ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2014; 10(10):1128-1137. doi:10.7150/ijbs.10359 This issue Cite
Research Paper
Defect in MAPK Signaling As a Cause for Monogenic Obesity Caused By Inactivating Mutations in the Melanocortin-4 Receptor Gene
Shan He1,2, Ya-Xiong Tao1 ![]()
1. Department of Anatomy, Physiology, and Pharmacology, College of Veterinary Medicine, Auburn University, Auburn, Alabama 36849-5519, USA.
2. Key Laboratory of Freshwater Animal Breeding, Ministry of Agriculture, College of Fisheries, Huazhong Agricultural University, Hubei Collaborative Innovation Center for Freshwater Aquaculture, Wuhan, Hubei 430070, China.
Abstract
The melanocortin-4 receptor (MC4R) is a Family A G protein-coupled receptor that plays an essential role in regulating energy homeostasis, including both energy intake and expenditure. Mutations leading to a reduced MC4R function confer a major gene effect for obesity. More than 170 distinct mutations have been identified in humans. In addition to the conventional Gs-stimulated cAMP pathway, the MC4R also activates MAPKs, especially ERK1/2. We also showed there is biased signaling in the two signaling pathways, with inverse agonists in the Gs-cAMP pathway acting as agonists for the ERK1/2 pathway. In the current study, we sought to determine whether defects in basal or agonist-induced ERK1/2 activation in MC4R mutants might potentially contribute to obesity pathogenesis in patients carrying these mutations. The constitutive and ligand-stimulated ERK1/2 activation were measured in wild type and 73 naturally occurring MC4R mutations. We showed that nineteen mutants had significantly decreased basal pERK1/2 level, and five Class V variants (where no functional defects have been identified previously), C40R, V50M, T112M, A154D and S295P, had impaired ligand-stimulated ERK1/2 activation. Our studies demonstrated for the first time that decreased basal or ligand-stimulated ERK1/2 signaling might contribute to obesity pathogenesis caused by mutations in the MC4R gene. We also observed biased signaling in 25 naturally occurring mutations in the Gs-cAMP and ERK1/2 pathways.
Keywords: Melanocortin-4 receptor, naturally occurring mutation, extracellular signal-regulated kinases 1 and 2 signaling, biased signaling.
Introduction
The melanocortin-4 receptor (MC4R) is a member of Family A G protein-coupled receptors (GPCRs) that has been shown to be involved in regulating energy homeostasis, including both energy intake and expenditure [1-3]. Human genetic studies demonstrated that functionally relevant mutations in the MC4R confer the commonest major gene form of obesity, characterized by its early-onset and severity [4]. About 170 MC4R mutations, including nonsense, missense, frameshift, and inframe deletions, have been identified, primarily from study groups of obese patients of different ethnic origins [5, 6]. We grouped MC4R mutations into five Classes [7]. Class I are null mutations that are defective in receptor biosynthesis. Class II mutants are synthesized but defective in trafficking onto the cell surface. Class III mutants are synthesized and transported onto the cell surface, but are defective in ligand binding. Class IV mutants have normal cell surface expression and ligand binding but are defective in Gs-mediated cAMP signaling. Class V variants have no known defects with normal cell surface expression, ligand binding and cAMP signaling [7]. The obesity observed in vivo in the patients could not be well explained by the in vitro cellular phenotype with Class V variants. Therefore, the analysis of the in vitro signaling defects of these mutations is necessary to understand its potential roles in obesity pathogenesis.
When the MC4R was cloned, it was shown to be coupled to the stimulatory G protein, increasing adenylyl cyclase activity and intracellular cAMP levels [8, 9] (reviewed in [10]). Indeed, almost all of the earlier studies on the MC4R used direct or indirect measures of cAMP levels as the indicator for receptor activation. However, like other GPCRs, the MC4R has been found to activate other G proteins, including the inhibitory G protein (Gi) and signaling pathways, including MAPK, such as ERK1/2, p38 MAPK, and c-Jun NH2-terminal kinase [3, 11, 12]. In vitro experiments in different cell lines expressing MC4R heterologously or GT1-1 cells that express mouse MC4R endogenously showed that agonist stimulation of MC4R activates ERK1/2, which can be mediated by cAMP-protein kinase A, phosphatidylinositol 3-kinase, calcium, and protein kinase C, depending on the cell lines used [13-16]. In vivo, activation of ERK1/2 by MC4R is also observed in the hypothalamus, and this signaling is involved in mediating melanotan II-induced inhibition of food intake [14, 17]. Therefore, defective ERK1/2 signaling might be involved in obesity pathogenesis caused by mutations in the MC4R gene.
Previous studies discovered biased activation of ERK1/2 in MC4R with several artificially generated and one naturally occurring MC4R mutations having divergent basal or agonist-stimulated cAMP and ERK1/2 signaling [12, 18-20]. We also showed that several inverse agonists at the Gs-cAMP pathway are indeed agonists at the ERK1/2 pathway, suggesting that these ligands are biased [16]. However, little is known about the potential dysfunction in ERK1/2 signaling in naturally occurring MC4R mutations, and their contributions to obesity pathogenesis in patients carrying these mutations. In the present study, we investigated the constitutive and ligand-stimulated activation of ERK1/2 in wild type (WT) and 73 naturally occurring MC4R mutations from all five Classes (Fig. 1). By comparing the signaling properties of the mutant receptors in the Gs-cAMP pathway previously reported and the ERK1/2 data obtained in the current study, we were interested in whether there is biased signaling in the naturally occurring MC4R mutations.
Schematic model of the human MC4R. The naturally occurring mutations characterized in this study are highlighted with gray background. The variants that had impaired ligand-stimulated ERK1/2 activation in Class V are highlighted with red background. The mutations that had biased signaling in the Gs-cAMP and ERK1/2 pathways are highlighted with blue background.
