Therefore, the goals of this in vitro study were to investigate whether DMT-induced contraction involves caldesmon phosphorylation and to determine the cellular signaling pathway involved.
Materials and Methods
Animal preparation
All experimental procedures and protocols were approved by the Institutional Animal Care and Use Committee at Gyeongsang National University. All experimental procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals prepared by the Institute for Laboratory Animal Research.
Preparation of aortic rings for tension measurements
The aortic rings were prepared for the tension measurements as previously described [16,17]. Male Sprague Dawley rats weighing 250-300 g each were anesthetized with an intramuscular injection of Zoletil 50 (125 mg tiletamine-HCl plus 125 mg zolazepam base/5 ml; 15 mg/kg; Virbac Laboratories, Carros, France). The descending thoracic aorta was removed and dissected from its surrounding connective tissue and fat under microscopic guidance in Krebs solution. The aorta was then cut into 2.5-mm rings, which were suspended on Grass isometric transducers (FT-03, Grass Instrument, Quincy, MA, USA) under a 3.0-g resting tension in a 10-mL Krebs bath at 37°C and continuously aerated with 95% O2 and 5% CO2 to maintain pH values within 7.35-7.45. The rings were equilibrated at a 3.0-g resting tension for 120 min, and the bath solution was changed every 30 min. The endothelium was removed from the aortic rings by inserting a 25-gauge needle into the lumens of the rings and gently rubbing the rings for a few seconds. When the contraction (in response to 10-8 M phenylephrine) stabilized, the removal of the endothelium was confirmed by observing < 10% relaxation in response to 10-5 M acetylcholine. The contractile response induced by isotonic 60 mM KCl was measured for all aortic rings. The isotonic 60 mM KCl solution was prepared by replacing the NaCl in the Krebs solution with an equimolar amount of KCl. After washing out the KCl from the organ bath and allowing the isometric tension to return to baseline, concentration-response curves to DMT, SP600125 and chelerythrine were obtained as described in the experimental protocols. A single ring was used for each concentration-response curve induced by DMT, SP600125 and chelerythrine. As DMT-induced contraction is attenuated by endothelial nitric oxide release, the nitric oxide synthase inhibitor Nω-nitro-L-arginine methyl ester (L-NAME, 10-5 M) and the cyclooxygenase inhibitor indomethacin (10-5 M) were included in the Krebs solution to prevent the release of endogenous nitric oxide and endogenous prostacyclin, respectively, from any residual endothelia [17,18].
Experimental protocols
The effect of rauwolscine (10-5 M), an alpha-2 adrenoceptor inhibitor, on the DMT-induced concentration (10-9 to 10-6 M)-response curves was assessed by comparing the DMT-induced contraction in endothelium-denuded aortas pretreated with or without rauwolscine. Rauwolscine was added to the organ bath 20 min prior to the addition of DMT.
Dose-response (10-6 to 3 × 10-5 M) curves of the JNK inhibitor SP600125 were generated in endothelium-denuded aortas pre-contracted with 10-6 M DMT or PKC activator phorbol 12,13-dibutyrate (PDBu, 10-6 M). After DMT (10-6 M) or PDBu (10-6 M) produced a sustained and stable contraction in the endothelium-denuded aortas, SP600125 was cumulatively added into the organ bath to generate SP600125 dose-response curves. The next higher concentration of SP600125 was added to the organ bath after a previous concentration of SP600125 had produced a sustained relaxant response. In addition, after DMT (10-6 M) produced vasoconstriction in isolated endothelium-denuded rat aortas, the PKC inhibitor chelerythrine (10-5 or 3 × 10-5 M) was added to DMT-induced vasoconstricted aortas, and isometric tension was continuously monitored for 90 min [19].
Cell culture
Cells were isolated from rat thoracic aortas by enzymatic dissociation and grown in Dulbecco's modified Eagle medium supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin [16]. Cells were sub-cultured twice per week by harvesting with trypsin/ethylenediaminetetraacetic acid and seeding into flasks at a density of 7.5 × 105 cells/mm2. For the experiments, cells between passage numbers 2 and 10 were seeded into dishes (107 cells/100-mm dish), fed every other day and used when confluent (6-7 days). Cells were serum deprived overnight prior to treatment.
