ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2016; 12(7):776-785. doi:10.7150/ijbs.15248 This issue Cite
Research Paper
Vitamin K2 Ameliorates Damage of Blood Vessels by Glucocorticoid: a Potential Mechanism for Its Protective Effects in Glucocorticoid-induced Osteonecrosis of the Femoral Head in a Rat Model
Yuelei Zhang, Junhui Yin, Hao Ding, Changqing Zhang ![]() , You-Shui Gao
, You-Shui Gao ![]()
Department of Orthopedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai, China.
Abstract

Glucocorticoid has been reported to decrease blood vessel number and harm the blood supply in the femoral head, which is recognized to be an important mechanism of glucocorticoid-induced osteonecrosis of the femoral head (ONFH). To prevent glucocorticoid-induced ONFH, medication that promotes both bone formation and angiogenesis would be ideal. Vitamin K2 has been revealed to play an important role in bone metabolism; however, few studies have focused on the effect of Vitamin K2 on new vascular formation. Thus, this study aimed to investigate whether Vitamin K2 promoted new blood vessel formation in the presence of glucocorticoids, both in vitro and in vivo. The effect of Vitamin K2 on viability, migration, in vitro tube formation, and VEGF, vWF, CD31, KDR, Flt and PDGFB in EAhy926 incubated with or without dexamethasone were elucidated. VEGF, TGF-β and BMP-2, angiogenesis-related proteins secreted by osteoblasts, were also detected in the osteoblast-like cell line of MG63. In addition, blood vessels of the femoral head in rats administered with or without methylprednisolone and Vitamin K2 were evaluated using angiography and CD31 staining. In vitro studies showed that Vitamin K2 significantly protected endothelial cells from dexamethasone-induced apoptosis, promoted endothelial cell migration and in vitro tube formation. Angiogenesis-related proteins both in EAhy926 and MG63 were also upregulated by Vitamin K2 when cotreated with dexamethasone. In vivo studies showed enhanced blood vessel volume and CD31-positive staining cells in rats cotreated with VK2 and methylprednisolone compared to rats treated with methylprednisolone only. Collectively, Vitamin K2 has the ability to promote angiogenesis in vitro and to ameliorate vessels of the femoral head in glucocorticoid-treated rats in vivo, indicating that Vitamin K2 is a promising drug that may be used to prevent steroid-induced ONFH.
Keywords: vitamin K2, Eahy926, osteoblast, angiogenesis, osteonecrosis of femoral heads.
Introduction
Glucocorticoid (GC) has been attributed to the most common cause of non-traumatic osteonecrosis of the femoral head (ONFH), while decreased blood vessel number and a reduced blood supply have been considered to be an important mechanism for GC-induced ONFH[1, 2]. Recent studies have revealed that glucocorticoids could directly harm endothelial cells, resulting in capillary rarefaction, disturbance of the coagulation-fibrinolysis system and thrombi formation in the femoral head, which consequently decreased blood supply of the trabecular bone in a severe manner[1, 3]. In addition, GCs decrease the expression of vascular endothelial growth factor (VEGF) protein, which is one of the most important angiogenic factors acting directly on endothelial cells to induce angiogenesis[4]. GCs have also been demonstrated to suppress collagen synthesis of myofibroblastic cells and destroy the repair process after necrosis[5, 6]. Furthermore, GC-induced osteocyte apoptosis disrupts the mechanosensory function of the osteocyte-lacunar-canalicular system and affects the perfusion of newly born vessels because increased osteocyte apoptosis is associated with decreases in VEGF and skeletal angiogenesis[2,7]. Moreover, GC decreased skeletal angiogenesis in vivo. MicroCT analysis showed that GC administration significantly decreased the blood vessel volume and blood vessel surface area of the vertebrae and femora in mice[7]. Given that blood vessels are critical in the biology of bone as nutrient providers and play an important role in bone remodeling[8], to prevent glucocorticoid-induced ONFH, the protection of vascularity and the formation of new blood vessels are of great importance.
Several studies have demonstrated that angiogenesis and osteogenesis are tightly coupled[9, 10]. Endothelial cells could enhance osteogenic differentiation of human mesenchymal stem cells (hMSCs) and osteoblasts via direct cell-cell contact[11,12]. Conversely, growth of the vascular network is also regulated by signals provided by bone cells, among which VEGF is well-understood[13]. VEGF modulates blood vessel development via the regulation of endothelial cell proliferation, migration and differentiation during osteogenesis, while VEGF secreted by osteoblasts and MSC could affect endothelial cell activities directly by binding to VEGF receptors, which predominantly exists on endothelial cells[14]. In addition, BMP-2, which is involved in osteogenic differentiation and bone formation, is also known to stimulate neovascularization in bone[15]. Cocultures of hMSC and human endothelial cells have demonstrated that endothelium-derived BMP-2 significantly enhanced osteogenic differentiation of BMSC in vitro[16], indicating an interaction of BMP-2 and VEGF and the regulation of osteogenesis and angiogenesis[16, 17].
Vitamin K2 (VK2) is essential for γ-carboxylation of Glu proteins and plays an important role in bone metabolism. Several studies have shown that VK2 promoted osteogenic differentiation of bone marrow stem cells and osteoblast-osteocyte transition in vitro[18, 19]. VK2 has also been reported to prevent bone loss and promote bone healing in vivo[20, 21]. Furthermore, several studies have revealed the effect of VK2 on endothelial cells. Gas6 is a vitamin K-dependent protein and carboxylated Gas6 can protect endothelial cells from serum starvation-induced apoptosis[22, 23]. Except acting as a cofactor, VK2 has been reported to exhibit biological functions by regulating mitochondrial function via its ability to transfer electrons in the electron transport chain[24]. Hegarty et al. discovered that VK2 was able to rescue endothelial cell apoptosis in ubiad1 mutant zebrafish, which exhibited cardiac edema, as well as vascular degeneration, due to defects in endothelial cell survival[25].