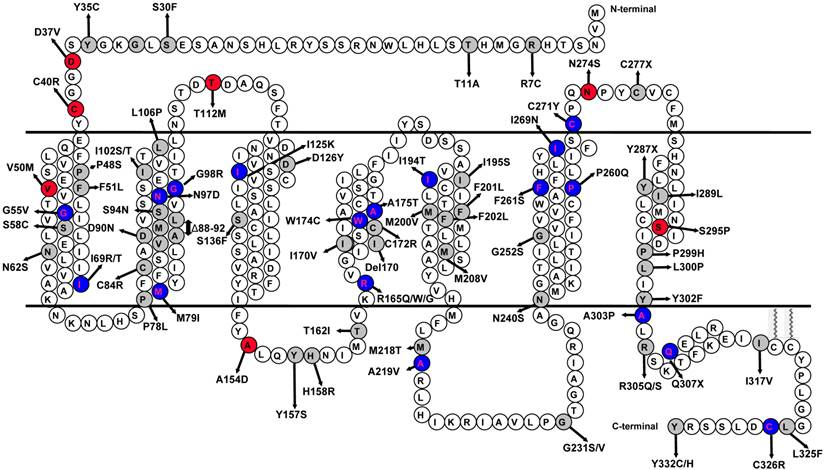
Biased signaling, also called functional selectivity, agonist-directed stimulus trafficking, or ligand-induced differential signaling, is a very actively studied area in GPCR field, representing a frontier in GPCR pharmacology and drug discovery [21-26]. Biased ligands with enhanced therapeutic potential and diminished side effects targeting several GPCRs are in various stages of clinical trials. The atomic basis of biased signaling is also beginning to be elucidated with crystal structure analysis [27-30]. Not only ligands can be biased, mutant receptors can also be biased [24, 31]. In addition to lab-generated mutations that show biased signaling (see [32] for an example), naturally occurring mutations in several GPCRs including glucagon-like peptide-1 receptor, calcium-sensing receptor, melanocortin-1 receptor, and MC4R, have also been shown to exhibit biased signaling [12, 33-36]. With the MC4R, only one mutation, D90N, was shown previously to have biased signaling [12]. Therefore our data on 73 mutations would expand this observation significantly.
Methods and Materials
Reagents and supplies
[Nle4,D-Phe7]-α-MSH (NDP-α-MSH) was purchased from Peptides International (Louisville, KY). Cell culture plates and flasks were purchased from Corning (Corning, NY). Cell culture media, newborn calf serum, antibiotics and reagents were obtained from Invitrogen (Carlsbad, CA).
Plasmids
The WT human MC4R in pcDNA3.1 was previously described [37]. Mutant MC4Rs used in this study were generated using QuikChangeTM site-directed mutagenesis kit (Stratagene, La Jolla, CA) and have been reported in our previous studies [7, 37-43]. Plasmids prepared were sequenced by the DNA Sequencing Facility of University of Chicago Cancer Research Center (Chicago, IL, USA) before used for transfection experiments.
Cell culture and DNA transfection
HEK293T cells were obtained from the American Type Culture Collection (Manassas, VA), and maintained at 5% CO2 in DMEM containing 10 mM HEPES, 10% newborn calf serum, 100 units/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin B and 50 μg/ml gentamicin. Cells were plated on gelatin-coated 100 mm dishes and transfected at 50-70% confluency using calcium phosphate transfection method. Then cells were washed twice and incubated with Waymouth/BSA (Waymouth's MB752/1 media (Sigma-Aldrich, St. Louis, MO) containing 1 mg/ml bovine serum albumin (BSA)) 24 h after transfection and starved for 18 h at 37 °C before ligand stimulation.
ERK1/2 phosphorylation assay
The phosphorylated ERK1/2 (pERK1/2) activity was measured as described previously [16, 19, 20]. Briefly, 48 h after transfection, starved cells (see above) were treated for 5 min with either buffer alone or 1 μM NDP-α-MSH. The time point in the experiments was chosen based on previous report of MC4R-mediated ERK1/2 signaling [16]. Cells were solubilized in lysis buffer and lysates were separated on 10% SDS-PAGE gel. Proteins were then transferred onto PVDF membrane. Phospho-ERK1/2 and β-tubulin were detected by immunoblotting with rabbit anti-pERK1/2 antibody (1:1000~1:2000, Cell Signaling Technology, Beverly, MA) and mouse anti-β-tubulin antibody (1:5000~1:10,000, Developmental Studies Hybridoma Bank at the University of Iowa, Iowa City, IA), respectively. Blots were probed with horseradish peroxidase-conjugated secondary donkey anti-rabbit (1:2000) and horseradish peroxidase-conjugated donkey anti-mouse (1:5000~1:10,000, both from Jackson ImmunoResearch Laboratories, West Grove, PA) at room temperature for 2 h. The membranes were then visualized using enhanced chemiluminescence reagent (Pierce, Rockford, IL) and quantified using ImageJ 1.44 software (National Institute of Health, Bethesda, MD) after densitometric scanning of the films. ERK1/2 phosphorylation was normalized according to the loading of proteins by expressing the data as a ratio of pERK1/2 over β-tubulin.
Statistics
The significance of differences in pERK1/2 activities between WT and mutant MC4Rs were analyzed using paired two-tailed Student's t-test with GraphPad Prism 4.0 software (La Jolla, CA).
Results
Ligand-stimulated activation of ERK1/2 in Class V MC4R variants
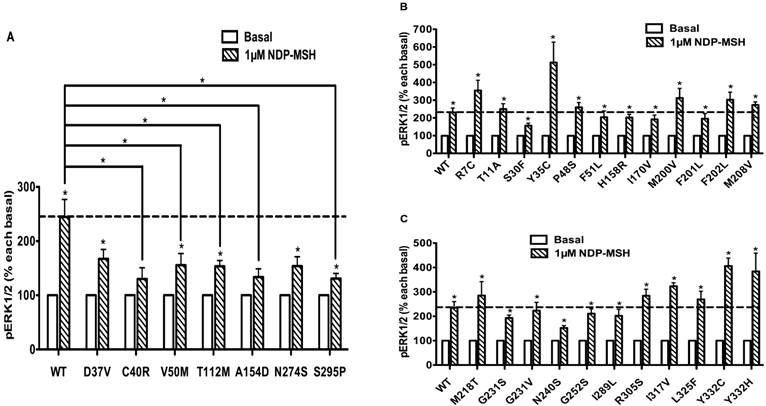
HEK293T cells, which do not express MC4R endogenously [37], have been widely used as a mammalian expression system in MC4R studies. We used HEK293T cells transiently transfected with the WT or 73 naturally occurring MC4R mutations to study the effect of NDP-α-MSH on MAPK pathway. No significant change of pERK1/2 level was observed upon NDP-α-MSH treatment in HEK293T cells transiently transfected with the empty vector pcDNA3.1 (data not shown). The pERK1/2 levels of the 30 Class V variant MC4Rs, where no functional defects were identified previously, including R7C, T11A, S30F, Y35C, D37V, C40R, P48S, V50M, F51L, T112M, A154D, H158R, I170V, M200V, F201L, F202L, M208V, M218T, G231S, G231V, N240S, G252S, N274S, I289L, S295P, R305S, I317V, L325F, Y332C and Y332H, were measured after 5 min stimulation with NDP-α-MSH. As shown in Figs. 2 and 3, we found that C40R and A154D did not respond to NDP-α-MSH stimulation in ERK1/2 activation. Although the pERK1/2 levels of V50M, T112M, and S295P MC4Rs were increased significantly upon NDP-α-MSH stimulation (P < 0.05), the stimulation was significantly lower than that of the WT MC4R (P < 0.05). The other 25 Class V variants showed similar agonist-stimulated ERK1/2 activation as the WT MC4R (P < 0.05) (Figs. 2 and 3).
