ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2015; 11(5):618-628. doi:10.7150/ijbs.11380 This issue Cite
Research Paper
Tamoxifen Inhibits ER-negative Breast Cancer Cell Invasion and Metastasis by Accelerating Twist1 Degradation
Gang Ma1, 2, Jianjun He1, Yang Yu2, Yixiang Xu2, 3, Xiaobin Yu2, Jarrod Martinez2, David M. Lonard2, Jianming Xu2, 4, ![]()
1. Department of Breast and Thyroid Cancer Surgery, The First Affiliated Hospital of Xi'an Jiaotong University Medical School, Xi'an, China;
2. Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, TX, USA;
3. Institute of Biosciences and Technology, Texas A&M University Health Science Center, Houston, TX, USA;
4. Institute for Cancer Medicine and School of Basic Medical Sciences, Luzhou Medical College, Sichuan, China
Abstract

Twist1 is a transcription factor driving epithelial-mesenchymal transition, invasion and metastasis of breast cancer cells. Mice with germ-line Twist1 knockout are embryonic lethal, while adult mice with inducible Twist1 knockout have no obvious health problems, suggesting that Twist1 is a viable therapeutic target for the inhibition of invasion and metastasis of breast cancer in adult patients. In this study, we expressed a luciferase protein or a Twist1-luciferase fusion protein in HeLa cells as part of a high throughput system to screen 1280 compounds in the Library of Pharmacologically Active Compounds (LOPAC) from Sigma-Aldrich for their effects on Twist1 protein expression. One of the most interesting compounds identified is tamoxifen, a selective estrogen receptor (ER) modulator used to treat ER-positive breast cancer. Tamoxifen treatment significantly accelerated Twist1 degradation in multiple cell lines including HEK293 human kidney cells, 4T1 and 168FARN mouse mammary tumor cells with either ectopically or endogenously expressed Twist1. Tamoxifen-induced Twist1 degradation could be blocked by the MG132 proteasome inhibitor, suggesting that tamoxifen induces Twist1 degradation through the ubiquitination-proteasome pathway. However, tamoxifen-induced Twist1 degradation was independent of Twist1 mRNA expression, estrogen signaling and MAPK-mediated Twist1 phosphorylation in these cells. Importantly, tamoxifen also significantly inhibited invasive behavior in Matrigel and lung metastasis in SCID-bg mice of ER-negative 4T1 mammary tumor cells, which depend on endogenous Twist1 to invade and metastasize. These results indicate that tamoxifen can significantly accelerate Twist1 degradation to suppress cancer cell invasion and metastasis, suggesting that tamoxifen can be used not only to treat ER-positive breast cancers but also to reduce Twist1-mediated invasion and metastasis in ER-negative breast cancers.
Keywords: Breast cancer, Twist1, tamoxifen, Twist1 degradation, lung metastasis
Introduction
Owing to early diagnosis and treatment and the development of new therapeutic drugs and technologies, the quality of life and survival times for breast cancer patients are continuously improving. However, because of its high incidence, unsatisfactory response to therapeutic reagents, and development of acquired drug resistance, breast cancer remains a major threat to woman's health. About 75% of breast cancers express estrogen receptor (ER) α, and these ER-positive breast cancers are typically treated with selective estrogen receptor modulators (SERMs) such as tamoxifen or aromatase inhibitors such as anastrozole and letrozole in combination with other commonly used chemo- or radio- therapies. However, nearly half of these cancers exhibit unsatisfactory responses and many that do respond well initially develop acquired resistance to these endocrine therapies. About 15-20% of breast cancers overexpress the HER2 oncogene, and these HER2-positive cancers can be treated with the HER2-neutralizing antibodies such as trastuzumab in combination with other commonly used chemo- or radio-therapies. However, the response rate and the long-term clinical outcome of HER2 antibody-based therapy remain unsatisfactory. Finally, about 15-20% of breast cancers are triple negative cancers, which do not express ER and progesterone receptor (PR) and do not overexpress HER2. These cancers still have no targeted therapy and can be only treated via conventional chemo- and/or radio-therapies. Therefore, it is important to identify other molecular targets and to develop new drugs against these targets to expand the range of therapeutic interventions to treat all three molecular subtypes of breast cancer.
Twist1 is a basic helix-loop-helix (bHLH) domain-containing transcription factor that binds the Nde1 E-box to activate or repress gene expression [1, 2]. Twist1 is essential for mesoderm specification and differentiation during embryonic development [1, 3]. Twist1 germ-line knockout mouse embryos die with unclosed cranial neural tubes and defective head mesenchyme, somites and limb buds [3]. Heterozygous loss-of-functional mutations of the human Twist1 gene cause Saethre-Chotzen syndrome [4, 5]. Interestingly, in adult mice Twist1 protein is only detected in a few cell types including the dermal papilla of the skin and fibroblasts in the mammary gland. Inducible knockout of Twist1 in mice older than 2 weeks significantly prolongs the hair growth cycle without causing any obvious health problem [6]. These findings indicate that although Twist1 is absolutely required for embryonic development, its function is not essential for maintaining a generally healthy condition of adult animal.
Importantly, Twist1 is expressed in many types of cancer cells including breast cancer cells, and its expression is usually associated with invasive and metastatic cancer phenotypes [2, 7]. Twist1 drives epithelial-mesenchymal transition (EMT), migration and invasion of cancer cells, and hence promotes cancer metastasis [2, 7-9]. Twist1 stability and function are enhanced by its phosphorylation mediated by MAPKs, one of the major cancer-driving pathways downstream of tyrosine receptor kinases and ras oncoproteins [10]. Twist1 promotes EMT in part by directly repressing E-cadherin and ER expression by recruiting the nucleosome remodeling and deacetylase (NuRD) complex for gene repression [8, 11] and by upregulating Bmi1, AKT2, YB-1 and WNT5A [2, 12-15]. Emerging evidence also suggests that Twist1 plays a role in cancer stem cells' expansion, chemotherapeutic resistance, and induction of cancer cell differentiation into endothelial cells [16-18]. Taken together, these crucial roles for Twist1 in cancer and the aforementioned non-essential role of Twist1 in adult animal suggest that Twist1 is an attractive molecular target for inhibiting cell invasion, metastasis and acquired drug resistance in breast cancers.