Given the coupling of osteogenesis and angiogenesis, as well as the biological functions of VK2, the purpose of this study was to determine whether VK2 protects the blood supply and promotes vascularity in femoral heads with GC treatment. Because endothelial cells mediate neo-angiogenesis in bone[13] and induce a bidirectional effect with osteoblasts, we observed the effect of VK2 on endothelial cells and angiogenic factors in osteoblasts with or without GC treatment, and we also examined the vascularity of femoral heads in GC-treated rats with and without VK2 administration in vivo.
Material and methods
In vitro studies
Cell culture and treatment
The human endothelial cell line EAhy926 and the human osteoblast-like cell line MG63 were obtained from KeyGENE BIOTECH (Nanjing, China). The cell lines were both maintained in DMEM medium supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in a humidified atmosphere of 5% CO2. To test the effect of dexamethasone (DEX) and VK2, EAhy926 and MG63 were incubated in the presence or absence of VK2 and 10-5M DEX, respectively. The concentration of VK2 was 10-6 M, which was comparable to the serum level when patients were treated with vitamin K[26] and was used in all of the following studies. While the concentration of DEX was determined according to previous studies, in which 10-5M DEX obviously suppressed the expression of VEGF[27, 28].
Endothelial cell proliferation assay
To detect the effect of DEX and VK2 on EAhy926 proliferation, the cell counting kit -8 (CCK-8) assay was performed according to the manufacturer's instructions. After the treatment period in 96 wells, 100 μL culture medium plus with 10 μL CCK-8 was added and incubated for 2 h at 37 °C. The absorbance was then measured at 450 nm. Values were expressed with the D-value between the absorbance detected at the appropriate period and initial absorbance, which was measured after cell adhesion.
Endothelial cell viability and apoptosis assay
Cell viability was measured using the neutral red uptake assay as previously described[29]. Briefly, EAhy926 cells were plated onto 96-well plate (20000 cells/well) for adhesion, and medium was changed with FBS-free medium with or without DEX and VK2. After the treatment period, the culture medium was changed with neutral red-containing medium for an additional 2 hours, and neutral red lysis buffer was used to extract the neutral red stain and measured at 570 nm.
The Annexin V-FITC cell apoptosis detection kit (Beyotime, Shanghai, China) was used to evaluate the effect of VK2 on apoptosis according to the manufacturer's instructions. Briefly, endothelial cells were incubated with FBS-free medium in the presence or absence of VK2 and DEX for 96 hours, and the cells were collected, washed with PBS twice, and later resuspended with 200 μL Annexin V-FITC and 10 μL propidium iodide. After incubation for 20 minutes at room temperature, flow cytometry was used to evaluate the cell apoptosis rate. Early apoptotic cells were labeled green and the dead and late apoptotic cells were labeled red, while the live cells were not stained.
Endothelial cell tube formation assay
EAhy926 cells were treated with different FBS-free condition medium for 48 hours, suspended with normal FBS-free medium, seeded (2 × 104 cells per well) onto solidified Matrigel in 96-well plates and incubated at 37°C for 6 hours. Tubular networks were visualized and analyzed automatically using WimTube software (Wimasis, Munich, Germany) .
Endothelial cell migration assay
To determine the effect of VK2 on endothelial cell migration, a migration chamber of 24 transwells with an 8-μm pore size was used. EAhy926 cells were pretreated with FBS-free medium in the presence or absence of VK2 and DEX for 48 hours. Next, 5×104 cells were resuspended with 200-μL FBS-free medium and plated on the top chamber, and 500-μL FBS containing medium was added in the bottom chamber to allow migration. After incubation at 37°C for 6 hours, cells on the top surface of the membrane were scraped with a cotton swab, while cells on the bottom surface of the membrane were fixed with 4% paraformaldehyde and stained with 0.2% crystal violet dye for 30 minutes. Next, 33% ethylic acid was used to extract the violet staining and measured at 570 nm.
Western blotting analysis
After propagating to a density of more than 80%, the medium was changed into FBS-free medium and incubated for an additional 24 hours, and EAhy926 cells were treated with different condition medium for 48 hours, while MG63 were incubated in medium with or without DEX and VK2 for one week. Cells were harvested and lysed with cell lysis buffer on ice for 30 minutes. Cell homogenates were centrifuged at 12000 rpm for 15 min at 4 °C and the supernatants were collected. Next, the protein concentrations were determined using a commercial BCA kit. Thirty µg of proteins were subjected to SDS-PAGE after denaturation at 95°C for 5 min and transferred onto a 0.45-μm polyvinylidine difluoride (PVDF) membrane. After blockade with 5% non-fat dry milk for 1 hour at 37 °C, the membranes were incubated overnight at 4 °C with rabbit anti-human primary antibody for VEGF (BOSTER, 1:400 dilution) or β-actin (CST, 1:1000 dilution). The membranes were subsequently immersed in rat anti-rabbit secondary antibody (CST, 1:1000) for 1 hour at 37°C. After chemiluminescence using a commercial assay, the target bands were detected using a gel image-processing system. Densitometric analysis of the target bands was performed using Quantity ONE software (BIO-RAD) and was normalized against β-actin.
Quantitative real-time polymerase chain reaction analysis (qRT-PCR)
One µg of total RNA extracted from EAhy926 and MG63 were reversely transcribed using TransScript All-in-One First-Strand cDNA Synthesis SuperMix (TransGen Biotech, Beijing, China) according to the manufacturer's instructions. Next, qRT-PCR of VEGF, von Willebrand factor (vWF), platelet-derived growth factor beta (PDGF-B), CD31, VEGF receptor-1 (Flt), VEGF receptor-2 (KDR) TGF-β and β-actin were performed using TransStart Tip Green qPCR SuperMix (TransGen Biotech). The qRT-PCR was performed for 40 cycles with denaturation at 95 °C for 30 s, annealing at 95 °C for 5 s and extension at 60 °C for 30 s followed by a 65 °C~95 °C solubility curve, which was constructed to analyze the fluorescence signal. The relative amount of mRNA was normalized against β-actin. The forward and reverse primers of each cDNA are shown in Table 1.