Ligand-stimulated activation of ERK1/2 in Class IV MC4R mutants
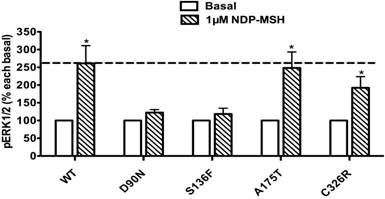
The pERK1/2 levels of the four mutant MC4Rs in Class IV (mutants that are expressed on the cell surface and can bind the ligand but are defective in Gs-cAMP signaling), including D90N, S136F, A175T and C326R, were examined after 5 min stimulation with NDP-α-MSH in HEK293T cells transiently transfected with these MC4R mutants. As shown in Fig. 4, A175T and C326R MC4Rs had significant ligand-stimulated ERK1/2 activation, similar to that of the WT MC4R (P < 0.05). D90N and S136F did not respond to NDP-α-MSH stimulation with ERK1/2 activation.
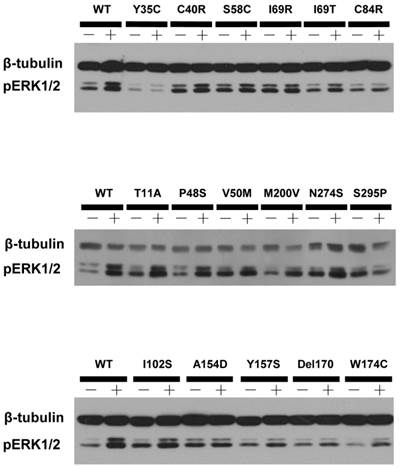
A representative blot of ligand-stimulated ERK1/2 activation in HEK293T cells transiently transfected with Class V variants.
The ligand-stimulated pERK1/2 levels in HEK293T cells transiently transfected with Class V variants. Results are expressed as percentage of the value obtained in non-stimulated cells and represent the mean ± SEM of five independent experiments. * indicates significant difference from basal pERK1/2 level or from the stimulation of the WT MC4R (P < 0.05).
The ligand-stimulated pERK1/2 levels in HEK293T cells transiently transfected with Class IV mutants. Results are expressed as percentage of the value obtained in non-stimulated cells and represent the mean ± SEM of five independent experiments. * indicates significant differences from basal pERK1/2 level (P < 0.05).
The ligand-stimulated pERK1/2 levels in HEK293T cells transiently transfected with Class III mutants. Results are expressed as percentage of the value obtained in non-stimulated cells and represent the mean ± SEM of five independent experiments. * indicates significant differences from basal pERK1/2 level (P < 0.05).
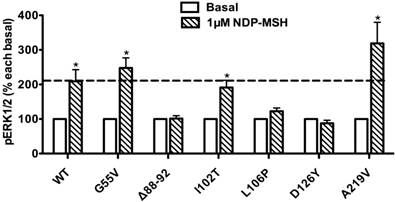
Ligand-stimulated activation of ERK1/2 in Class III MC4R mutants
The pERK1/2 levels of six Class III mutant MC4Rs (mutants that are expressed on the cell surface but have defects in ligand binding), including G55V, Δ88-92, I102T, L106P, D126Y, and A219V, were measured after 5 min stimulation with NDP-α-MSH in HEK293T cells transiently transfected with these MC4R mutants. As shown in Fig. 5, we found that G55V, I102T and A219V MC4Rs had significantly increased pERK1/2 levels, similar to that of the WT MC4R (P < 0.05). Δ88-92, L106P and D126Y did not respond to NDP-α-MSH stimulation with ERK1/2 activation.
Ligand-stimulated activation of ERK1/2 in Class II MC4R mutants
The pERK1/2 levels of 29 Class II mutant MC4Rs (mutants that are translated but are defective in trafficking onto the cell surface), including S58C, N62S, I69R, I69T, P78L, C84R, N97D, G98R, I102S, I125K, Y157S, T162I, R165G, R165W, R165Q, C172R, W174C, I194T, I195S, P260Q, F261S, I269N, C271Y, P299H, L300P, Y302F, A303P, R305Q, and Q307X, were measured after 5 min stimulation with NDP-α-MSH in HEK293T cells transiently transfected with these mutants. As shown in Fig. 6, seven mutants, including I69T, N97D, G98R, I125K, W174C, I194T and F261S, had significantly increased pERK1/2 levels, similar to the WT MC4R (P < 0.05). Eight mutants, including S58C, I195S, P260Q, C271Y, L300P, Y302F, R305Q and Q307X, although responded to NDP-α-MSH stimulation with increased pERK1/2 levels, had lower responses than the WT MC4R. The other fourteen Class II mutants had no response to NDP-α-MSH stimulation in ERK1/2 activation.
Ligand-stimulated activation of ERK1/2 in Class I MC4R mutants
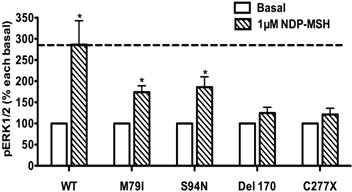
The pERK1/2 levels of four Class I mutant MC4Rs (mutants that have decreased expression levels), including M79I, S94N, Del170, and C277X, were measured after 5 min stimulation with NDP-α-MSH in HEK293T cells transiently transfected with MC4R mutants. As shown in Fig. 7, we found that M79I and S94N had significantly increased pERK1/2 levels (P < 0.05), whereas Del170 and C277X MC4Rs exhibited impaired ERK1/2 activation.