In this study, we developed a luciferase-based high throughput screening system to identify small molecular inhibitors that can induce Twist1 degradation in cancer cells from Sigma's Library of Pharmacologically Active Compounds (LOPAC). We report that tamoxifen strongly accelerates Twist1 degradation through the proteasome pathway in an estrogen signaling independent manner, resulting in a significant inhibition of breast cancer cell invasion and metastasis.
Materials and Methods
Cell culture
The HEK293 cell line with doxycycline-inducible Flag-tagged Twist1 expression was described previously [8, 10]. This HEK293 cell line, the 168FARN and 4T1 mouse mammary tumor cell lines and the HeLa and MDA-MB-435 human cancer cell lines were cultured in Dulbecco's Modified Eagle's Medium (DMEM), supplemented with 10% fetal calf serum (FCS) at 37oC in a tissue culture incubator with 21% of O2 and 5% of CO2.
Plasmid construction
We used pQCXIH plasmid (Clontech, Mountain View, CA) to construct the expression vectors for the Twist1-luciferase (Twist1-Luc) fusion protein and the luciferase (Luc) control. To construct the pQCXIH-Twist1-Luc vector, the coding region of the human Twist1 cDNA was amplified by PCR using the 5'-ttgcggccgccaccatgatgcaggacgtgtc primer with a NotI site and the Kozak sequence and the 5'-ttaccggtgtgggacgcggacatggaccagg primer with an AgeI site. The luciferase-coding region was amplified by PCR using the 5'-taccggtatggaagacgccaaaaac primer with an AgeI site and the 5'-ccttaattaattacacggcgatctttc primer with a PacI site. These two amplified DNA fragments were cloned into the pQCXIH plasmid by using the NotI, AgeI and PacI sites. To construct the pQCXIH-Luc vector, the luciferase coding region was amplified by PCR from the pGL3-basic vector using the 5'-gaccggtgccaccatggaagacgccaaaaacat primer with an AgeI site and a Kozak sequence and the 5'-ccttaattaattacacggcgatctttc primer with a PacI site. The amplified DNA was cloned into the pQCXIH plasmid by using the AgeI and PacI sites. Both expression vectors were validated by DNA sequencing.
Screening the Library of Pharmacologically Active Compounds (LOPAC), cell transfection and luciferase assays
HeLa cells were seeded in 96-well plate at a density of 9000 cells/well and cultured in DMEM with 10% of FCS overnight. Cells were transfected with pQCXIH-Twist1-Luc or pQCXIH-Luc plasmid (250 ng/well) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA; 0.75 μl/well), and cultured overnight. Then, these transfected cells in 96-well plates with 100 μl medium were treated with 1 μl of DMSO (vehicle) or 1 μl of one of the 1280 compound stock solutions (1 mM in DMSO) made from the LOPAC library (Sigma-Aldrich, St. Louis, MO). After treating cells for 24 hours at 37oC in a tissue culture incubator, cells were washed with PBS and lysed for luciferase assay as described previously [19]. To confirm the screening result from tamoxifen treatment, HeLa cells in 24-well plate were transfected with pQCXIH-Twist1-Luc or pQCXIH-Luc plasmid and a β-galactosidase expression plasmid. After cells were treated with vehicle (ethanol) or tamoxifen (10 μM) for 24 hours, their luciferase and β-galacrtosidase activities were measured. The relative luciferase activity was normalized to the β-galactosidase activity in each sample. Transfection experiments were performed in duplicates with at least one additional repeat.
Western blot
Cells treated with tamoxifen, 17β-estradiol, ICI 182,780, 4-hydroxytamoxifen, cycloheximide and/or MG132 (Sigma-Aldrich, St. Louis, MO) in different experiments were harvested and lysed on ice for 20 minutes in a lysis buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.5 mM EDTA, 0.1% SDS, 1% Triton-X 100, 1 mM DTT, 100 μM Na3VO3, 100 μM sodium pyrophosphate, and 100 μM sodium fluoride. Protein concentrations were measured by using the Bradford Protein Assay reagent (Bio-Rad, Hercules, CA). Processed cell lysates with 30 μg of protein in each were separated by electrophorysis in a 10% SDS-PAGE gel. The protein in the gel was blotted onto a nitrocellulose membrane. The membrane was blocked with 5% fat-free milk or 5% BSA in PBST (PBS with 0.1% Tween-20) at room temperature for 1 hour, and then incubated with a primary antibody at 4oC overnight. This study used the primary antibodies against Twist1 (Abcam, Cambridge MA; 1:200 dilution), phospho-Twist1 (1:1000 dilution)[10], MAPKs and phospho-MAPKs (Cell Signaling, Danvers, MA; 1:1000 dilution) as well as β-actin (Santa Cruz Biotechnology, Santa Cruz, CA; 1:5000 dilution). After washing 3 times with PBST, the membrane was incubated with an appropriate secondary antibody conjugated to horseradish peroxidase at room temperature for 1 hour. The membrane was washed 3 times in PBST and then incubated with a substrate solution in an ECL kit to develop a chemiluminescence signal. The X-Ray film was exposed at least for 30 seconds before development.
RNA isolation and quantitative RT-PCR (Q-PCR)
Total RNA was extracted from tamoxifen-treated HEK293 cells with inducible Twist1 expression and tamoxifen-treated 4T1 and 168FARN cells with endogenous Twist1 expression using Trizol reagent (Life Technologies, Grand Island, NY) according to the manufacturer's protocol. The isolated total RNA was further purified using RNeasy columns (Qiagen, Valencia, CA). For TaqMan Q-PCR analysis, total RNA was reversely transcribed. Sequence-specific primers and fluorescence labeled probes for human Twist1 and β-actin cDNAs were designed and matched by using the Universal Probe Library Center Software (Roche, Indianapolis, IN). Relative concentrations of Twist1 mRNA were obtained by normalization to β-actin mRNA.