In vivo studies
Animal grouping and treatment
A total of 20 Sprague-Dawley (SD) rats were divided into four groups: the control group with normal feed; the methylprednisolone (MP) group treated with 20 mg/kg MP intramuscular injection 3 days a week for three continuous weeks (9 injections in all); the MP+VK2 group in which the rats were fed with 30 mg/kg/d VK2 5 days a week in addition to a MP injection as the MP group until sampling; and the VK2 group with 30 mg/kg/d VK2 oral administration 5 days a week until sampling. The animals were sacrificed 6 weeks after the first MP injection and the bilateral femoral heads of each rat were obtained. All procedures adhered to the recommendations of the U.S. Department of Health for the care and use of laboratory animals and were approved by the Ethics Committee of Shanghai Jiao Tong University.
The quantitative real-time polymerase chain reaction primer sequences
| Gene | Forward primer | Reverse Primer |
|---|---|---|
| VEGF | GAACTTTCTGCTGTCTTG | TTCTTGTCTTGCTCTATCT |
| KDR | GAGGGGAACTGAAGACAGGC | GGCCAAGAGGCTTACCTAGC |
| Flt | TGTCATCGTTTCCAGACCCG | AAGTCACACCTTGCTTCGGA |
| vWF | TTGACGGGGAGGTGAATGTG | ATGTCTGCTTCAGGACCACG |
| CD31 | GTCCCTGATGCCGTGGAAAG | GGAGCAGGGCAGGTTCATAA |
| PDGF-B | GAGTCGGCATGAATCGCTGC | CCCATCTTCCTCTCCGGGGT |
| TGF-β | GGAAATTGAGGGCTTTCGCC | CCGGTAGTGAACCCGTTGAT |
| BMP-2 | CTGCGGTCTCCTAAAGGTCG | AGCAGCAACGCTAGAAGACA |
| β-actin | GTCGAGTCGCGTCCACC | GTCATCCATGGCGAACTGGT |
Angiography and micro-CT scanning
Six weeks after the first MP injection, the rats were anesthetized, and the abdominal aorta and vein were dissected. The proximal aorta was ligated and abdominal vein was cut off. Heparinized saline, 4% paraformaldehyde and Microfil (MV-112, Flow Tech, Inc., Carver, MA) were injected through the distal abdominal aorta, respectively, until a constant outflow was observed to exit from the abdominal vein. Next, the rats were stored in 4°C overnight, and the bilateral femoral heads were extracted. For micro-CT scanning of the vessels, the femoral heads were fixed and then decalcified with 10% EDTA solution. Micro-CT scanning was performed at a voxel of 9 microns, and 2-D images were disposed with CTVol and reconstructed 3-D images of vasculature were obtained. The total vessel volume was calculated using CTAn.
Immunohistochemical staining
After micro-CT scanning and paraffin embedding, the femoral heads were sectioned at 5-µm thickness in the coronal plane. The slices were deparaffinized, antigen retrieved, incubated with anti-CD31 primary antibody and the appropriate biotinylated secondary antibody, and sections were visualized with DAB precipitate and counterstained with hematoxylin. Photomicrographs were acquired using a LEICA DM 4000 (Leica microsystems, Germany).
Statistical methods
The data were expressed as the mean ± standard deviation (SD) with three independent experiments. Statistical analysis of multiple comparisons was performed using one-way analysis of variance (ANOVA) with Tukey's post hoc test. Significant differences were considered when P < 0.05 (*) and P < 0.01 (**).
Results
Effects of DEX and VK2 on EAhy926 proliferation
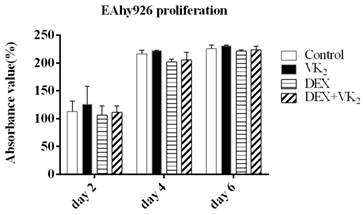
In this study, we observed that EAhy926 cells proliferated rapidly particularly within 4 days and maintained a specific cell number after 4 days in the presence of FBS, however, we did not detect significant difference between the proliferation of EAhy926 in these four groups. (Figure 1).
Effect of DEX and VK2 on EAhy926 proliferation. DEX inhibited EAhy926 proliferation (P > 0.05) and a larger absorbance value was detected when treated with DEX plus VK2 compared to treatment with DEX only (P > 0.05). VK2 alone also promoted EAhy926 proliferation to a certain extent (P > 0.05).
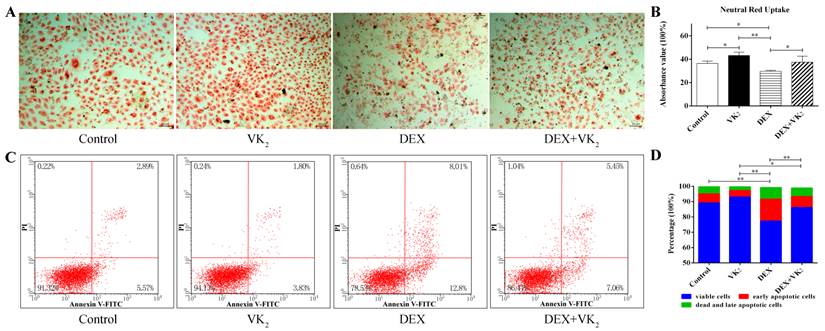
VK2 enhanced EAhy926 viability and survival
After incubation in FBS-free condition medium for 48 hours, more neutral red-stained cells were present in groups with VK2 treatment alone compared to the control group. Moreover, DEX significantly decreased the number of red-stained cells, and VK2 had an obvious protective effect on EAhy926 cells treated with DEX. We further demonstrated this phenomenon using a quantitative method. (Figure 2A)
From the flow cytometric analysis on Annexin-V/PI double-labeled cells, exposure of DEX significantly increased the number of cells with Annexin-V-positive staining in EAhy926 (21.87±1.83%). Conversely, VK2 supplementation attenuated the apoptotic effect of DEX in EAhy926 cells (12.80±0.30%). Furthermore, EAhy926 cells treated with VK2 alone resulted in less apoptotic cells compared to the control group (6.66 ± 1.21% vs. 10.62 ± 2.21%) (Figure 2B).