Western blot analysis
Western blot analysis was performed using the method described by Lee et al. [20]. Cells were lysed in PRO-PREP protein extract solution to obtain total cell lysates, and the lysates were centrifuged at 13,000 rpm for 20 min at 4°C. The protein concentrations were determined using the Bradford method. For sample loading, equal volumes of 2× sodium dodecyl sulfate sample buffer (0.1 mol/L Tris-HCI, 20% glycerol, 4% sodium dodecyl sulfate and 0.01% bromophenol blue) and supernatant fractions from the lysates were mixed. Proteins (60 µg) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis for 90 min at 110 V. The separated proteins were transferred to polyvinylidene difluoride membranes for 2 h at 20 mA using SD Semi-dry Transfer Cells (Bio-Rad Laboratories, Hercules, CA, USA). After blocking the membranes with 5% nonfat milk in Tris-buffered saline (pH 7.0), the membranes were incubated overnight at 4°C with specific antibodies at a dilution of 1:500 in 5% skim milk in Tris-buffered saline containing Tween-20. Bound antibody was detected with horseradish peroxidase-conjugated anti-goat or anti-rabbit IgG. The membranes were washed and developed using a western blotting luminol reagent system (iNtRON Biotechnology, Houston, TX, USA) and autoradiography.
Drugs
All drugs were of the highest commercially available purity. L-NAME, indomethacin, acetylcholine, SP600125, rauwolscine, GF109203X, PDBu and chelerythrine were obtained from Sigma Aldrich (St. Louis, MO, USA). Anti-phospho-JNK, anti-JNK, anti-phospho-PKC and anti-PKC antibodies were obtained from Cell Signaling Technology (Beverly, MA, USA). DMT was donated by Orion Pharma (Turku, Finland). Anti-phospho-caldesmon and anti-caldesmon antibodies were obtained from Millipore (Billeria, MA, USA) and Abcam (Cambridge Science Park, Cambridge, England), respectively. SP600125, PDBu and GF109203X were dissolved in dimethyl sulfoxide (final organ bath concentration, < 0.1%). Unless otherwise stated, all other drugs were dissolved and diluted in distilled water.
Data analysis
The values are expressed as the means ± SD. Vasodilation induced by SP600125 or chelerythrine is expressed as a percentage of the maximum contraction induced by PDBu (10-6 M) or DMT (10-6 M). DMT-induced contraction is expressed as a percentage of the maximum contraction induced by isotonic 60 mM KCl. The effect of SP600125 or chelerythrine on the pre-contraction induced by DMT or PDBu was analyzed using a two-way repeated-measure analysis of variance (ANOVA) followed by Bonferroni's post-test (Prism 5.0, GraphPad Software, San Diego, USA). The effect of rauwolscine on DMT-induced contraction was analyzed using a two-way repeated-measure ANOVA followed by Bonferroni's post-test. The DMT-induced contraction or SP600125-induced relaxation at each concentration was analyzed using a repeated-measure ANOVA followed by Bonferroni's post-test. Band intensities from western blot analyses were assessed using scanning densitometry. A one-way ANOVA followed by Bonferroni's post-test was employed to analyze the effects of the inhibitors on the DMT-induced caldesmon phosphorylation and on the DMT- or PDBu-induced JNK phosphorylation. The effects of the incubation period on the DMT-induced caldesmon phosphorylation were analyzed using an unpaired Student's t-test. DMT-induced PKC phosphorylation and PDBu-induced caldesmon phosphorylation were analyzed using an unpaired Student's t-test. P-values < 0.05 were considered statistically significant.