Cell invasion assays
Cell invasion assay was carried out using the BD BioCoat Matrigel Invasion Chamber (BD Biosciences, Bedford, MA). 4T1 cells cultured in DMEM with 10% FCS were treated with vehicle (ethanol) or 10 uM tamoxifen for 24 hours, and then seeded into the upper chambers (25000 cells/chamber) with serum-free DMEM. The lower chamber was filled with DMEM containing 1% FCS as chemoattractant. After culturing for 24 hours, the cells and Matrigel above the membrane were removed with a cotton swab, and the cells adherent to the lower surface of the membrane were stained with DAPI. The stained cells on the air-dried membrane were examined and counted at 20X magnification under a fluorescence microscope.
Analysis of 4T1 cell-derived xenograft mammary tumor growth and lung metastasis in mice
4T1 cells were cultured in DMEM with 10% FCS to confluence and harvested by trypsin digestion. Cells were washed in PBS and resuspended in serum-free DMEM at 1 × 106 cells/ml. Female SCID-bg mice aged 6-8 weeks were anesthetized with Avertin (50 mg/kg, i.p.). 4T1 Cells (1×105 in 0.1 ml volume) were injected into the right second mammary fat pad of each mouse. Four days after the injection, mice were randomly divided into two groups. One group was treated with 50 μl of corn oil (once every 2 days, i.p.) for 5 weeks, while another group was treated with tamoxifen (5 mg/kg body weight in 50 μl of corn oil, once every 2 days, i.p.) for 5 weeks. Tumor length and width were measured once a week and tumor volume was estimated by (length × width2)/2 as described previously [20]. At the experimental end point, mice were euthanized, and their lungs were dissected out and examined under a stereoscope for imaging and counting of metastatic foci seen on the lung surface. Then, the lungs were fixed in 4% paraformaldehype in PBS and embedded in paraffin blocks. Coronal lung sections of 5 μm thickness were prepared. Five sections with at least a 200 μm interval with respect to each other were stained with hematoxylin and eosin (H&E) and imaged under a microscope. The total lung area and metastasis tumor area on the lung section were measured and the ratio of tumor area to lung area were calculated and used as a lung metastasis index as described previously [21].
Results
Tamoxifen treatment drastically decreased Twist1 protein in cells
To establish a high throughput screening system for identifying bioactive small molecules that reduce Twist1 protein in cancer cells, we constructed a pQCXIH-Twist1-Luc vector to express the Twist1-luciferase fusion protein and a pQCXIH-Luc vector to express only the luciferase protein as a control (Fig. 1A). The relative cellular amounts of both Twist1-Luc and luciferase proteins can be conveniently measured by their luciferase activities. We equally transfected HeLa cells in 96-well plates with either pQCXIH-Twist1-Luc or pQCXIH-Luc plasmid, followed by treatment with one of the 1280 pharmacologically active compounds in the LOPAC library. Measurements of luciferase activities in all transfected and treated cells led to the identification of several small compounds that selectively reduced the luciferase activity in cells transfected with pQCXIH-Twist1-Luc plasmid, but had no significant effects on the luciferase activity in cells transfected with the pQCXIH-Luc plasmid. These compounds include tamoxifen citrate (tamoxifen), sertraline, indatraline, CGS-12066A, methiothepin, GR 127935, parthenolide and MK-886. tamoxifen is a well-known SERM for treating ER-positive breast cancer. Sertraline, indatraline and CGS-12066A are a selective serotonin reuptake inhibitor (SSRI), a serotonin receptor agonist and a serotonin antagonist, respectively. Methiothepin and GR-127935 are 5-HT1B/1D receptor antagonists. Parthenolide is an inhibitor of serotonin release from platelets. MK-886 is a potent and specific inhibitor of leukotriene biosynthesis. In this study, tamoxifen was selected for further characterization because of its known therapeutic role in ER-positive breast cancers and the crucial role of Twist1 in driving epithelial-mesenchymal transition and metastasis in breast cancer cells [2].
To validate these screening results, we transiently expressed Twist1-Luc fusion protein or Luc protein in HeLa cells in 24-well plates and treated these cells with tamoxifen or vehicle (ethanol) for 24 hours, and measured their luciferase activities. The results demonstrated that tamoxifen treatment indeed drastically decreased the luciferase activity in cells expressing Twist1-Luc protein when compared with that in cells expressing the Luc protein only (Fig. 1B). These results indicate that tamoxifen treatment can selectively reduce Twist1-Luc protein versus the luciferase protein in transfected HeLa cells. Furthermore, we tested how tamoxifen affected the level of Twist1 protein expressed in a stable HEK293 cell line with inducible Twist1 expression [8]. Constant Twist1 expression in these cells was induced by doxycycline treatment and the cells were then treated with tamoxifen for different time periods. Western blot analysis shows that Twist1 protein remained relatively constant in doxycycline-treated cells without tamoxifen treatment. However, Twist1 protein was significantly reduced in doxycycline-treated cells that were also treated with tamoxifen for 6, 10 and 24 hours (Fig. 1C). Consistently, Twist1 protein in these cells was decreased in a tamoxifen dose-dependent manner (Fig. 1D). Moreover, we treated 168FARN mouse mammary tumor cells, MDA-MB-435 human cancer cells, and 4T1 mouse mammary tumor cells that express endogenous Twist1 with tamoxifen and found that tamoxifen treatment also significantly decreased Twist1 protein in these cells in both a tamoxifen-dependent and a time-dependent manner (Fig. 1E and F). These results clearly indicated that tamoxifen treatment can effectively decrease Twist1 protein in cells.