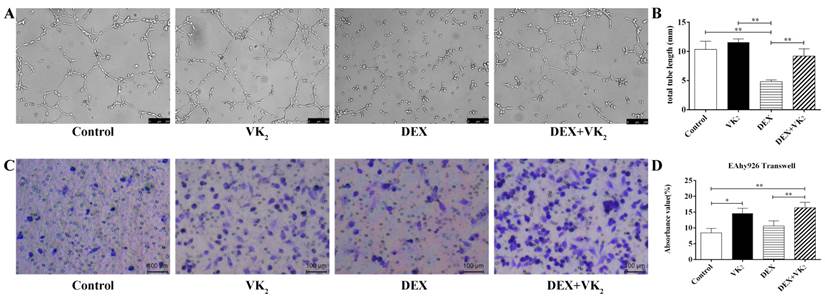
Effects of DEX and VK2 on tube formation and migration of EAhy926 in vitro
To quantify tube formation, images from triplicate samples were measured and the total tube length were calculated. DEX showed strong anti-angiogenesis activity in the angiogenesis assay model with no loop formation and a smaller tube length, which was 4.81±0.30 mm. However, more visible tubes and longer tube length (9.20±1.26 mm) were observed in the DEX+VK2 group (P < 0.01). We also observed that EAhy926 cells treated with VK2 alone had stronger angiogenesis activity than cells in the control group (11.58±0.58 mm vs. 10.36±0.40 mm, P > 0.05 ) (Figure 3A, B).
After migration for 6 hours, the Transwell assays showed that the control group had fewer violet-stained cells than the other three groups on the bottom surface of the membrane. From the qualitative analysis, we further determined that both DEX and VK2 stimulated the migration of endothelial cells with VK2 more significantly (Figure 3C, D).
Effect of DEX and VK2 on EAhy926 viability and apotosis. A. Neutral red uptake assay showed fewer neutral red-stained cells in wells treated with DEX only compared to the control group. However, more red staining cells were observed in EAhy926 cells incubated with DEX plus VK2. VK2 alone also significantly enhanced EAhy926 viability. B. Flow cytometry showed that DEX induced apoptosis in EAhy926 cells, whereas supplementation of VK2 significantly decreased the apoptotic effect significantly. (* indicated P < 0.05; ** indicated P < 0.01).
Effect of DEX and VK2 on EAhy926 tube formation and migration. A. Representative microscopic images are shown after incubation for 6 hours in matrigel with or without DEX and VK2. B. Quantification of total tube length of endothelial cells. DEX clearly inhibited endothelial cell tube formation with no loop formation and more tube shaped cells were observed in EAhy926 treated with DEX plus VK2. C. Representative images of the transwell assay at 6 hours showed a higher proportion of spreading cells (violet-stained) after DEX or VK2 incubation compared to the control group. Moreover, the migration stimulation effect of VK2 was stronger than DEX treatment. D. Absorbance value of the violet staining extracted from migrated cells in each group (* indicated P < 0.05; ** indicated P < 0.01).
VEGF expression analysis in EAhy926 and MG63. A. Western blotting analysis of total cell lysates in EAhy926, β-actin was used as the loading control. B. VEGF mRNA expression in EAhy926 treated with or without DEX and VK2, each condition was performed in triplicate (* indicated P < 0.05; ** indicated P < 0.01). C. Western blotting analysis of total cell lysates in MG63. β-actin was used as the loading control. D. VEGF mRNA expression in MG63 cells treated with or without DEX and VK2, each condition was performed in triplicate (* indicated P < 0.05; ** indicated P < 0.01).
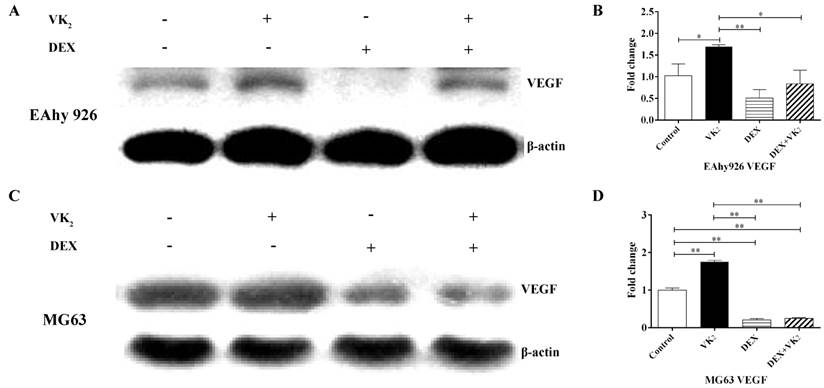
VK2 upregulated VEGF expression in both EAhy926 and MG63 cells
VEGF expression in the cell lysate of EAhy926 was measured using western-blotting analyses, and the results showed that the relative expression of VEGF in cells treated with DEX was significantly lower compared to the control group. However, compared with DEX, the VEGF content in cells treated with DEX and VK2 was clearly higher. Furthermore, upregulation of VEGF protein in VK2 was also observed compared with the control group according to the western blotting analysis (Figure 4A). In addition, VEGF mRNA expression was also detected using qRT-PCR, and lower mRNA expression was detected in DEX. Higher mRNA expression was found in cells treated with DEX and VK2, which is consistent with the results obtained from western blotting analyses. Furthermore, more mRNA expression was detected in cells when incubated with VK2 alone (Figure 4B).