The ligand-stimulated pERK1/2 levels in HEK293T cells transiently transfected with Class II mutants. Results are expressed as percentage of the value obtained in non-stimulated cells and represent the mean ± SEM of five independent experiments. * indicates significant differences from basal pERK1/2 level (P < 0.05).
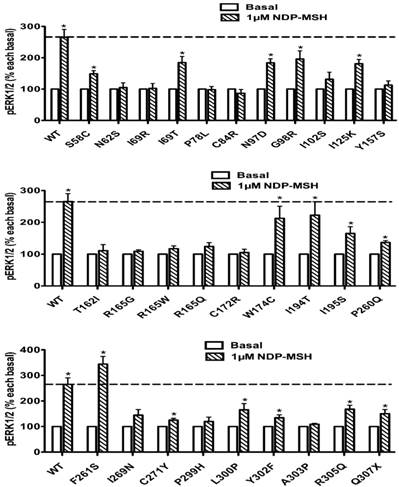
The ligand-stimulated pERK1/2 levels in HEK293T cells transiently transfected with Class I mutants. Results are expressed as percentage of the value obtained in non-stimulated cells and represent the mean ± SEM of five independent experiments. * indicates significant differences from basal pERK1/2 level (P < 0.05).
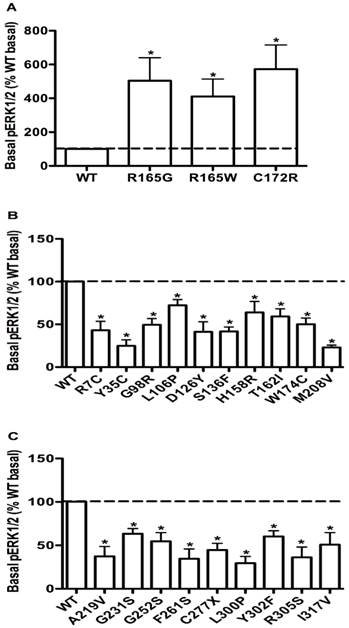
The basal pERK1/2 levels in HEK293T cells transiently transfected with WT or mutant MC4Rs with increased basal pERK1/2 levels (A), or decreased basal pERK1/2 levels (B and C). Results are expressed as percentage of the WT basal pERK1/2 level and represent the mean ± SEM of five independent experiments. * indicates significant differences (P < 0.05).
Constitutive activities in ERK1/2 signaling in WT and mutant MC4Rs
To evaluate the obesity pathogenesis caused by mutations in the MC4R gene through constitutive MAPK signaling, we compared the basal pERK1/2 levels of these 73 mutants with the WT MC4R. Three mutants, R165G, R165W and C172R, displayed significantly increased basal pERK1/2 signaling (P < 0.05) (Fig. 8A) compared with the basal level of the WT MC4R. The basal pERK1/2 levels of nineteen mutants, including R7C, Y35C, G98R, L106P, D126Y, S136F, H158R, T162I, W174C, M208V, A219V, G231S, G252S, F261S, C277X, L300P, Y302F, R305S and I317V, were significantly decreased compared with that of the WT MC4R (P < 0.05) (Fig. 8B and 8C). The other 51 mutant MC4Rs had similar basal pERK1/2 levels as the WT MC4R (data not shown).
Discussion
In addition to the conventional Gs-stimulated adenylyl cyclase pathway, the MC4R also activates MAPKs, especially ERK1/2. The activation of ERK1/2 pathway is one cellular mechanism that may underlie the regulation of energy homeostasis mediated by the MC4R. In this study, we investigated the constitutive and ligand-stimulated activation (pERK1/2) in WT and 73 naturally occurring MC4R mutations, including mutations from all five classes. Our hypothesis was that decreased basal or ligand-stimulated ERK1/2 signaling might contribute to obesity pathogenesis caused by mutations in the MC4R gene. We were also interested in investigating whether there was biased signaling in these naturally occurring mutations in the Gs-cAMP and ERK1/2 pathway.
In previous functional characterization studies, Class V variants behave similarly as the WT MC4R in heterologous expression systems in all parameters studied, including cell surface expression, ligand binding, and agonist-stimulated cAMP [7, 37]. Whether and how these variants cause energy imbalance and therefore obesity was unclear. We hypothesized that defect in ERK1/2 signaling might contribute to obesity pathogenesis. We measured pERK1/2 levels of 30 Class V variants after 5 min stimulation with NDP-α-MSH. The cell surface expression, ligand binding, and cAMP accumulation (both basal and stimulated) of five variants (C40R, V50M, T112M, A154D, S295P) are similar to the WT MC4R [7, 37, 39, 44]. In the present study, we found that these five variants had impaired ligand-stimulated ERK1/2 activation (Figs. 2 and 3). We propose that defective ERK1/2 signaling in these Class V variants might be a cause of obesity observed in patients harboring these variants.
Class IV mutants are expressed on the cell surface and bind ligand with normal affinity, but are defective in agonist-stimulated signaling (decreased efficacy and/or potency) at the Gs-cAMP pathway, including D90N, S136F, A175T and C326R. The expression of A175T on the plasma membrane is similar to that of the WT receptor, but its ability to generate cAMP in response to ligand is reduced [44, 45]. C326R also has impaired cAMP signaling [46]. Our results indicated that A175T and C326R MC4Rs, although defective in Gs-cAMP signaling, retained normal ERK1/2 signaling in response to NDP-α-MSH stimulation as the WT MC4R, hence exhibiting biased signaling in the Gs-cAMP and ERK1/2 pathways (Fig. 4). In contrast, D90N and S136F MC4Rs, with defective Gs-cAMP signaling, also had decreased ERK1/2 activation, exhibiting no biased signaling (Fig. 4). Interestingly, of all the naturally occurring mutations in the MC4R reported, D90N and S136F are the only mutations that exert dominant negative effect on the WT MC4R [47, 48], very different from the studies in other GPCRs where intracellularly retained GPCRs frequently exert dominant negative effect on the cognate WT receptors when co-expressed [49, 50]. We cannot discern a causal relationship between the lack of biased signaling and dominant negative effect in these two mutations.