Results
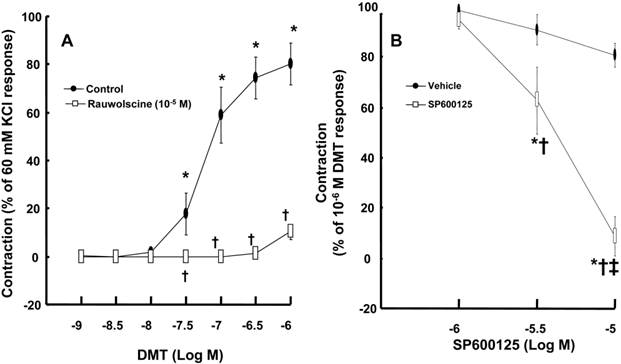
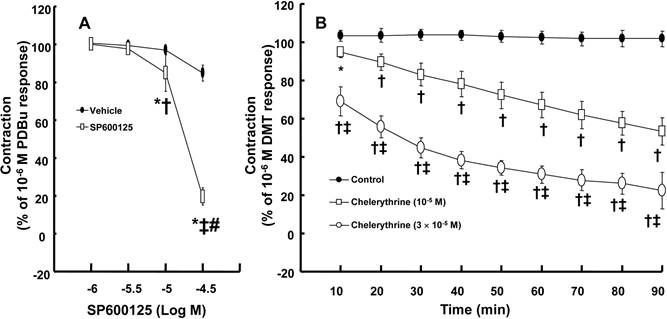
DMT (3 × 10-8 to 10-6 M) produced vasoconstriction (P < 0.001) (Fig. 1A). Rauwolscine (10-6 M), an alpha-2 adrenoceptor inhibitor, nearly abolished DMT-induced contraction (P < 0.001 versus the control) (Fig. 1A), suggesting that DMT-induced contraction is mediated by the alpha-2 adrenoceptor. SP600125 (3 × 10-6 and 10-5 M), a JNK inhibitor, produced vasodilation in endothelium-denuded aortas pre-contracted with 10-6 M DMT in a concentration-dependent manner (Fig. 1B) (P < 0.001 versus the vehicle), suggesting that the JNK-mediated pathway contributes to DMT-induced contraction. The JNK inhibitor SP600125 (10-5 and 3 × 10-5 M) induced relaxation in endothelium-denuded aortas that were pre-contracted with the PKC activator PDBu (10-6 M) in a concentration-dependent manner (P < 0.001) (Fig. 2A), suggesting that PKC-dependent contraction is mediated by JNK. The PKC inhibitor chelerythrine (10-5 and 3 × 10-5 M) caused vasodilation in endothelium-denuded aortas pre-contracted with DMT in a concentration-dependent manner (P < 0.001) (Fig. 2B), suggesting that the PKC-mediated pathway contributes to DMT-induced contraction.
A: The effect of rauwolscine on dexmedetomidine (DMT)-induced contraction of endothelium-denuded aorta. B: SP600125 dose-response curves in endothelium-denuded aortas pre-contracted with 10-6 M DMT. All data are expressed as the mean ± SD (A, N = 6, B, N = 7) and as the percentage of maximal contraction induced by isotonic 60 mM KCl or 10-6 M DMT. N indicates the number of rat thoracic aortic rings. A: *P < 0.001 versus 10-9 M DMT in the control group. †P < 0.001 versus the control. B: *P < 0.001 versus the vehicle. †P < 0.001 versus 10-6 M SP600125. ‡P < 0.001 versus 3 × 10-6 M SP600125.
A: SP600125 dose-response curves in endothelium-denuded aortas pre-contracted with 10-6 M phorbol 12,13-dibutyrate (PDBu, 10-6 M). All data are expressed as the mean ± SD (N = 5) and as the percentage of maximal contraction induced by 10-6 M PDBu. N indicates the number of rats from which the descending thoracic aortic rings were derived. *P < 0.001 versus the vehicle. †P < 0.01 and ‡P < 0.001 versus 10-6 M SP600125. #P < 0.001 versus 10-5 M SP600125. B: The effect of chelerythrine in endothelium-denuded aorta pre-contracted with 10-6 M dexmedetomidine (DMT). All data are expressed as the mean ± SD and as the percentage of maximal contraction induced by DMT (10-6 M), where N indicates the number of rat thoracic aortic rings: control, N = 7; 10-5 M chelerythrine, N = 7; 3 × 10-5 M chelerythrine, N = 7; *P < 0.05 and †P < 0.001 versus control, ‡P < 0.001 versus 10-5 M chelerythrine.