Because VEGF secreted by osteoblasts plays an important role in the regulation of angiogenesis, we further investigated the expression of VEGF in osteoblasts. The results showed that DEX obviously downregulated VEGF expression in MG63 and supplementation of VK2 weakened the downregulation on both protein and mRNA level. (Figure 4C, D)
VK2 promoted angiogenesis-related mRNA expression in EAhy926 cells
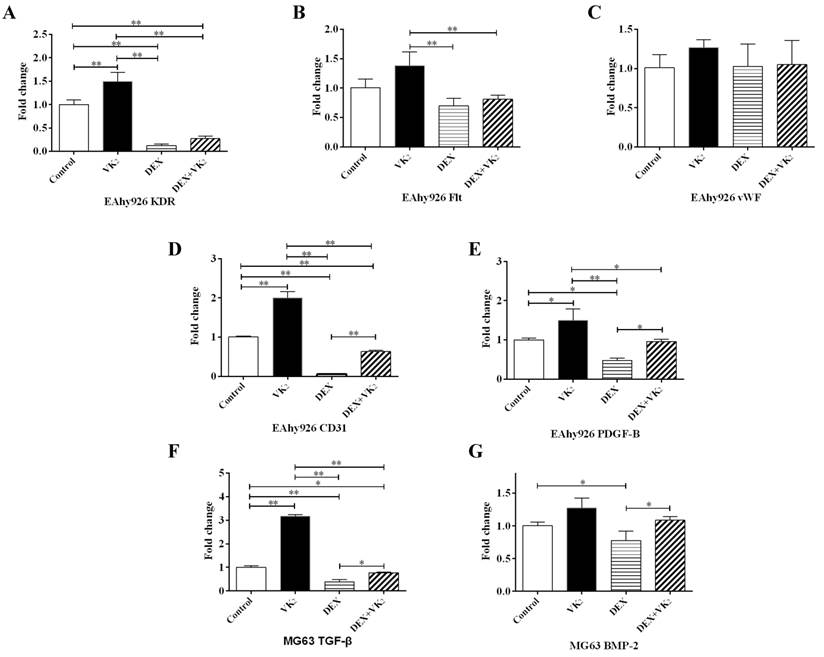
VEGF is a key regulator of angiogenesis. We further investigated the gene expression of two main VEGF receptors (KDR and Flt-1) in EAhy926 cells. The expression of KDR was obviously lower in cells treated with DEX and when cotreated with VK2. VK2 alone significantly promoted KDR expression of endothelial cells incubated with medium in the absence of DEX. Similar results were observed with Flt-1 expression, although it was not robust. (Figure 5A, B).
Furthermore, vWF is specifically produced by endothelial cells. In this study, we did not detect a significant effect of DEX and VK2 on vWF expression. CD31 is an endothelial marker, which is expressed mainly by endothelial cells, PDGF-B is secreted by endothelial cells to stimulate maturation of the vessel wall. qRT-PCR analysis showed similar results between CD31 and PDGF-B. DEX suppressed the expression of both CD31 and PDGF-B, while VK2 upregulated their expression significantly when either administered alone or together with DEX. (Figure 5C-E)
VK2 promoted angiogenesis-related mRNA expression in MG63 cells
To identify whether DEX and VK2 affected osteoblast-secreted proteins, which promote angiogenesis, TGF-β and BMP-2 were detected using qRT-PCR. DEX significantly suppressed TGF-β and BMP-2 expression in osteoblasts, while obvious upregulation of TGF-β and BMP-2 in MG63 was observed when treated with DEX plus VK2. In addition, VK2 clearly promoted TGF-β mRNA expression in MG63 compared with the control (Figure 5F, G).
VK2 ameliorated blood vessels of the femoral head
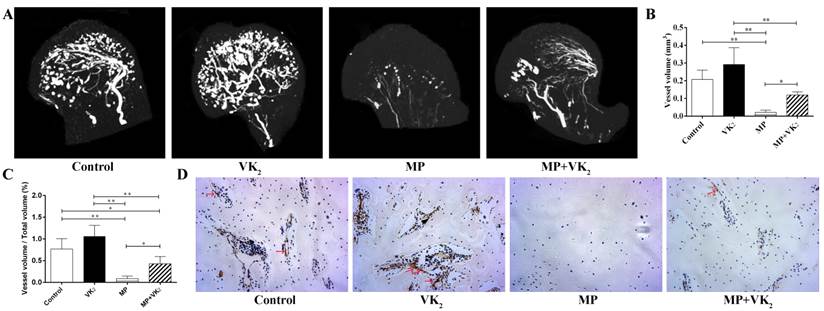
After treatment with the respective protocol for six weeks, the femoral heads of all rats were obtained. Micro-CT analysis showed that MP significantly damaged the blood supply of femoral heads and co-administration with VK2 had a notable protective effect on blood vessels in femoral heads, which were demonstrated on both reconstructed images and total vessel volume. Furthermore, rats with VK2 administration alone tended to have more blood vessels than rats without any intervention (Figure 6A-C).
CD31 is a marker of endothelial cells and is often used to reflect angiogenesis. Immunohistochemical staining showed similar results as shown using angiography. Femoral heads in the MP group also showed fewer vessel-like structures, and conversely, many vacuole cells occupied a large area of the subchondral region. Interestingly, around those residual vessels, few CD-31-positive cells were observed. However, more vessels with brown-stained cells in the periphery were observed in the femoral heads of rats in the MP+VK2 group compared to the MP group (Figure 6D).
Effect of DEX and VK2 on angiogenic factors in EAhy926 and MG63. (A-E) Quantitative RT-PCR analysis of KDR, Flt, vWF, CD31 and PDGF-B in EAhy926. (F,G) TGF-β and BMP-2 mRNA expression in MG63. (Each condition was performed in triplicate, * indicated P < 0.05, ** indicated P < 0.01).