Tamoxifen treatment decreases the cellular levels of Twist1 protein. A. In pQCXIH-Luc and pQCXIH-Twist1-Luc plasmids, the expression of luciferase or Twist1-Luciferase was driven by the CMV promoter. B. HeLa cells transfected with pQCXIH-Luc or pQCXIH-Twist1-Luc plasmids and with a β-galactosidase expression plasmid were treated with vehicle (ethanol) or tamoxifen (10 μM) for 24 hours. Then, the luciferase activity was measured and normalized to the β-galactosidase activity for each sample. Relative luciferase activity was calculated by (Luc activity in tamoxifen-treated cells/Luc activity in vehicle-treated cells) × 100. Data are presented as mean ± SD. ***, P < 0.0001 by t test. C. HEK293 cells with an inducible Twist1 expression system were treated with 1 μg/ml of doxycycline for 12 hours to induce stable Twist1 expression and then co-treated with 1 μg/ml of doxycycline and vehicle or 10 μM of tamoxifen for the time periods as indicated. HEK293 cells without inducible Twist1 expression served as a negative control (Lane 1). Western blot analyses were performed with Twist1 and β-actin antibodies. D. HEK293 cells with inducible Twist1 expression were treated with 1 μg/ml of doxycycline for 12 hours, and then co-treated with 1 μg/ml of doxycycline and different concentrations of tamoxifen as indicated for 24 hours before Western blotting was performed. E. Western blot analyses of Twist1 in 168FARN and 4T1 cells treated with vehicle or tamoxifen (10 μM) for different time periods as indicated. F. Western blot analyses of Twist1 in 168FARN and MDA-MB-435 cells treated with the indicated concentrations of tamoxifen for 24 hours. Results shown in panels B to F are representative results of at least three repeat assays.
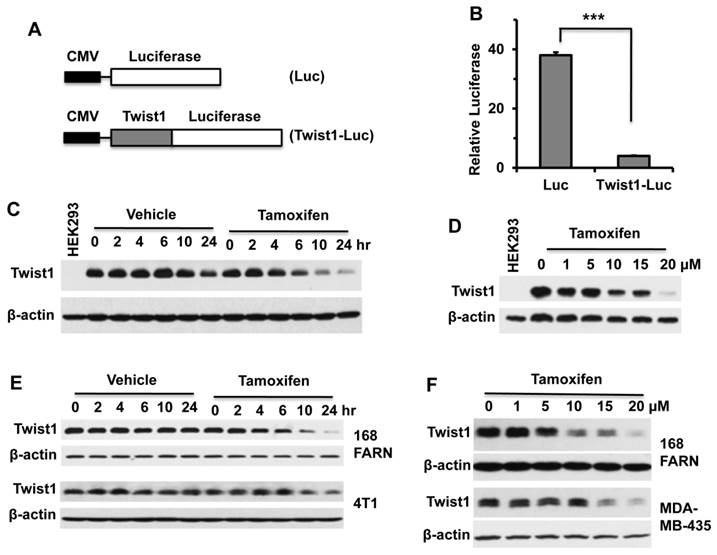
Tamoxifen significantly accelerated Twist1 protein degradation in cells
To determine whether tamoxifen could change Twist1 mRNA levels, we treated HEK293 cells with inducible Twist1 expression and 168FARN and 4T1 mammary tumor cells with endogenous Twist1 expression with different concentrations of tamoxifen for 24 hours and measured Twist1 mRNA by qPCR. Our results demonstrate that tamoxifen treatment had no effect on Twist1 mRNA levels in HEK293 and 4T1 cells and even slightly increased Twist1 mRNA levels in 168FARN cells (Fig. 2A). These results indicate that tamoxifen-induced decreases in Twist1 protein in these cells are not due to changes at its mRNA level.
To determine whether tamoxifen could alter Twist1 protein stability, we treated HEK293 cells with inducible Twist1 expression and 168FARN cells with endogenous Twist1 expression with either cycloheximide or cycloheximide and tamoxifen, and then examined Twist1 protein degradation at different time points. The results from Western blotting show that Twist1 protein was degraded much faster in tamoxifen-treated cells versus cells without tamoxifen treatment upon inhibition of protein synthesis by cycloheximide; the difference in Twist1 protein levels was particularly evident after tamoxifen treatment for 6 hours (Fig. 2B and C). These results demonstrated that tamoxifen treatment significantly accelerates Twist1 protein degradation.
Tamoxifen induces Twist1 degradation through the proteasome-dependent pathway in an estrogen signaling independent manner
To elucidate the mechanism of tamoxifen-induced Twist1 degradation, we examined whether inhibition of proteasome-mediated protein degradation pathway could diminish the effect of tamoxifen on Twist1 protein stability. We treated HEK293 cells with inducible Twist1 expression and 168FARN cells with endogenous Twist1 expression with the proteasome inhibitor MG132 or with MG132 and tamoxifen, followed by analysis of Twist1 protein levels at different time points. We found that Twist1 protein was accumulated in a time-dependent manner in cells treated with MG132 alone, suggesting that Twist1 was normally degraded through the proteasome-mediated pathway. Interestingly, Twist1 protein was maintained at the same level through all the time points in cells treated with both MG132 and tamoxifen, suggesting that tamoxifen-accelerated Twist1 degradation is mostly mediated by the proteasome pathway (Fig. 3A and B). These results also suggest that tamoxifen can prevent Twist1 accumulation in proteasome-inhibited cells through other unknown pathways.
Next, we examined whether tamoxifen-induced Twist1 degradation is related to estrogen signaling. First, we measured ERα and ERβ mRNA levels by Q-PCR and their protein levels by Western blot in HEK293 cells with inducible Twist1 expression and 4T1 and 168FARN cells with endogenous Twist1 expression. However, we did not detect any obvious ERα or ERβ expression in these cells (data not shown). We also treated these three cell lines with 17β-estradiol, ICI 182780 (a pure ER antagonist), 4-hydroxytamoxifen and tamoxifen. We found that 17β-estradiol or ICI 182780 treatment did not change Twist1 protein levels, indicating that either activation or inhibition of estrogen signaling pathways was not related to the tamoxifen-induced Twist1 degradation observed in these cells (Fig. 3C). We also found that 4-hydroxytamoxifen played a similar role as tamoxifen to downregulate Twist1 protein in these cells, which was not surprising since they are similar compounds (Fig. 3C).
We have previously reported that phosphorylation of serine 68 by MAPKs stabilizes Twist1 protein [10]. Therefore, we examined MAPK activities, phosphorylated serine 68-Twist1 and total Twist1 in tamoxifen-treated 168FARN cells. Our results demonstrated that there were no significant changes in either total or activated ERK1/2, p38 and JNK MAP kinases in 168FARN cells treated with or without tamoxifen (Fig. 3D). Consistently, tamoxifen treatment also did not change the level of phosphorylated serine 68-Twist1, although it significantly reduced the total Twist1 level in 168FARN cells (Fig. 3E). These results suggest that tamoxifen-promoted Twist1 degradation is independent of phosphorylation of the serine 68 residue in Twist1.