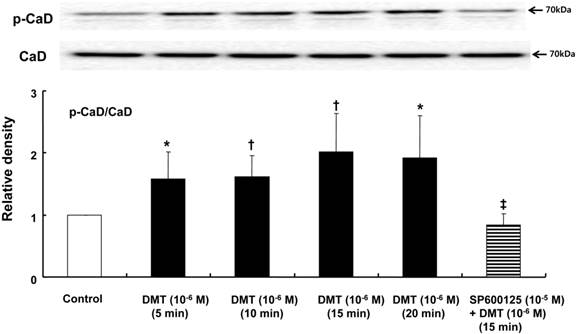
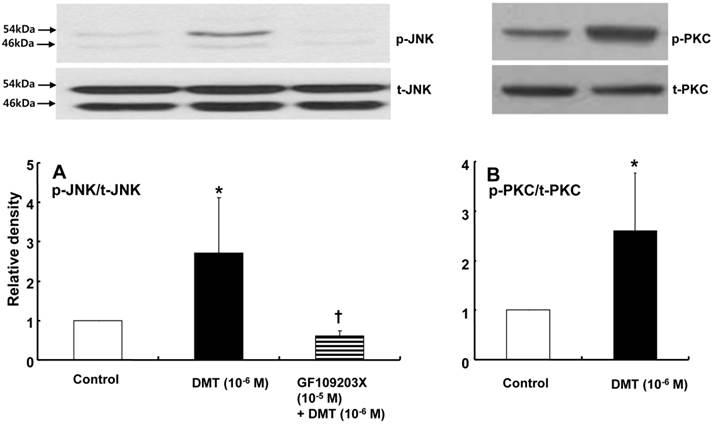
DMT (10-6 M) induced caldesmon phosphorylation at 5, 10, 15 and 20 min after pretreatment with DMT (P < 0.05 versus the control) (Fig. 3). The JNK inhibitor SP600125 (10-5 M) and the alpha-2 adrenoceptor inhibitor rauwolscine (10-5 M) inhibited DMT-induced caldesmon phosphorylation (P < 0.01 versus 10-6 M DMT alone) (Figs. 3 and 4A), suggesting that DMT-induced caldesmon phosphorylation appears to be mediated by the alpha-2 adrenoceptor and JNK. The PKC activator PDBu (10-6 M) induced caldesmon phosphorylation (P < 0.05) (Fig. 4B). The PKC inhibitor GF109203X (10-5 M) inhibited JNK phosphorylation induced by the PKC activator PDBu (10-6 M) (P < 0.001 versus 10-6 M PDBu alone) (Fig. 4C). The PKC inhibitor GF109203X (10-5 M) attenuated DMT-induced JNK phosphorylation (P < 0.01 versus 10-6 M DMT alone) (Fig. 5A), suggesting that DMT-induced JNK phosphorylation is mediated by PKC. DMT induced PKC phosphorylation (Fig. 5B) (P < 0.05 versus control).
The effect of SP600125 on dexmedetomidine (DMT)-induced caldesmon (CaD) phosphorylation (N = 5) in rat aortic vascular smooth muscle cells (VSMCs). VSMCs were treated with 10-6 M DMT alone for 5, 10, 15 and 20 min or with 10-6 M DMT for 15 min after pretreatment with 10-5 M SP600125 for 1 hr. CaD phosphorylation was examined by western blot analysis as described in the Methods section. The data are presented as the mean ± SD. N indicates the number of independent experiments. p-CaD: phosphorylated CaD. *P < 0.05 and †P < 0.01 versus the control. ‡P < 0.01 versus 10-6 M DMT (15 min) alone.