Analysis of blood vessels in the femoral heads of rats treated with MP or VK2. A. Representative 3-D microangiographic images of blood vessels in the femoral heads of rats in each group, white lines represent blood vessels. (B,C). Quantification of total blood vessel volume and the rate of blood vessel volume in femoral heads (* indicated P < 0.05; ** indicated P < 0.01). D. Representative images of CD31 staining in the femoral heads of each group; the arrow indicates a CD31-positive endothelial cell.
Discussion
Robust inhibition of GC on angiogenesis has been illustrated both in vitro and in vivo, as well as decreased bone formation and increased bone resorption. Furthermore, GC administration has been attributed to the most common cause of nontraumatic ONFH and secondary osteoporosis[1-6]. Thus, medication that promotes both bone formation and angiogenesis would be optimal for the prevention of GC-induced bone disease. Vitamin K is essential for γ-carboxylation of Glu proteins. This vitamin also has an important role in bone metabolism. Several studies have focused on the effect of vitamin K-dependent proteins on angiogenesis[18,19]. A study by Burstyn-Cohen showed that Protein S, which is a vitamin K-dependent protein, acts as a positive regulator in vascular development and homeostasis[30]; Hasanbasic et al. also reported that only carboxylated gas6 protects endothelial cells from serum starvation-induced apoptosis[23]. Furthermore, Hegar et al. concluded that VK2 synthesis is required for vascular endothelial cell survival and development[25]. In this study, we found that VK2 protected endothelial cells from GC-induced apoptosis. Moreover, VK2 promoted cell migration, in vitro angiogenesis and the expression of angiogenesis-related proteins in endothelial cells. More importantly, the positive effect of VK2 on angiogenesis has also been observed in GC-treated rats. Taken together, these findings indicate that VK2 is a promising method for the prevention of GC-induced bone loss.
In our in vitro study, we further demonstrated that GC can directly injure endothelial cells, inducing decreased cell viability and cell apoptosis, which has been reported in previous studies[3, 31]. In addition, GC inhibited in vitro angiogenesis in endothelial cells; however, we observed that GC promoted endothelial migration, which is inconsistent with findings obtained in previous studies[32, 33]. A study by Zarrinpashneh showed that lack of serum and GC inducible kinase 1 (SGK1) resulted in defective endothelial cell migration[34], while SGK1 is transcriptionally induced by GCs and serum[35], which may explain the differences between our results and those of other previous studies. Furthermore, we observed the protective effect of VK2 on endothelial cell apoptosis and tube formation impaired by DEX, which confirmed the protective effect of VK2 on angiogenesis.
VEGF has been demonstrated as a key inducer in angiogenesis by regulating endothelial cell proliferation, migration and vascularization[14, 36]. Several studies have also shown that VEGF couples angiogenesis and osteogenesis via its effect on endothelial cells and modulation of osteoblasts, which both express VEGF receptors[37]. On the one hand, VEGF production by osteoblasts stimulate differentiation and vessel formation of endothelial cells; on the other hand, VEGF is also involved in various aspects of bone development, including osteoblast differentiation and osteoclast recruitment[38-40]. In the present study, DEX significantly decreased VEGF expression, which was consistent with findings obtained in a previous study[4]. We observed that VK2 promoted VEGF expression in both endothelial cells and osteoblasts. Furthermore, VEGF receptors (KDR and Flt-1) were also upregulated in endothelial cells, and this phenomenon indicated that VEGF secretion may be an important facet of VK2 in promoting angiogenesis and osteogenesis as shown in previous studies[41].
We also investigated the expression of several factors involved in angiogenesis. CD31, also known as platelet endothelial cell adhesion molecule-1, is a marker for vascular endothelium and is mainly expressed in endothelial cells[42]. vWF, a marker that stains newly formed vessels, is specifically produced by endothelial cells and mediates platelet adhesion to the vascular wall[43]. In our study, mRNA expression of both CD31 and vWF was substantially higher in the DEX+VK2 group compared to the DEX group, and VK2 alone also enhanced their expression. PDGF-B, which is secreted by endothelial cells, was also downregulated by DEX and upregulated by VK2. PDGF-B functions to recruit perivascular cells to stimulate maturation of the vessel wall and stabilizes sprouting microvessels[44, 45]. In addition, PDGF-B was demonstrated to make a significant contribution to bone repair[44], indicating that VK2 contributes to both vessel maturation and bone formation. TGF-β is also an important growth factor involved in vessel structure maturation and osteogenic cell proliferation, and indirect cocultures of endothelial cell and osteoblasts significantly contribute to TGF-β release from osteoblasts[46]. Our PCR results showed notable TGF-β upregulation by VK2 both in endothelial cells and osteoblasts, which strengthened the finding that VK2 promotes both angiogenesis and osteogenesis.
Successful in vivo osteogenesis and bone defect healing is dependent on a sufficient blood supply, resulting from neo-angiogenesis. Previous studies have reported that GC significantly decreased skeletal angiogenesis and vascularity combined with decreased cancellous bone volume[7], which has been proposed as one mechanism of GC-induced ONFH[1]. Our study demonstrated not only damage of GC on the femoral head vascularity but also angiogenesis stimulation of VK2 in vivo. Furthermore, using immunohistochemical techniques, we observed more vessels with endothelial marker expression in rats with GC and VK2 administration, which further affirmed our hypothesis.
VK2 has been used for osteoporosis in Asia countries for years, studies showed that VK2 could increase bone mineral density and prevent the incidence of fracture in patients with osteoporosis[47, 48]. In vitro studies further confirmed the important role of VK2 in bone metabolism including the γ-carboxylation of osteocalcin, promotion of osteogenesis and inhibition of osteoclastogenesis[18, 49]. Considering the mechanism of GC-induced ONFH[1] and the advantageous effect of VK2 on angiogenesis, VK2 might yield beneficial effects on GC-induced ONFH.
Conclusions
In this study, we demonstrated that VK2 reduced the negative effects of GC on endothelial cells, including viability, tube formation and angiogenesis related proteins expression. We also observed significant improvement of blood vessels by VK2 administration in the femoral head of GC-treated rats. In conclusion, VK2 has the potential to ameliorate damage of blood vessels in a GC-treated rat model, which indicates its clinical significance for the prevention of GC-induced ONFH.