Class III mutant MC4Rs are expressed on the cell surface, but are defective in ligand binding per se, with either decreased binding capacity and/or affinity, resulting in impairments in hormone stimulated signaling, including G55V, Δ88-92, I102T, L106P, D126Y and A219V. We found that G55V and A219V had increased pERK1/2 levels upon NDP-α-MSH stimulation, although they have significant decrease in binding capacity [40, 43] (Fig. 5), likely due to the presence of spare receptors [49]. Δ88-92, L106P and D126Y MC4Rs, with defective ligand binding, also had decreased ERK1/2 activation (Fig. 5).
Class II mutant receptors are produced but are retained intracellularly, most likely in the endoplasmic reticulum due to misfolding being detected by the cell's quality control system [51]. This class comprises the largest set of MC4R mutations reported to date. In this study, we investigated the ERK1/2 signaling of 29 Class II mutants. Our results indicated that seven mutants, I69T, N97D, G98R, I125K, W174C, I194T and F261S, showed increased pERK1/2 levels upon NDP-α-MSH stimulation (Fig. 6). I69T MC4R has only 34% of the cell surface expression and 5% of cAMP signaling of the WT MC4R [52]. N97D and I125K have defective cell surface expression and are unable to generate cAMP in response to ligand stimulation [44, 52], but its MAPK signaling could be stimulated by NDP-α-MSH. G98R has negligible cell surface expression [37]. W174C and F261S have significantly reduced cell surface expression levels compared to the WT MC4R [40]. I194T has decreased cell surface expression, with no measurable ligand binding or signaling to either α-MSH or β-MSH stimulation [42]. However, these mutant MC4Rs could respond to NDP-α-MSH stimulation with increased ERK1/2 signaling, suggesting that these Class II mutants with partial defect in cell surface expression exhibited biased signaling. In the follicle-stimulating hormone receptor, a naturally occurring mutation results in diminished expression at the cell surface and lack of detectable Gs-cAMP signaling but retains ERK1/2 signaling [53]. In addition, we found that the other 22 Class II mutants had decreased pERK1/2 levels, consistent with the impaired cell surface expression levels in these mutants (Fig. 6).
Due to defective protein synthesis and/or accelerated protein degradation, decreased receptor proteins of Class I mutants are present in the cell. Mutants such as M79I, S94N, Del170, C277X and R305Q likely belong to this class. Although M79I and S94N have relatively normal cell surface expression and ligand binding, they have significantly decreased total expression and partially defective signaling [42]. In the present study, the pERK1/2 levels of M79I and S94N MC4Rs were significantly increased upon NDP-α-MSH stimulation, suggesting that these mutants with partial defect in protein expression could stimulate ERK1/2 signaling (Fig. 7).
Table 1 summarizes the cAMP signaling data previously reported in the literature (together with the original references) and ERK1/2 signaling data reported in this paper. By comparing the two sets of data, we can tentatively identify the biased mutant receptors.
The WT MC4R has some basal activity in cAMP production [54]. Previously, it was suggested that defects in basal cAMP signaling contribute to obesity pathogenesis caused by mutations in the MC4R gene [55]. We asked whether defects in basal MAPK signaling might also cause obesity. We compared the basal pERK1/2 levels of the 73 mutants with the WT MC4R. Nineteen mutants, including R7C, Y35C, G98R, L106P, D126Y, S136F, H158R, T162I, W174C, M208V, A219V, G231S, G252S, F261S, C277X, L300P, Y302F, R305S and I317V, had significantly decreased basal pERK1/2 levels compared with that of the WT MC4R (Fig. 8B,C). We suggest that decreased basal ERK1/2 signaling might contribute to obesity pathogenesis caused by mutations in the MC4R gene, especially for mutants with no defects identified so far, such as Y35C, H158R, G231S, R305S, and I317V. In addition, we found that three mutants, R165G, R165W and C172R, had significantly increased basal pERK1/2 signaling compared with that of the WT MC4R (Fig. 8A). We have observed constitutive activation of ERK1/2 signaling pathway in the MC4R before in both naturally occurring and laboratory-generated mutations [10, 16, 20]. Because the basal activities in the Gs-cAMP pathway for the three mutations, R165G, R165W and C172R, are not increased compared to the WT MC4R [43, 56], but they were constitutively active in the ERK1/2 signaling pathway, therefore they had biased constitutive activity. Previously, in the melanocortin-1 receptor, a mutant receptor constitutively active in the Gs-cAMP pathway is not constitutively active in the ERK1/2 pathway, also representing a case of biased constitutive signaling [57].
Ligand-induced cAMP and ERK1/2 signaling in mutant MC4Rs.