A: The effect of rauwolscine on dexmedetomidine (DMT)-induced caldesmon (CaD) phosphorylation (N = 4) in rat aortic vascular smooth muscle cells (VSMCs). B: The effect of phorbol 12, 13-dibutyrate (PDBu) on CaD phosphorylation (N = 4) in rat aortic VSMCs. VSMCs were treated with 10-6 M DMT alone for 15 min or 10-6 M DMT for 15 min after pretreatment with 10-5 M rauwolscine for 1 hr and 10-6 M PDBu alone for 10 min. CaD phosphorylation was examined by western blot analysis as described in the Methods. The data are expressed as the mean ± SD. N indicates the number of independent experiments. p-CaD: phosphorylated CaD. A: *P < 0.001 versus the control. †P < 0.01 versus 10-6 M DMT alone. B: *P < 0.05 versus the control. C: The effect of GF109203X on PDBu-induced c-Jun NH2-terminal kinase (JNK) phosphorylation (N = 3) in rat aortic VSMCs. VSMCs were treated with 10-6 M PDBu alone for 10 min or 10-6 M PDBu for 10 min after pretreatment with 10-5 M GF109203X for 1 hr. JNK phosphorylation was examined by western blot analysis as described in the Methods section. The data are expressed as the mean ± SD. N indicates the number of independent experiments. p-JNK: phosphorylated JNK. t-JNK: total JNK. *P < 0.001 versus the control. †P < 0.001 versus 10-6 M PDBu alone.
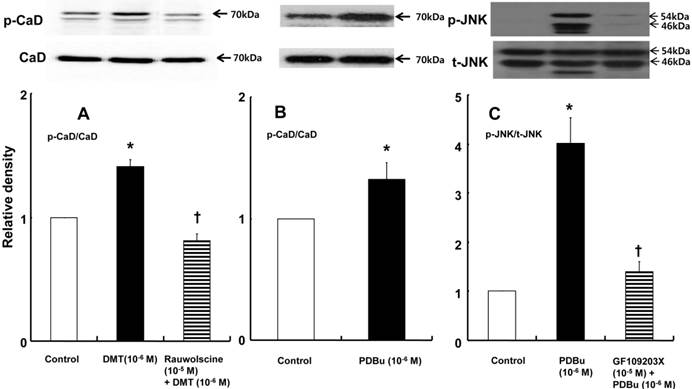
A: The effects of GF109203X on dexmedetomidine (DMT)-induced c-Jun NH2-terminal kinase (JNK) phosphorylation (N = 4) in rat aortic vascular smooth muscle cells (VSMCs). VSMCs were treated with 10-6 M DMT alone for 10 min or with 10-6 M DMT for 10 min after pretreatment with 10-5 M GF109203X for 1 hr. JNK phosphorylation was examined by western blot analysis as described in the Methods section. The data are presented as the mean ± SD. N indicates the number of independent experiments. p-JNK: phosphorylated JNK. t-JNK: total JNK. *P < 0.05 versus the control. †P < 0.01 versus 10-6 M DMT alone. B: The effect of DMT on protein kinase C (PKC) phosphorylation (N = 4) in rat aortic VSMCs. VSMCs were treated with 10-6 M DMT alone for 10 min. PKC phosphorylation was examined by western blot analysis as described in the Methods section. The data are presented as the mean ± SD. N indicates the number of independent experiments. p-PKC: phosphorylated PKC, t-PKC: total PKC. *P < 0.05 versus the control.
Discussion
This study is the first to suggest that DMT-induced contraction involves caldesmon phosphorylation mediated by JNK activation via the alpha-2 adrenoceptor. The major findings of this in vitro study are as follows: 1) SP600125 and rauwolscine attenuated DMT-induced caldesmon phosphorylation; 2) GF109203X attenuated DMT-induced JNK phosphorylation; 3) GF109203X attenuated PDBu-induced JNK phosphorylation; 4) SP600125 and chelerythrine produced vasodilation in endothelium-denuded aortas pre-contracted with DMT or PDBu; and 5) rauwolscine inhibited DMT-induced contraction.