Acknowledgements
The current research was funded by the National Natural Science Foundation of China (No. 81301572) and the SMC-Chen Xing Plan for Splendid Young Teachers of Shanghai Jiao Tong University. We would like to thank Shanghai Institute of Microsurgery for Extremities for providing experimental equipment and all staff for their instructions.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kerachian MA, Seguin C, Harvey EJ. Glucocorticoids in osteonecrosis of the femoral head: a new understanding of the mechanisms of action. J Steroid Biochem Mol Biol. 2009;114:121-8
2. Weinstein RS. Glucocorticoid-induced osteonecrosis. Endocrine. 2012;41:183-90
3. Kerachian MA, Harvey EJ, Cournoyer D, Chow TY, Seguin C. Avascular necrosis of the femoral head: vascular hypotheses. Endothelium. 2006;13:237-44
4. Harada S, Rodan SB, Rodan GA. Expression and regulation of vascular endothelial growth factor in osteoblasts. Clin Orthop Relat Res. 1995;313:76-80
5. Li X, Jin L, Cui Q, Wang GJ, Balian G. Steroid effects on osteogenesis through mesenchymal cell gene expression. Osteoporos Int. 2005;16:101-8
6. Harada I. The effects of glucocorticoids on angiogenesis in vitro. Nihon Seikeigeka Gakkai zasshi. 1992;66:763-70
7. Weinstein RS, Wan C, Liu Q, Wang Y, Almeida M, O'Brien CA. et al. Endogenous glucocorticoids decrease skeletal angiogenesis, vascularity, hydration, and strength in aged mice. Aging cell. 2010;9:147-61
8. Wan C, Shao J, Gilbert SR, Riddle RC, Long F, Johnson RS. et al. Role of HIF-1alpha in skeletal development. Ann N Y Acad Sci. 2010;1192:322-6
9. Coolbaugh CC. Effects of reduced blood supply on bone. Am J Physiol. 1952;169:26-33
10. Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 1999;5:623-8
11. Tsai TL, Wang B, Squire MW, Guo LW, Li WJ. Endothelial cells direct human mesenchymal stem cells for osteo- and chondro-lineage differentiation through endothelin-1 and AKT signaling. Stem Cell Res Ther. 2015;6:88
12. Stahl A, Wenger A, Weber H, Stark GB, Augustin HG, Finkenzeller G. Bi-directional cell contact-dependent regulation of gene expression between endothelial cells and osteoblasts in a three-dimensional spheroidal coculture model. Biochem Biophys Res Commun. 2004;322:684-92
13. Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature. 2014;507:323-8
14. Eshkar-Oren I, Viukov SV, Salameh S, Krief S, Oh CD, Akiyama H. et al. The forming limb skeleton serves as a signaling center for limb vasculature patterning via regulation of Vegf. Development. 2009;136:1263-72
15. Chen D, Zhao M, Mundy GR. Bone morphogenetic proteins. Growth factors. 2004;22:233-41
16. Li H, Xue K, Kong N, Liu K, Chang J. Silicate bioceramics enhanced vascularization and osteogenesis through stimulating interactions between endothelia cells and bone marrow stromal cells. Biomaterials. 2014;35:3803-18
17. Deckers MM, van Bezooijen RL, van der Horst G, Hoogendam J, van Der Bent C, Papapoulos SE. et al. Bone morphogenetic proteins stimulate angiogenesis through osteoblast-derived vascular endothelial growth factor A. Endocrinology. 2002;143:1545-53
18. Atkins GJ, Welldon KJ, Wijenayaka AR, Bonewald LF, Findlay DM. Vitamin K promotes mineralization, osteoblast-to-osteocyte transition, and an anticatabolic phenotype by {gamma}-carboxylation-dependent and -independent mechanisms. Am J Physiol Cell Physiol. 2009;297:C1358-67
19. Azuma K, Ouchi Y, Inoue S. Vitamin K: novel molecular mechanisms of action and its roles in osteoporosis. Geriatr Gerontol Int. 2014;14:1-7
20. Iwamoto J, Seki A, Sato Y, Matsumoto H, Tadeda T, Yeh JK. Vitamin K-2 Promotes Bone Healing in a Rat Femoral Osteotomy Model with or without Glucocorticoid Treatment. Calcif Tissue Int. 2010;86:234-41
21. Sasaki N, Kusano E, Takahashi H, Ando Y, Yano K, Tsuda E. et al. Vitamin K2 inhibits glucocorticoid-induced bone loss partly by preventing the reduction of osteoprotegerin (OPG). J Bone Miner Metab. 2005;23:41-7
22. Shea MK, Holden RM. Vitamin K status and vascular calcification: evidence from observational and clinical studies. Adv Nutr. 2012;3:158-65
23. Hasanbasic I, Rajotte I, Blostein M. The role of gamma-carboxylation in the anti-apoptotic function of gas6. J Thromb Haemost. 2005;3:2790-7
24. Vos M, Esposito G, Edirisinghe JN, Vilain S, Haddad DM, Slabbaert JR. et al. Vitamin K2 is a mitochondrial electron carrier that rescues pink1 deficiency. Science. 2012;336:1306-10
25. Hegarty JM, Yang H, Chi NC. UBIAD1-mediated vitamin K2 synthesis is required for vascular endothelial cell survival and development. Development. 2013;140:1713-9
26. Yoshiji H, Kuriyama S, Noguchi R, Yoshii J, Ikenaka Y, Yanase K. et al. Combination of vitamin K2 and the angiotensin-converting enzyme inhibitor, perindopril, attenuates the liver enzyme-altered preneoplastic lesions in rats via angiogenesis suppression. J Hepatol. 2005;42:687-93