Tamoxifen accelerates Twist1 protein degradation. A. Q-PCR analyses of Twist1 mRNA in HEK293 cells with inducible Twist1 expression and 4T1 and 168FARN cells with endogenous Twist1 expression. These cells were treated with different concentrations of tamoxifen for 24 hours. B. Western blot analysis of Twist1 in HEK293 cells with inducible Twist1 expression. Cells were treated with 1 μg/ml of doxycycline for 12 hours to induce Twist1 expression, then treated with 50 μM of cycloheximide to inhibit protein synthesis and with vehicle (ethanol) or 10 μM of tamoxifen for different time periods as indicated. Analysis of β-actin served as a loading control. HEK293 parent cells without Twist1 expression (Lane 1) served as a negative control. Band intensities were measured by densitometry and presented in the right panel. The half-life time of Twist1 protein is 4.41 hours in the vehicle-treated cells or 3.80 hours in the tamoxifen-treated cells. C. Western blot analysis of Twist1 in 168FARN cells treated with vehicle (ethanol) or 10 μM of tamoxifen and with 50 μM of cycloheximide for different time periods as indicated. Band intensities were measured by densitometry and presented in the right panel. The half-life time of Twist1 protein is 4.91 hours in the vehicle-treated cells or 4.26 hours in the tamoxifen-treated cells. Results shown in panels B and C are representative results of at least three repeat assays.
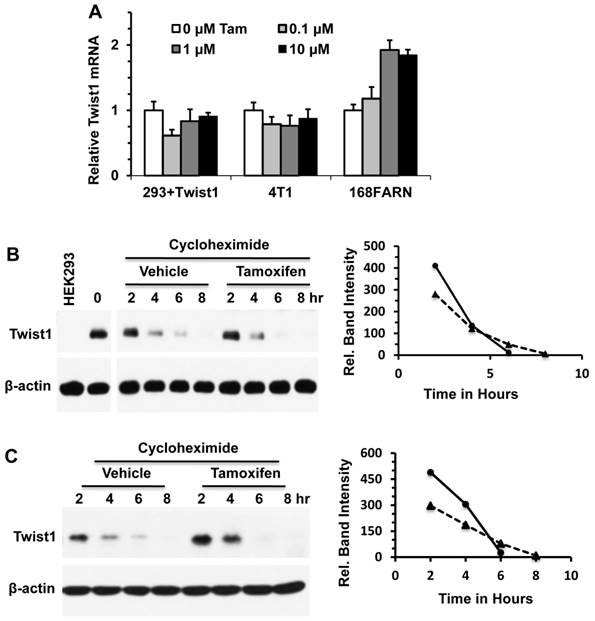
Tamoxifen-accelerated Twist1 degradation is dependent on the proteasome pathway, but is independent of estrogen and MAPK signaling pathways. A. Western blot analysis of Twist1 in HEK293 cells with inducible Twist1 expression (HEK293+Twist1) after cells were treated with 20 μM of MG132 and vehicle (ethanol) or 10 μM of tamoxifen for different time periods as indicated. HEK293 parent cells without Twist1 expression served as a negative control. B. Western blot analysis of Twist1 in 168FARN cells treated with 20 μM of MG132 and vehicle (ethanol) or 10 μM of tamoxifen. C. Western blot analysis of Twist1 in HEK293+Twist1, 4T1 and 168FARN cells treated for 24 hours with vehicle (V, ethanol), or 10 μM of 17β-estradiol (E2), ICI 182,780 (ICI), 4-hydroxytamoxifen (4OH) and tamoxifen (Tam) as indicated. D. Western blot analysis of active phospho-Erk1/2, total Erk1/2, active phospho-p38, total p38, active phospho-JNK, and total JNK in 168FARN cells treated with different concentrations of tamoxifen for 6 hours. E. Western blot analysis of phospho-Ser68-Twist1 and total Twist1 in 168FARN cells treated with different concentrations of tamoxifen for 6 hours. Results shown in all panels are representative results of at least three repeat assays.
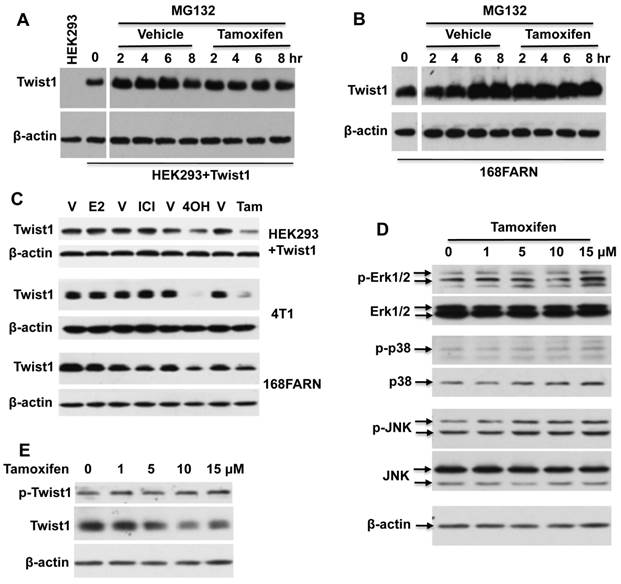
Tamoxifen treatment reduces the invasive capability of mammary tumor cells
Twist1 is known to increase breast cancer cell invasiveness [2, 7, 8]. Therefore, we treated 4T1 mammary tumor cells with endogenous Twist1 expression with tamoxifen and measured the impact of tamoxifen treatment on cell invasion. We found that tamoxifen treatment significantly reduced the number of 4T1 cells that could invade through a layer of Matrigel in a transwell cell invasion assay system (Fig. 4A and B). These results suggest that tamoxifen-induced Twist1 degradation can partially inhibit EMT and invasiveness of breast cancer cells.