Among the mitogen-activated protein kinases, DMT-induced contraction is primarily mediated by JNK [10]. Consistent with a previous report, SP600125 (a JNK inhibitor) produced vasodilation in endothelium-denuded aortas that were pre-contracted with DMT, suggesting that DMT-induced contraction involves JNK activation [10]. Consistent with previous reports, rauwolscine (an alpha-2 adrenoceptor inhibitor) attenuated DMT-induced contraction [10,11,18]. In agreement with a previous report, chelerythrine (a PKC inhibitor) inhibited the DMT-mediated contraction of endothelium-denuded aortas (Fig. 2B) [11]. SP600125 (a JNK inhibitor) also attenuated the contraction evoked by the PKC activator PDBu (Fig. 2A). Together, these findings suggest that JNK is a downstream signal effector of PKC that mediates DMT-induced contraction. In addition, mitogen-activated protein kinases, including p38 mitogen-activated protein kinase and extracellular signal-regulated kinase, are downstream effectors of PKC that mediate vascular smooth muscle contraction [12,13]. Together, these results suggest that alpha-2 adrenoceptor-mediated, DMT-induced contraction involves JNK activation by PKC [12,13]. In terms of the amount of phosphorylated 20-kDa myosin regulatory light chain required for the actin-myosin interaction, inhibition of DMT-induced contraction by the PKC inhibitor chelerythrine appears to be associated with the attenuated inhibition of the myosin light chain phosphatase [11].
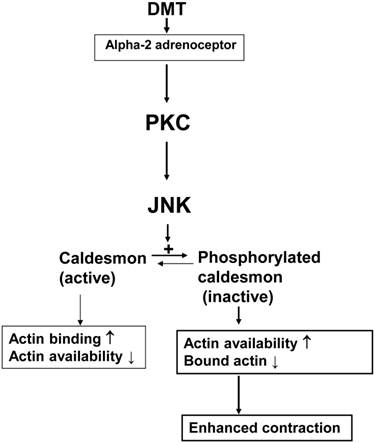
Caldesmon binds to actin and inhibits the interaction between actin and myosin, thereby regulating smooth muscle contraction [14]. The inhibitory effect of the heavy isoform of caldesmon on actin-myosin interactions is reversed by either Ca2+/calmodulin or caldesmon phosphorylation [21.22]. In addition, caldesmon phosphorylation induced by the PKC stimulant phorbol ester, which results in smooth muscle contraction, is mediated by an extracellular signal-regulated kinase [23]. Similar to a previous report, SP600125 attenuated DMT-induced caldesmon phosphorylation (Fig. 3), and PDBu also induced caldesmon phosphorylation [23]. Rauwolscine attenuated the caldesmon phosphorylation induced by DMT (Fig. 4A). Together, DMT-induced caldesmon phosphorylation, which increases actin availability for interaction with myosin, appears to be mediated by a pathway involving the alpha-2 adrenoceptor and JNK in rat aortic smooth muscle. GF109203X attenuated JNK phosphorylation (Fig. 4C and 5A) induced by DMT or PDBu, and these results suggest that PKC is an upstream effector of JNK that mediates DMT-induced caldesmon phosphorylation. Thus, together with results from isometric tension measurements, SP600125- and chelerythrine-induced inhibition of DMT-mediated contraction appears to be partially associated with the decreased availability of unbound actin. Consistent with a previous study and the results of current tension study, DMT induced PKC phosphorylation, suggesting the involvement of PKC in DMT-induced contraction [11]. Further research regarding the involvement of PKC in DMT-induced caldesmon phosphorylation is needed to confirm the involvement of a PKC-JNK axis in this phosphorylation event. Combined with the results of previous studies, the putative cellular mechanism responsible for DMT-induced caldesmon phosphorylation is as follows: alpha-2 adrenoceptor-mediated, DMT-induced contraction involves caldesmon phosphorylation through the activation of JNK, which is mediated by PKC (Fig. 6) [10,11]. Further studies regarding the effects of other signaling pathways (e.g., calcium/calmodulin or p21-activated protein kinase) on the caldesmon-mediated inhibition of actin availability during DMT-induced contraction and detailed information on the pathway downstream of caldesmon phosphorylation are warranted.