27. Xia Q, Yang S, Zhang SQ, Chen B, Wang DB, Zhu QX. et al. The effect of mizolastine on expression of vascular endothelial cell growth factor, tumour necrosis factor-alpha and keratinocyte-derived chemokine in murine mast cells, compared with dexamethasone and loratadine. Clin Exp Dermatol. 2005;30:165-70
28. Park SK, Kim HI, Yang YI. Roles of vascular endothelial growth factor, Angiopoietin 1, and Angiopoietin 2 in nasal polyp. Laryngoscope. 2009;119:409-13
29. Abud AP, Zych J, Reus TL, Kuligovski C, de Moraes E, Dallagiovanna B. et al. The use of human adipose-derived stem cells based cytotoxicity assay for acute toxicity test. Regul Toxicol Pharmacol. 2015;73:992-8
30. Burstyn-Cohen T, Heeb MJ, Lemke G. Lack of protein S in mice causes embryonic lethal coagulopathy and vascular dysgenesis. J Clin Invest. 2009;119:2942-53
31. Vogt CJ, Schmid-Schonbein GW. Microvascular endothelial cell death and rarefaction in the glucocorticoid-induced hypertensive rat. Microcirculation. 2001;8:129-39
32. Jang YH, Shin HS, Sun Choi H, Ryu ES, Jin Kim M, Ki Min S. et al. Effects of dexamethasone on the TGF-beta1-induced epithelial-to-mesenchymal transition in human peritoneal mesothelial cells. Lab Invest. 2013;93:194-206
33. Shikatani EA, Trifonova A, Mandel ER, Liu ST, Roudier E, Krylova A. et al. Inhibition of proliferation, migration and proteolysis contribute to corticosterone-mediated inhibition of angiogenesis. PloS one. 2012;7:e46625
34. Zarrinpashneh E, Poggioli T, Sarathchandra P, Lexow J, Monassier L, Terracciano C. et al. Ablation of SGK1 impairs endothelial cell migration and tube formation leading to decreased neo-angiogenesis following myocardial infarction. PloS one. 2013;8:e80268
35. Webster MK, Goya L, Ge Y, Maiyar AC, Firestone GL. Characterization of sgk, a novel member of the serine/threonine protein kinase gene family which is transcriptionally induced by glucocorticoids and serum. Mol Cell Biol. 1993;13:2031-40
36. Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M. et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435-9
37. Maes C, Goossens S, Bartunkova S, Drogat B, Coenegrachts L, Stockmans I. et al. Increased skeletal VEGF enhances beta-catenin activity and results in excessively ossified bones. EMBO J. 2010;29:424-41
38. Yang YQ, Tan YY, Wong R, Wenden A, Zhang LK, Rabie AB. The role of vascular endothelial growth factor in ossification. Int J Oral Sci. 2012;4:64-8
39. Dai J, Rabie AB. VEGF: an essential mediator of both angiogenesis and endochondral ossification. J Dent Res. 2007;86:937-50
40. Jabalee J, Franz-Odendaal TA. Vascular endothelial growth factor signaling affects both angiogenesis and osteogenesis during the development of scleral ossicles. Dev Biol. 2015;406:52-62
41. Hamidi MS, Gajic-Veljanoski O, Cheung AM. Vitamin K and bone health. J Clin Densitom. 2013;16:409-13
42. Muraoka N, Shum L, Fukumoto S, Nomura T, Ohishi M, Nonaka K. Transforming growth factor-beta3 promotes mesenchymal cell proliferation and angiogenesis mediated by the enhancement of cyclin D1, Flk-1, and CD31 gene expression during CL/Fr mouse lip fusion. Birth Defects Res A Clin Mol Teratol. 2005;73:956-65
43. Uras N, Oguz SS, Zergeroglu S, Akdag A, Polat B, Dizdar EA. et al. CD31 and Factor VIII in angiogenesis of normal and pre-eclamptic human placentas. J Obstet Gynaecol. 2012;32:533-6
44. Hannink M, Donoghue DJ. Structure and function of platelet-derived growth factor (PDGF) and related proteins. Biochim Biophys Acta. 1989;989:1-10
45. Mittermayr R, Slezak P, Haffner N, Smolen D, Hartinger J, Hofmann A. et al. Controlled release of fibrin matrix-conjugated platelet derived growth factor improves ischemic tissue regeneration by functional angiogenesis. Acta Biomater. 2016;29:11-20
46. Herzog DP, Dohle E, Bischoff I, Kirkpatrick CJ. Cell communication in a coculture system consisting of outgrowth endothelial cells and primary osteoblasts. Biomed Res Int. 2014;2014:320123
47. Fang Y, Hu C, Tao X, Wan Y, Tao F. Effect of vitamin K on bone mineral density: a meta-analysis of randomized controlled trials. J Bone Miner Metab. 2012;30:60-8
48. Shiraki M, Shiraki Y, Aoki C, Miura M. Vitamin K2 (menatetrenone) effectively prevents fractures and sustains lumbar bone mineral density in osteoporosis. J Bone Miner Res. 2000;15:515-21
49. Takeuchi Y, Suzawa M, Fukumoto S, Fujita T. Vitamin K(2) inhibits adipogenesis, osteoclastogenesis, and ODF/RANK ligand expression in murine bone marrow cell cultures. Bone. 2000;27:769-76
Author contact
![]() Corresponding authors: Chang-Qing Zhang (E-mail: zhangcqedu.cn) You-Shui Gao (E-mail:acerzhang5233edu.cn) Department of Orthopedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, 600 Yishan Road, Shanghai 200233, China.Telephone: +86-1891-7233480 FAX: 021-64701361.
Corresponding authors: Chang-Qing Zhang (E-mail: zhangcqedu.cn) You-Shui Gao (E-mail:acerzhang5233edu.cn) Department of Orthopedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, 600 Yishan Road, Shanghai 200233, China.Telephone: +86-1891-7233480 FAX: 021-64701361.
Received 2015-2-12
Accepted 2016-3-25
Published 2016-4-28