Tamoxifen treatment significantly reduced the lung metastasis derived from an ER-negative mammary tumor cells in mice
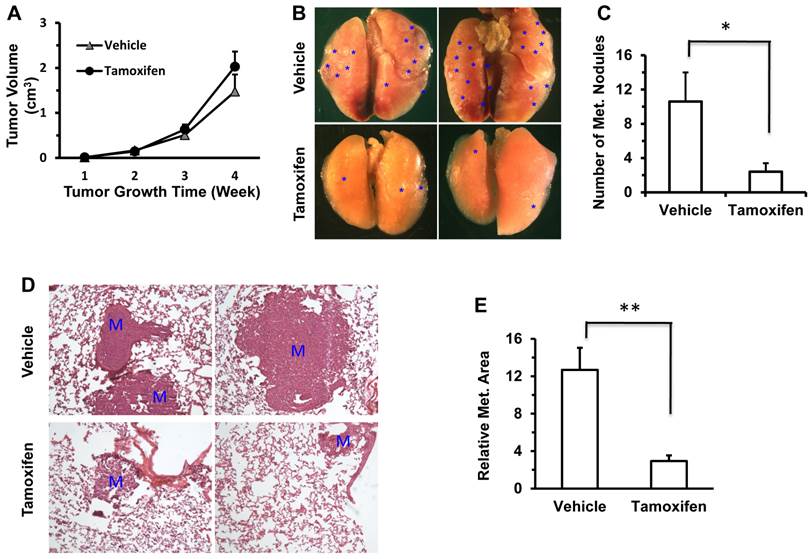
It has been previously reported that knockdown of Twist1 by expressing shRNAs in 4T1 mammary tumor cells significantly inhibits its metastasis to lung in mice [7, 8]. To test whether a tamoxifen-induced decrease in Twist1 protein could reduce lung metastasis in ER-negative 4T1 mammary tumor cells, we injected 4T1 cells into the mammary fat pads of SCID mice, followed by tamoxifen or vehicle (core oil) treatment and examination of primary tumor weights and lung metastases. We found that tamoxifen treatment had no significant influence on primary tumor weight (Fig. 5A), which is consistent with previous studies showing knockdown Twist1 had no obvious effect on primary tumor growth in mice [8]. However, tamoxifen treatment significantly reduced the number of metastatic foci observed on the lung surface (Fig. 5B and C) and the lung metastasis index calculated based on the metastasis area versus the lung area (Fig. 5D and E). These results indicate that tamoxifen treatment can significantly inhibit the lung metastasis derived from the xenograft tumors of the ER-negative 4T1 mammary tumor cells in the mammary gland.
Tamoxifen treatment decreases cell invasiveness of 4T1 mammary tumor cells. 4T1 cells pre-treated with vehicle (ethanol) or 10 μM tamoxifen for 24 hours were seeded in the upper chambers of the transwell invasion assay system (2.5 × 104 cells/chamber). The upper chamber contains serum-free medium with vehicle (ethanol) or 10 μM tamoxifen, while the lower chamber contains medium with 1% serum and vehicle (ethanol) or 10 μM tamoxifen. After culturing for 24 hours, invaded cells on the lower side of the membrane were stained with DAPI, and imaged (Panel A) and counted (Panel B) under a fluorescence microscope. **, P < 0.01 by two-tailed Student's t test, n = 5 for each.
Discussion
The crucial role of Twist1 in cancer metastasis and drug resistance and the nonessential role of Twist1 in adult general health make Twist1 a promising target for inhibiting cancer with tolerable adverse effects in adult patients. With the primary goal to identify small molecule inhibitors that can induce Twist1 degradation, we screened the LOPAC library with 1280 pharmacologically active compounds. This library was chosen because the in vivo metabolic kinetics and toxicity of these compounds have been well characterized. Once an inhibitor is identified, it can be rapidly used in translational studies and possibly even in clinical trials. Among the several compounds identified from our screening, we selected tamoxifen as a leading compound to characterize its role in Twist1 degradation because tamoxifen has been extensively used in the treatment of ER-positive breast cancers.
Our results demonstrated that tamoxifen treatment significantly accelerated Twist1 protein degradation in multiple cell types, including HEK293 kidney epithelial cells with ectopically expressed Twist1 and 4T1 and 168FARN mammary tumor cells with endogenously expressed Twist1. Furthermore, the tamoxifen-induced Twist1 degradation could be blocked by the proteasome inhibitor MG132, indicating that tamoxifen stimulates Twist1 degradation via the proteasome pathway. Importantly, tamoxifen treatment also significantly reduced the invasive behavior and metastasis capability of the 4T1 mammary tumor cells in cell culture and mice. 4T1 cells are ER-negative mammary tumor cells and tamoxifen showed no obvious effects on 4T1 cell proliferation in culture and xenograft tumor growth in the mouse mammary glands. It was also demonstrated that knockdown of Twist1 in 4T1 cells significantly inhibited the invasion and metastasis of 4T1 cells in culture and 4T1 xenograft tumors in mice without significantly affecting the proliferation and xenograft tumor growth of 4T1 cells [7, 8]. Therefore, the phenotypes of tamoxifen-treated 4T1 cells and tumors closely copy the phenotypes of 4T1 tumor cells with Twist1 knockdown, suggesting that tamoxifen-decreased Twist1 is responsible, at least in part, for the reduced invasive behavior and metastasis of 4T1 cells and tumors.