The clinical relevance of vasoconstriction mediated by DMT-induced caldesmon phosphorylation could be attenuated by the following facts. First, DMT produces endothelial nitric oxide release [18,24]. Thus, the DMT-induced contraction observed in this in vitro study using isolated endothelium-denuded aortas would be inhibited in an in vivo state. Second, in this study, we used rat aortas, which are considered to be a conduit vessel, whereas the small resistance arterioles regulating organ blood flow mainly determine blood pressure [25]. Although this in vitro study has the above mentioned limitations, vasoconstriction induced by a supraclinical dose of DMT (3 × 10-8 M), which exceeds the clinically relevant concentration of DMT (10-8 M) required for sedation, may contribute to hypertension due to the high DMT doses or concentrations observed in humans [7-9,26].
These results suggest that alpha-2 adrenoceptor-mediated, DMT-induced contraction involves the phosphorylation of caldesmon by JNK, which is activated by PKC.
The putative cellular signal transduction pathway responsible for the caldesmon phosphorylation involved in dexmedetomidine (DMT)-induced contraction of rat aortic smooth muscle cells. PKC: protein kinase C; JNK: c-Jun NH2-terminal kinase.
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2013R1A1A2057459).
Competing Interests
Ju-Tae Sohn is currently funded by a grant (2013R1A1A2057459) from the National Research Foundation of Korea (RNF). The remaining authors have declared that no competing interest exists.
References
1. Gertler R, Brown HC, Mitchell DH, Silvius EN. Dexmedetomidine: a novel sedative-analgesic agent. Proc (BaylUniv Med Cent). 2001;14:13-21
2. Peltonen JM, Pihlavisto M, Scheinin M. Subtype-specific stimulation of[35S]GTPgammaS binding by recombinant alpha2-adrenoceptors. Eur J Pharmacol. 1998;355:275-279
3. Guimarães S, Moura D. Vascular adrenoceptors: an update. Pharmacol Rev. 2001;53:319-356
4. Schmeling WT, Kampine JP, Roerig DL, Warltier DC. The effects of the stereoisomers of the alpha 2-adrenergic agonist medetomidine on systemic and coronary hemodynamics in conscious dogs. Anesthesiology. 1991;75:499-511
5. Hall JE, Uhrich TD, Barney JA, Arain SR, Ebert TJ. Sedative, amnestic, and analgesic properties of small-dose dexmedetomidine infusions. Anesth Analg. 2000;90:699-705
6. Bloor BC, Ward DS, Belleville JP, Maze M. Effects of intravenous dexmedetomidine in humans. II. Hemodynamic changes. Anesthesiology. 1992;77:1134-1142
7. Erkonen G, Lamb F, Tobias JD. High-dose dexmedetomidine-induced hypertension in a child with traumatic brain injury. Neurocrit Care. 2008;9:366-369
8. Mason KP, Zurakowski D, Zgleszewski S, Prescilla R, Fontaine PJ, Dinardo JA. Incidence and predictors of hypertension during high-dose dexmedetomidine sedation for pediatric MRI. Paediatr Anaesth. 2010;20:516-523
9. Shah S, Sangari T, Qasim M, Martin T. Severe hypertension and bradycardia after dexmedetomidine for radiology sedation in a patient with acute transverse myelitis. Paediatr Anaesth. 2008;18:681-682
10. Ok SH, Jeong YS, Kim JG, Lee SM, Sung HJ, Kim HJ, Chang KC, Kwon SC, Sohn JT. c-Jun NH2-terminal kinase contributes to dexmedetomidine-induced contraction in isolated rat aortic smooth muscle. Yonsei Med J. 2011;52:420-428
11. Kim JG, Sung HJ, Ok SH, Kwon SC, Cheon KS, Kim HJ, Chang KC, Shin IW, Lee HK, Chung YK, Sohn JT. Calcium sensitization involved in dexmedetomidine-induced contraction of isolated rat aorta. Can J Physiol Pharmacol. 2011;89:681-689