| hMC4R | Class | cAMP | Ref. | pERK1/2 | Bias |
|---|---|---|---|---|---|
| R7C | V | ↑ | [40] | ↑ | No |
| T11A | V | ↑ | [7] | ↑ | No |
| S30F | V | ↑ | [56] | ↑ | No |
| Y35C | V | ↑ | [39] | ↑ | No |
| D37V | V | ↑ | [37] | ↓ | Yes |
| C40R | V | ↑ | [39] | ↓ | Yes |
| P48S | V | ↑ | [37] | ↑ | No |
| V50M | V | ↑ | [37] | ↓ | Yes |
| F51L | V | ↑ | [7] | ↑ | No |
| G55V | III | ↓ | [43] | ↑ | Yes |
| S58C | II | ↑ | [37] | ↑ | No |
| N62S | II | ↓ | [37, 44, 52] | ↓ | No |
| I69R | II | ↓ | [42] | ↓ | No |
| I69T | II | ↓ | [52] | ↑ | Yes |
| P78L | II | ↓ | [37, 60, 61] | ↓ | No |
| M79I | I | ↓ | [42] | ↑ | Yes |
| C84R | II | ↓ | [40] | ↓ | No |
| Δ88-92 | III | ↓ | [38] | ↓ | No |
| D90N | IV | ↓ | [47] | ↓ | No |
| S94N | I | ↑ | [42] | ↑ | No |
| N97D | II | ↓ | [44] | ↑ | Yes |
| G98R | II | ↓ | [62] | ↑ | Yes |
| I102S | II | ↓ | [7] | ↓ | No |
| I102T | III | ↑ | [7] | ↑ | No |
| L106P | III | ↓ | [44] | ↓ | No |
| T112M | V | ↑ | [37] | ↓ | Yes |
| I125K | II | ↓ | [44, 52] | ↑ | Yes |
| D126Y | III | ↓ | [42] | ↓ | No |
| S136F | IV | ↓ | [40] | ↓ | No |
| A154D | V | ↑ | [7] | ↓ | Yes |
| Y157S | II | ↓ | [37] | ↓ | No |
| H158R | V | ↑ | [63] | ↑ | No |
| T162I | II | ↓ | [52] | ↓ | No |
| R165G | II | ↑ | [43] | ↓ | Yes |
| R165W | II | ↓ | [52, 60, 61] | ↓ | No |
| R165Q | II | ↓ | [44, 52, 60] | ↓ | No |
| I170V | V | ↑ | [37] | ↑ | No |
| Del170 | I | ↓ | [42] | ↓ | No |
| C172R | II | ↓ | [43] | ↓ | No |
| W174C | II | ↓ | [40] | ↑ | Yes |
| A175T | IV | ↓ | [44] | ↑ | Yes |
| I194T | II | ↓ | [42] | ↑ | Yes |
| I195S | II | ↑ | [42] | ↑ | No |
| M200V | V | ↑ | [7] | ↑ | No |
| F201L | V | ↑ | [42] | ↑ | No |
| F202L | V | ↑ | [43] | ↑ | No |
| M208V | V | ↑ | [43] | ↑ | No |
| M218T | V | ↑ | [39] | ↑ | No |
| A219V | III | ↓ | [40] | ↑ | Yes |
| G231S | V | ↑ | [64] | ↑ | No |
| G231V | V | ↑ | [42] | ↑ | No |
| N240S | V | ↑ | [7] | ↑ | No |
| G252S | V | ↑ | [56] | ↑ | No |
| P260Q | II | ↓ | [42] | ↑ | Yes |
| F261S | II | ↓ | [40] | ↑ | Yes |
| I269N | II | ↑ | [43] | ↓ | Yes |
| C271Y | II | ↓ | [37, 44, 52] | ↑ | Yes |
| N274S | V | ↑ | [37] | ↓ | Yes |
| C277X | I | ↓ | [65] | ↓ | No |
| I289L | V | ↑ | [42] | ↑ | No |
| S295P | V | ↑ | [7] | ↓ | Yes |
| P299H | II | ↓ | [66] | ↓ | No |
| L300P | II | ↑ | [42] | ↑ | No |
| Y302F | II | ↑ | [41] | ↑ | No |
| A303P | II | ↑ | [43] | ↓ | Yes |
| R305Q | II | ↑ | [46] | ↑ | No |
| R305S | V | ↑ | [42] | ↑ | No |
| Q307X | II | ↓ | [42] | ↑ | Yes |
| I317V | V | ↑ | [40] | ↑ | No |
| L325F | V | ↑ | [40] | ↑ | No |
| C326R | IV | ↓ | [46] | ↑ | Yes |
| Y332C | V | ↑ | [42] | ↑ | No |
| Y332H | V | ↑ | [42] | ↑ | No |
“↑”: denotes normal ligand-stimulated response as WT MC4R. “↓”: denotes defective ligand-stimulated response compared to WT MC4R. “Bias” column is designed to demonstrate whether ligand-stimulated cAMP and pERK1/2 signaling pathways were divergent. “Yes” denotes biased activation of either signaling pathway; “No” denotes balanced cAMP and pERK1/2 signaling.
Although this is the first comprehensive study on biased signaling in naturally occurring mutations in the MC4R, these findings need to be further confirmed in neuronal cells since the MC4R is expressed primarily in the central nervous system in vivo. Our recent studies on the pharmacoperones for the MC4R compared the rescuing efficacy in HEK293 cells and neuronal cells [58, 59]. Our results showed that although the results are very similar, a few differences indeed are observed. More importantly, future studies need to confirm the in vivo relevance of mutant receptors with biased signaling properties in regulating energy homeostasis using knockin mice. For the MC4R, proximal (conformational changes in the receptor upon binding of biased ligand or receptor mutation), intermediate (from conformational change into downstream signaling), and distal (effects of differential signaling on the gene expression and physiology) mechanisms of biased signaling [23] all remain to be further explored.
In conclusion, the results of the present study demonstrated that nineteen mutants had significantly decreased basal pERK1/2 level, and five Class V variants had impaired ligand-stimulated ERK1/2 activation. The decreased basal or ligand-stimulated ERK1/2 signaling might contribute to obesity pathogenesis caused by mutations in the MC4R gene. We also observed biased signaling in these naturally occurring mutations in the Gs-cAMP and ERK1/2 pathway.
Abbreviations
ERK1/2: extracellular signal-regulated kinases 1 and 2; GPCR: G protein-coupled receptor; MAPK: mitogen-activated protein kinas; MC4R: melanocortin-4 receptor; NDP-α-MSH: [Nle4,D-Phe7]-α-melanocyte stimulating hormone; pERK1/2: phosphorylated ERK1/2; WT: wild type.