After being administered to patients, tamoxifen is metabolized into 4-OH-tamoxifen, which binds to ER and results in inhibition of its transcriptional activity in certain tissues such as the breast tissue but activation of its activity in certain other tissues such as the uterus. Due to its inhibitory role in the growth of ER-positive breast cancers that rely on estrogen signaling to survive and grow, tamoxifen has been extensively used to treat ER-positive breast cancers [22]. Tamoxifen treatment can inhibit breast cancer relapse after surgery, reduce the risk of invasive breast cancer progression from ductal carcinoma in situ (DCIS) and stop or even shrink some of the metastatic breast tumors. It is conceivable that some of these pharmaceutical effects of tamoxifen may be independent of estrogen signaling. In this study, we demonstrated that tamoxifen-induced Twist1 degradation is independent of estrogen signaling, supporting the notion that additional tamoxifen targets exist in breast cancer cells including ER-negative breast cancer cells. Previous studies also showed that tamoxifen displays chemopreventative activity against ER-negative breast cancer and other cancers [23-25]. Specifically, pharmacological concentrations of tamoxifen induce apoptosis of ER-negative breast cancer cells by modulating the activities of PI3K/AKT and ERK protein kinases [26], and tamoxifen also suppresses both tumor growth and metastasis derived from an ER-negative basal-like breast cancer cell line in Balb/c mice [27]. On the other hand, tamoxifen exhibits certain detrimental effects on cancer therapy. For example, tamoxifen inhibits the immune response by reducing the expression of IL-18, the numbers of CD4+ T cells, the activity of natural killer cells, the function of monocytes, the development of antibodies and the differentiation and activation of dendritic cells and by increasing the expression of TGF-β1 [28-34]. Taken together, these studies suggest that tamoxifen acts not only by binding to ER to inhibit ER-positive breast cancers but also by affecting other yet unknown molecular targets, so called “off targets”, to either facilitate or deduct from overall therapeutic outcomes of tamoxifen. Therefore, identification and characterization of all direct molecular targets of tamoxifen and their regulated pathways may help to maximize its therapeutic efficacy for ER-positive breast cancers and extend its applications to ER-negative and other unrelated cancers.
Our results show that tamoxifen-induced Twist1 degradation happened after cells were treated for a relatively long period of time, suggesting an indirect molecular mechanism responsible for tamoxifen-accelerated degradation of Twist1. We have previously shown that MAPK-mediated phosphorylation of Twist1 at serine 68 stabilizes Twist1 protein to enhance its cellular function [10]. Thus, we examined whether tamoxifen could inhibit the activities of ERK1/2, p38 and JNK kinases and decrease the phosphorylation level of Twist1 in ER-negative mammary tumor cells with Twist1 expression. However, we found no significant changes. We also examined any possible influences of estrogen signaling on Twist1 protein degradation in these cells and found no obvious effect of estrogen signaling on Twist1 degradation. Therefore, despite the above efforts that were made, the direct target(s) of tamoxifen linked to Twist1 degradation in ER-negative breast cancer cells is still unclear.
Tamoxifen treatment inhibits lung metastasis derived from the 4T1 cell xenograft tumors in mice. A. There was no significant difference in the growth of 4T1 cell xenograft tumors in the mammary fat pads of vehicle (corn oil) or tamoxifen-treated SCID mice (n = 5 for each). B. Representative images taken from the lungs of vehicle or tamoxifen-treated SCID mice carrying 4T1 xenograft mammary tumors. *, indicate the metastasis foci on the lung surface. C. The average numbers of the metastasis foci observed in the vehicle-treated or tamoxifen-treated SCID mice (n = 5 each) carrying 4T1 xenograft mammary tumors. *, P < 0.05 by two-tailed Student's t test. D. H&E-stained lung sections prepared from vehicle- or tamoxifen-treated SCID mice carrying 4T1 xenograft mammary tumors. M, metastasis developed in the lung tissue. E. The average value of the relative lung metastasis areas is larger in the vehicle-treated versus tamoxifen-treated SCID mice (n = 5 each) with 4T1 xenograft mammary tumors. **, P < 0.01 by two tailed Student's t test.
It should be of interesting to compare the tamoxifen doses used to induce Twist1 degradation and inhibit cell invasion and metastasis in ER-negative breast cancer cells and that used to inhibit ER-positive breast cancer cells in vitro and in vivo. We used 10 μM tamoxifen to induce Twist1 degradation in ER-negative breast cancer cells in this study, which was about 10 times higher than the 4-OH-tamoxifen concentration used to inhibit the growth of ER-positive breast cancer cells such as MCF-7 cells in culture. In mice bearing xenograft tumors derived from ER-negative breast cancer cells, administration of 5 mg/kg tamoxifen every other day (2.5 mg/kg/day) significantly reduced their metastasis. This dosage is similar to that used to inhibit the growth of xenograft tumors derived from the MCF-7 ER-positive breast cancer cells. For example, it has been shown that tamoxifen doses at 5, 25, 50 or 100 μg/mouse/day, which are equal to 0.25, 1.25, 2.5 or 5 mg/kg/day for a 20 g mouse, could effectively inhibit the growth of MCF-7 cell-derived xenograft tumors in mice [35]. Furthermore, it has previously estimated that the drug dose used in mice is about 12.3 fold higher than the drug dose used in human patients according to their body surface areas [36]. Accordingly, a tamoxifen dose of 2.5 mg/kg/day for a 20 g mouse can be translated to a dose of 0.20 mg/kg/day for a 60 kg human being. Currently, ER-positive breast cancer patients are treated with 20 mg of tamoxifen (P.O. per day), which is equal to a dose of 0.333 mg/kg/day for a 60 kg patient. Therefore, the relative tomoxifen dose used to treat our mice with ER-negative xenograft tumors is very close to what used to treat ER-positive breast cancer patients.
In summary, we have identified tamoxifen as a small molecular inhibitor that accelerates Twist1 degradation and suppresses Twist1-mediated invasion and metastasis of ER-negative breast cancer cells. These findings not only provide a molecular explanation for the tamoxifen-mediated inhibition of certain ER-negative cancer metastasis but also suggest important implications for the use of tamoxifen in the treatment of ER-negative breast cancers with high levels of Twist1 expression.