12. Yamaguchi H, Igarashi M, Hirata A, Sugae N, Tsuchiya H, Jimbu Y, Tominaga M, Kato T. Altered PDGF-BB-induced p38 MAP kinase activation in diabetic vascular smooth muscle cells: roles of protein kinase C-delta. Arterioscler Thromb Vasc Biol. 2004;24:2095-2101
13. Ginnan R, Singer HA. PKC-delta-dependent pathways contribute to PDGF-stimulated ERK1/2 activation in vascular smooth muscle. Am J Physiol Cell Physiol. 2005;288:C1193-C1201
14. Kim HR, Appel S, Vetterkind S, Gangopadhyay SS, Morgan KG. Smooth muscle signalling pathways in health and disease. J Cell Mol Med. 2008;12:2165-2180
15. Akata T. General anesthetics and vascular smooth muscle: direct actions of general anesthetics on cellular mechanisms regulating vascular tone. Anesthesiology. 2007;106:365-391
16. Ok SH, Kwon SC, Yeol Han J, Yu J, Shin IW, Lee HK, Chung YK, Choi MJ, Sohn JT. Mepivacaine-induced contraction involves increased calcium sensitization mediated via Rho kinase and protein kinase C in endothelium-denuded rat aorta. Eur J Pharmacol. 2014;723:185-193
17. Ok SH, Bae SI, Shim HS, Sohn JT. Dexmedetomidine-induced contraction of isolated rat aorta is dependent on extracellular calcium concentration. Korean J Anesthesiol. 2012;63:253-259
18. Kim HJ, Sohn JT, Jeong YS, Cho MS, Kim HJ, Chang KC, Shin MK, Park CS, Chung YK. Direct effect of dexmedetomidine on rat isolated aorta involves endothelial nitric oxide synthesis and activation of the lipoxygenase pathway. Clin Exp Pharmacol Physiol. 2009;36:406-412
19. Jin L, Teixeira CE, Webb RC, Leite R. Comparison of the involvement of protein kinase C in agonist-induced contractions in mouse aorta and corpus cavernosum. Eur J Pharmacol. 2008;590:363-368
20. Lee HM, Ok SH, Sung HJ, Eun SY, Kim HJ, Lee SH, Kang S, Shin IW, Lee HK, Chung YK, Choi MJ, Bae SI, Sohn JT. Mepivacaine-induced contraction involves phosphorylation of extracellular signal-regulated kinase through activation of the lipoxygenase pathway in isolated rat aortic smooth muscle. Can J Physiol Pharmacol. 2013;91:285-294
21. Horiuchi KY, Miyata H, Chacko S. Modulation of smooth muscle actomyosin ATPase by thin filament associated proteins. Biochem Biophys Res Commun. 1986;136:962-968
22. Adam LP, Hathaway DR. Identification of mitogen-activated protein kinase phosphorylation sequences in mammalian h-Caldesmon. FEBS Lett. 1993;322:56-60
23. Kordowska J, Huang R, Wang CL. Phosphorylation of caldesmon during smooth muscle contraction and cell migration or proliferation. J Biomed Sci. 2006;13:159-172
24. Shafaroudi MM, McBride M, Deighan C, Wokoma A, Macmillan J, Daly CJ, McGrath JC. Two "knockout" mouse models demonstrate that aortic vasodilatation is mediated via alpha2a-adrenoceptors located on the endothelium. J Pharmacol Exp Ther. 2005;314:804-810
25. Christensen KL, Mulvany MJ. Location of resistance arteries. J Vasc Res. 2001;38:1-12
26. Ebert TJ, Hall JE, Barney JA, Uhrich TD, Colinco MD. The effects of increasing plasma concentrations of dexmedetomidine in humans. Anesthesiology. 2000;93:382-394
Author contact
![]() Corresponding author: Ju-Tae Sohn, Department of Anesthesiology and Pain Medicine, Gyeongsang National University Hospital, Jinju, Republic of Korea. Fax: +82-55-750-8142, Tel.: +82-55-750-8586, E-mail: jtsohngsnu.ac.kr
Corresponding author: Ju-Tae Sohn, Department of Anesthesiology and Pain Medicine, Gyeongsang National University Hospital, Jinju, Republic of Korea. Fax: +82-55-750-8142, Tel.: +82-55-750-8586, E-mail: jtsohngsnu.ac.kr
Received 2014-6-3
Accepted 2014-9-7
Published 2014-9-21