Acknowledgements
We thank M. Klymkowsky for providing the E7 beta-tubulin antibody, which was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242. This study was funded by the American Diabetes Association Grant 1-12-BS212, and Auburn University Intramural Grant Program and Interdisciplinary Grant of College of Veterinary Medicine at Auburn University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR. et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131-41
2. Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8:571-8
3. Tao YX. The melanocortin-4 receptor: Physiology, pharmacology, and pathophysiology. Endocr Rev. 2010;31:506-43
4. Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O'Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085-95
5. Tao YX. Mutations in melanocortin-4 receptor and human obesity. Prog Mol Biol Transl Sci. 2009;88:173-204
6. Hinney A, Volckmar AL, Knoll N. Melanocortin-4 receptor in energy homeostasis and obesity pathogenesis. Prog Mol Biol Transl Sci. 2013;114:147-91 doi:10.1016/B978-0-12-386933-3.00005-4
7. Tao YX, Segaloff DL. Functional analyses of melanocortin-4 receptor mutations identified from patients with binge eating disorder and nonobese or obese subjects. J Clin Endocrinol Metab. 2005;90:5632-8
8. Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ. et al. Molecular cloning, expression, and gene localization of a fourth melanocortin receptor. J Biol Chem. 1993;268:15174-9
9. Mountjoy KG, Mortrud MT, Low MJ, Simerly RB, Cone RD. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol Endocrinol. 1994;8:1298-308
10. Tao YX. Constitutive activity in melanocortin-4 receptor: biased signaling of inverse agonists. Adv Pharmacol. 2014;70:135-54
11. Breit A, Büch TR, Boekhoff I, Solinski HJ, Damm E, Gudermann T. Alternative G protein coupling and biased agonism: new insights into melanocortin-4 receptor signalling. Mol Cell Endocrinol. 2011;331:232-40
12. Büch TR, Heling D, Damm E, Gudermann T, Breit A. Pertussis toxin-sensitive signaling of melanocortin-4 receptors in hypothalamic GT1-7 cells defines agouti-related protein as a biased agonist. J Biol Chem. 2009;284:26411-20
13. Chai B, Li JY, Zhang W, Newman E, Ammori J, Mulholland MW. Melanocortin-4 receptor-mediated inhibition of apoptosis in immortalized hypothalamic neurons via mitogen-activated protein kinase. Peptides. 2006;27:2846-57
14. Daniels D, Patten CS, Roth JD, Yee DK, Fluharty SJ. Melanocortin receptor signaling through mitogen-activated protein kinase in vitro and in rat hypothalamus. Brain Res. 2003;986:1-11
15. Vongs A, Lynn NM, Rosenblum CI. Activation of MAP kinase by MC4-R through PI3 kinase. Regul Pept. 2004;120:113-8
16. Mo XL, Tao YX. Activation of MAPK by inverse agonists in six naturally occurring constitutively active mutant human melanocortin-4 receptors. Biochim Biophys Acta. 2013;1832:1939-48
17. Sutton GM, Duos B, Patterson LM, Berthoud HR. Melanocortinergic modulation of cholecystokinin-induced suppression of feeding through extracellular signal-regulated kinase signaling in rat solitary nucleus. Endocrinology. 2005;146:3739-47
18. Patten CS, Daniels D, Suzuki A, Fluharty SJ, Yee DK. Structural and signaling requirements of the human melanocortin 4 receptor for MAP kinase activation. Regul Pept. 2007;142:111-22
19. Huang H, Tao YX. Pleiotropic functions of the transmembrane domain 6 of human melanocortin-4 receptor. J Mol Endocrinol. 2012;49:237-48
20. Mo XL, Yang R, Tao YX. Functions of transmembrane domain 3 of human melanocortin-4 receptor. J Mol Endocrinol. 2012;49:221-35
21. Kenakin T. Agonist-receptor efficacy. II. Agonist trafficking of receptor signals. Trends Pharmacol Sci. 1995;16:232-8
22. Kenakin T. Inverse, protean, and ligand-selective agonism: matters of receptor conformation. FASEB J. 2001;15:598-611
23. Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H. et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1-13
24. Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov. 2010;9:373-86
25. Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol. 2012;52:179-97
26. Violin JD, Crombie AL, Soergel DG, Lark MW. Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol Sci. 2014;35:308-16
27. Liu JJ, Horst R, Katritch V, Stevens RC, Wuthrich K. Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science. 2012;335:1106-10
28. Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E. et al. Structural features for functional selectivity at serotonin receptors. Science. 2013;340:615-9
29. Fenalti G, Giguere PM, Katritch V, Huang XP, Thompson AA, Cherezov V. et al. Molecular control of δ-opioid receptor signalling. Nature. 2014;506:191-6
30. Shukla AK, Westfield GH, Xiao K, Reis RI, Huang LY, Tripathi-Shukla P. et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature. 2014;512:218-22
31. Lefkowitz RJ. A brief history of G-protein coupled receptors (Nobel Lecture). Angew Chem Int Ed Engl. 2013;52:6366-78
32. Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM. et al. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci U S A. 2003;100:10782-7
33. Grasberger H, Van Sande J, Hag-Dahood Mahameed A, Tenenbaum-Rakover Y, Refetoff S. A familial thyrotropin (TSH) receptor mutation provides in vivo evidence that the inositol phosphates/Ca2+ cascade mediates TSH action on thyroid hormone synthesis. J Clin Endocrinol Metab. 2007;92:2816-20
34. Herraiz C, Jimenez-Cervantes C, Zanna P, Garcia-Borron JC. Melanocortin 1 receptor mutations impact differentially on signalling to the cAMP and the ERK mitogen-activated protein kinase pathways. FEBS Lett. 2009;583:3269-74
35. Leach K, Wen A, Davey AE, Sexton PM, Conigrave AD, Christopoulos A. Identification of molecular phenotypes and biased signaling induced by naturally occurring mutations of the human calcium-sensing receptor. Endocrinology. 2012;153:4304-16
36. Sbai O, Monnier C, Dode C, Pin JP, Hardelin JP, Rondard P. Biased signaling through G-protein-coupled PROKR2 receptors harboring missense mutations. FASEB J. 2014;28:3734-44
37. Tao YX, Segaloff DL. Functional characterization of melanocortin-4 receptor mutations associated with childhood obesity. Endocrinology. 2003;144:4544-51
38. Donohoue PA, Tao YX, Collins M, Yeo GSH, O'Rahilly S, Segaloff DL. Deletion of codons 88-92 of the melanocortin-4 receptor gene: a novel deleterious mutation in an obese female. J Clin Endocrinol Metab. 2003;88:5841-5