Acknowledgements
We thank Dr. Jing Yang for making 168FARN cells available for this study. G.M. was supported by a pre-doctoral scholarship from the China Scholarship Council. This study was partially supported by NIH grants CA112403 and DK058242, the Cancer Prevention and Research Institute of Texas grant RP120730-P5, and subproject funding from the Susan G. Komen for the Cure Foundation Promise Grant PG12221410 to J. Xu.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Thisse B, el Messal M, Perrin-Schmitt F. The twist gene: isolation of a Drosophila zygotic gene necessary for the establishment of dorsoventral pattern. Nucleic Acids Res. 1987;15:3439-53
2. Qin Q, Xu Y, He T. et al. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012;22:90-106
3. Chen ZF, Behringer RR. twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes Dev. 1995;9:686-99
4. Howard TD, Paznekas WA, Green ED. et al. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat Genet. 1997;15:36-41
5. Krebs I, Weis I, Hudler M. et al. Translocation breakpoint maps 5 kb 3' from TWIST in a patient affected with Saethre-Chotzen syndrome. Hum Mol Genet. 1997;6:1079-86
6. Xu Y, Liao L, Zhou N. et al. Inducible knockout of Twist1 in young and adult mice prolongs hair growth cycle and has mild effects on general health, supporting Twist1 as a preferential cancer target. Am J Pathol. 2013;183:1281-92
7. Yang J, Mani SA, Donaher JL. et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927-39
8. Fu J, Qin L, He T. et al. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res. 2011;21:275-89
9. Qin L, Liu Z, Chen H. et al. The steroid receptor coactivator-1 regulates twist expression and promotes breast cancer metastasis. Cancer Res. 2009;69:3819-27
10. Hong J, Zhou J, Fu J. et al. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011;71:3980-90
11. Fu J, Zhang L, He T. et al. TWIST represses estrogen receptor-alpha expression by recruiting the NuRD protein complex in breast cancer cells. Int J Biol Sci. 2012;8:522-32
12. Yang MH, Hsu DS, Wang HW. et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat Cell Biol. 2010;12:982-92
13. Cheng GZ, Zhang W, Wang LH. Regulation of cancer cell survival, migration, and invasion by Twist: AKT2 comes to interplay. Cancer Res. 2008;68:957-60
14. Kalra J, Sutherland BW, Stratford AL. et al. Suppression of Her2/neu expression through ILK inhibition is regulated by a pathway involving TWIST and YB-1. Oncogene. 2010;29:6343-56
15. Shi J, Wang Y, Zeng L. et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell. 2014;25:210-25
16. Mani SA, Guo W, Liao MJ. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704-15
17. Pham CG, Bubici C, Zazzeroni F. et al. Upregulation of Twist-1 by NF-kappaB blocks cytotoxicity induced by chemotherapeutic drugs. Mol Cell Biol. 2007;27:3920-35
18. Chen HF, Huang CH, Liu CJ. et al. Twist1 induces endothelial differentiation of tumour cells through the Jagged1-KLF4 axis. Nat Commun. 2014;5:4697
19. Wang Y, Lonard DM, Yu Y. et al. Small molecule inhibition of the steroid receptor coactivators, SRC-3 and SRC-1. Mol Endocrinol. 2011;25:2041-53
20. Wang S, Yuan Y, Liao L. et al. Disruption of the SRC-1 gene in mice suppresses breast cancer metastasis without affecting primary tumor formation. Proc Natl Acad Sci U S A. 2009;106:151-6
21. Kuang SQ, Liao L, Zhang H. et al. AIB1/SRC-3 deficiency affects insulin-like growth factor I signaling pathway and suppresses v-Ha-ras-induced breast cancer initiation and progression in mice. Cancer Res. 2004;64:1875-85
22. Osborne CK. Tamoxifen in the treatment of breast cancer. N Engl J Med. 1998;339:1609-18
23. Gelmann EP. Tamoxifen induction of apoptosis in estrogen receptor-negative cancers: new tricks for an old dog?. J Natl Cancer Inst. 1996;88:224-6
24. Perry RR, Kang Y, Greaves B. Effects of tamoxifen on growth and apoptosis of estrogen-dependent and -independent human breast cancer cells. Ann Surg Oncol. 1995;2:238-45
25. Ferlini C, Scambia G, Marone M. et al. Tamoxifen induces oxidative stress and apoptosis in oestrogen receptor-negative human cancer cell lines. Br J Cancer. 1999;79:257-63
26. Lam L, Hu X, Aktary Z. et al. Tamoxifen and ICI 182,780 increase Bcl-2 levels and inhibit growth of breast carcinoma cells by modulating PI3K/AKT, ERK and IGF-1R pathways independent of ERalpha. Breast Cancer Res Treat. 2009;118:605-21
27. Sun X, Jiang R, Przepiorski A. et al. "Iron-saturated" bovine lactoferrin improves the chemotherapeutic effects of tamoxifen in the treatment of basal-like breast cancer in mice. BMC Cancer. 2012;12:591
28. Coskun U, Gunel N, Sancak B. et al. Effect of tamoxifen on serum IL-18, vascular endothelial growth factor and nitric oxide activities in breast carcinoma patients. Clin Exp Immunol. 2004;137:546-51
29. Robinson E, Rubin D, Mekori T. et al. In vivo modulation of natural killer cell activity by tamoxifen in patients with bilateral primary breast cancer. Cancer Immunol Immunother. 1993;37:209-12
30. Janis K, Hoeltke J, Nazareth M. et al. Estrogen decreases expression of chemokine receptors, and suppresses chemokine bioactivity in murine monocytes. Am J Reprod Immunol. 2004;51:22-31
31. Nagy E, Berczi I. Immunomodulation by tamoxifen and pergolide. Immunopharmacology. 1986;12:145-53
32. Nalbandian G, Paharkova-Vatchkova V, Mao A. et al. The selective estrogen receptor modulators, tamoxifen and raloxifene, impair dendritic cell differentiation and activation. J Immunol. 2005;175:2666-75
33. Casale TB, Bernstein IL, Busse WW. et al. Use of an anti-IgE humanized monoclonal antibody in ragweed-induced allergic rhinitis. J Allergy Clin Immunol. 1997;100:110-21
34. Thompson AM, Kerr DJ, Steel CM. Transforming growth factor beta 1 is implicated in the failure of tamoxifen therapy in human breast cancer. Br J Cancer. 1991;63:609-14
35. Osborne CK, Hobbs K, Clark GM. Effect of estrogens and antiestrogens on growth of human breast cancer cells in athymic nude mice. Cancer Res. 1985;45:584-90
36. Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659-61
Author contact
![]() Corresponding author: Jianming Xu, PhD, Department of Molecular and Cellular Biology, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030. Phone: 713-798-6199; E-mail: jxuedu
Corresponding author: Jianming Xu, PhD, Department of Molecular and Cellular Biology, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030. Phone: 713-798-6199; E-mail: jxuedu
Received 2014-11-17
Accepted 2015-1-9
Published 2015-4-11