39. Rong R, Tao YX, Cheung BM, Xu A, Cheung GC, Lam KS. Identification and functional characterization of three novel human melanocortin-4 receptor gene variants in an obese Chinese population. Clin Endocrinol. 2006;65:198-205
40. Fan ZC, Tao YX. Functional characterization and pharmacological rescue of melanocortin-4 receptor mutations identified from obese patients. J Cell Mol Med. 2009;13:3268-82
41. Roth CL, Ludwig M, Woelfle J, Fan ZC, Brumm H, Biebermann H. et al. A novel melanocortin-4 receptor gene mutation in a female patient with severe childhood obesity. Endocrine. 2009;36:52-9
42. Wang ZQ, Tao YX. Functional studies on twenty novel naturally occurring melanocortin-4 receptor mutations. Biochim Biophys Acta. 2011;1812:1190-9
43. Hohenadel MG, Thearle MS, Grice BA, Huang H, Dai MH, Tao YX. et al. Brain-derived neurotrophic factor in human subjects with function-altering melanocortin-4 receptor variants. Int J Obes (Lond). 2014;38:1068-74
44. Yeo GS, Lank EJ, Farooqi IS, Keogh J, Challis BG, O'Rahilly S. Mutations in the human melanocortin-4 receptor gene associated with severe familial obesity disrupts receptor function through multiple molecular mechanisms. Hum Mol Genet. 2003;12:561-74
45. Alfieri A, Pasanisi F, Salzano S, Esposito L, Martone D, Tafuri D. et al. Functional analysis of melanocortin-4-receptor mutants identified in severely obese subjects living in Southern Italy. Gene. 2010;457:35-41
46. Moore BS, Mirshahi UL, Yost EA, Stepanchick AN, Bedrin MD, Styer AM. et al. Long-term weight-loss in gastric bypass patients carrying melanocortin 4 receptor variants. PLoS One. 2014;9:e93629
47. Biebermann H, Krude H, Elsner A, Chubanov V, Gudermann T, Gruters A. Autosomal-dominant mode of inheritance of a melanocortin-4 receptor mutation in a patient with severe early-onset obesity is due to a dominant-negative effect caused by receptor dimerization. Diabetes. 2003;52:2984-8
48. Tarnow P, Rediger A, Brumm H, Ambrugger P, Rettenbacher E, Widhalm K. et al. A heterozygous mutation in the third transmembrane domain causes a dominant-negative effect on signaling capability of the MC4R. Obes Facts. 2008;1:155-62
49. Tao YX. Molecular mechanisms of the neural melanocortin receptor dysfunction in severe early onset obesity. Mol Cell Endocrinol. 2005;239:1-14
50. Tao YX. Inactivating mutations of G protein-coupled receptors and diseases: Structure-function insights and therapeutic implications. Pharmacol Ther. 2006;111:949-73
51. Tao YX, Conn PM. Chaperoning G protein-coupled receptors: From cell biology to therapeutics. Endocr Rev. 2014;35:602-47
52. Rene P, Le Gouill C, Pogozheva ID, Lee G, Mosberg HI, Farooqi IS. et al. Pharmacological chaperones restore function to MC4R mutants responsible for severe early-onset obesity. J Pharmacol Exp Ther. 2010;335:520-32
53. Tranchant T, Durand G, Gauthier C, Crepieux P, Ulloa-Aguirre A, Royere D. et al. Preferential β-arrestin signalling at low receptor density revealed by functional characterization of the human FSH receptor A189 V mutation. Mol Cell Endocrinol. 2011;331:109-18
54. Tao YX, Huang H, Wang ZQ, Yang F, Williams JN, Nikiforovich GV. Constitutive activity of neural melanocortin receptors. Methods Enzymol. 2010;484:267-79
55. Srinivasan S, Lubrano-Berthelier C, Govaerts C, Picard F, Santiago P, Conklin BR. et al. Constitutive activity of the melanocortin-4 receptor is maintained by its N-terminal domain and plays a role in energy homeostasis in humans. J Clin Invest. 2004;114:1158-64
56. Hinney A, Hohmann S, Geller F, Vogel C, Hess C, Wermter AK. et al. Melanocortin-4 receptor gene: case-control study and transmission disequilibrium test confirm that functionally relevant mutations are compatible with a major gene effect for extreme obesity. J Clin Endocrinol Metab. 2003;88:4258-67
57. Benned-Jensen T, Mokrosinski J, Rosenkilde MM. The E92K melanocortin 1 receptor mutant induces cAMP production and arrestin recruitment but not ERK activity indicating biased constitutive signaling. PLoS One. 2011;6:e24644
58. Huang H, Tao YX. A small molecule agonist THIQ as a novel pharmacoperone for intracellularly retained melanocortin-4 receptor mutants. Int J Biol Sci. 2014;10:817-24
59. Tao YX, Huang H. Ipsen 5i is a novel potent pharmacoperone for intracellularly retained melanocortin-4 receptor mutants. Front Endocrinol (Lausanne). 2014;5:131
60. Nijenhuis WA, Garner KM, VanRozen RJ, Adan RA. Poor cell surface expression of human melanocortin-4 receptor mutations associated with obesity. J Biol Chem. 2003;278:22939-45
61. Lubrano-Berthelier C, Durand E, Dubern B, Shapiro A, Dazin P, Weill J. et al. Intracellular retention is a common characteristic of childhood obesity-associated MC4R mutations. Hum Mol Genet. 2003;12:145-53
62. Kobayashi H, Ogawa Y, Shintani M, Ebihara K, Shimodahira M, Iwakura T. et al. A novel homozygous missense mutation of melanocortin-4 receptor (MC4R) in a Japanese woman with severe obesity. Diabetes. 2002;51:243-6
63. Piechowski CL, Rediger A, Lagemann C, Muhlhaus J, Muller A, Pratzka J. et al. Inhibition of melanocortin-4 receptor dimerization by substitutions in intracellular loop 2. J Mol Endocrinol. 2013;51:109-18
64. Xiang Z, Proneth B, Dirain ML, Litherland SA, Haskell-Luevano C. Pharmacological characterization of 30 human melanocortin-4 receptor polymorphisms with the endogenous proopiomelanocortin-derived agonists, synthetic agonists, and the endogenous agouti-related protein antagonist. Biochemistry. 2010;49:4583-600
65. Yang JJ, Tang SS, Hu C, Zhang R, Song ZC, Wang B. et al. Screening for melanocortin 4 receptor mutations in Chinese extremely obese individuals. Biomed Environ Sci. 2013;26:611-3
66. Lubrano-Berthelier C, Dubern B, Lacorte JM, Picard F, Shapiro A, Zhang S. et al. Melanocortin 4 receptor mutations in a large cohort of severely obese adults: prevalence, functional classification, genotype-phenotype relationship, and lack of association with binge eating. J Clin Endocrinol Metab. 2006;91:1811-8
Author contact
![]() Corresponding author: Ya-Xiong Tao, PhD. Department of Anatomy, Physiology and Pharmacology, 212 Greene Hall, College of Veterinary Medicine, Auburn University, Auburn, AL 36849, United States. Tel: 01-334-844-5396; FAX: 01-334-844-5388 Email: taoyaxiedu.
Corresponding author: Ya-Xiong Tao, PhD. Department of Anatomy, Physiology and Pharmacology, 212 Greene Hall, College of Veterinary Medicine, Auburn University, Auburn, AL 36849, United States. Tel: 01-334-844-5396; FAX: 01-334-844-5388 Email: taoyaxiedu.
Received 2014-8-18
Accepted 2014-9-15
Published 2014-10-9