ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2016; 12(7):872-883. doi:10.7150/ijbs.15641 This issue Cite
Research Paper
Glycosphingolipid GM3 is Indispensable for Dengue Virus Genome Replication
Kezhen Wang1#, Juanjuan Wang1#, Ta Sun1, Gang Bian1, Wen Pan1, Tingting Feng1, Penghua Wang2, Yunsen Li1 ![]() , Jianfeng Dai1
, Jianfeng Dai1 ![]()
1. Institutes of Biology and Medical Sciences, Jiangsu Key Laboratory of Infection and Immunity, Soochow University, Suzhou 215123, P.R. China
2. Department of Microbiology and Immunology, New York Medical College, Valhalla, NY 10595, USA.
# These two authors contribute equally.
Abstract

Dengue virus (DENV) causes the most prevalent arthropod-borne viral disease of humans worldwide. Glycosphingolipids (GSLs) are involved in virus infection by regulating various steps of viral-host interaction. However, the distinct role of GSLs during DENV infection remains unclear. In this study, we used mouse melanoma B16 cells and their GSL-deficient mutant counterpart GM95 cells to study the influence of GSLs on DENV infection. Surprisingly, GM95 cells were highly resistant to DENV infection compared with B16 cells. Pretreatment of B16 cells with synthetase inhibitor of GM3, the most abundant GSLs in B16 cells, or silencing GM3 synthetase T3GAL5, significantly inhibited DENV infection. DENV attachment and endocytosis were not impaired in GM95 cells, but DENV genome replication was obviously inhibited in GM95 cells compared to B16 cells. Furthermore, GM3 was colocalized with DENV viral replication complex on endoplasmic reticulum (ER) inside the B16 cells. Finally, GM3 synthetase inhibitor significantly reduced the mortality rate of suckling mice that challenged with DENV by impairing the viral replication in mouse brain. Taken together, these data indicated that GM3 was not required for DENV attachment and endocytosis, however, essential for viral genome replication. Targeting GM3 could be a novel strategy to inhibit DENV infection.
Keywords: Flavivirus, Dengue Virus, Glycolipid, Glycosphingolipids, Lipid Synthesis, Viral Replication
Background
Dengue virus (DENV) is a mosquito-borne pathogen responsible for outbreaks of dengue fever (DF), dengue hemorrhagic fever (DHF), and dengue shock syndrome (DSS). DENV is widely distributed in subtropical and tropical areas of the world and infect 50-100 million individuals every year[1, 2]. However, there is no specific antiviral drug against DENV[3, 4].
Like other flaviviruses, DENV is internalized via receptor-mediated endocytosis[5-7]. Then the viral genome is released and translated at the rough endoplasmic reticulum (ER) to produce a polyprotein, which is cleaved into three structural proteins (the capsid protein C, membrane protein M, and the envelope protein E), and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5)[6, 8]. After initial translation, viral RNA replication takes place in a structure called replication complex (RC) on the ER membranes. RC is composed of viral RNA, nonstructural proteins and other host factors[9-11]. DENV NS4A induced ER membrane alterations, which may serve as a scaffold to anchor the viral RC [12]. Some flaviviruses have been reported to exploit cellular lipids to assemble their RC[13]. For example, HCV RNA replication occurs on the membranous web (MW) containing the high levels of cellular lipid phosphatidylinositol-4-phosphate (PI4P)[14]. DENV NS3 recruited fatty acid synthase (FASN) to the sites of DENV replication, resulting in lipid biosynthesis[15]. West Nile virus (WNV) redistributes cholesterol to the sites of viral replication[16]. However, mechanisms of cellular lipids involved in DENV replication complex formation remain largely unclear.
Glycosphingolipids (GSLs), amphipathic molecules consisting of a ceramide and sugar residues, are ubiquitous cellular components of eukaryotic plasma membranes[17-19]. Besides of the roles in cell adhesion and signal transduction, GSLs have been shown to participate in various stages of life cycle of distinct viruses[20, 21]. Most of the current reports focused on the role of GSLs and their metabolites on the virus attachment and entry of specific cells[20, 21]. For example, galactosylceramide (GalCer) and sulfatide interact with HIV gp120 or gp41, and act as an alternate receptor for HIV in CD4-negative cells[22]; GD1a and GT1b are used as entry receptors by murine polyoma virus[23]; Gangliosides are ligands for human noroviruses (NoVs)[24], rotaviruses[25] and parvovirus B19[26]. Gangliosides are required for productive cell entry of rotaviruses[25], and ceramide also plays roles in entry and egress processes of Japanese encephalitis virus (JEV)[27].
As GSLs have biophysical properties of membrane bending and stretching, they are also involved in viral maturation and budding[20, 21]. For example, HIV-1 selectively assemble and bud from GSL-enriched lipid microdomains, resulting in incorporation of GSL into the viral membrane[28]. A recent study by Konan and colleagues showed that a GSL -binding protein, FAPP2, recruited GSLs to the HCV replication complex, and critical for modulation of HCV genome replication[14]. However, the distinct role of GSLs on DENV infection is unclear.
GM95 is a mutant cell line of B16 cells that do not synthesize GSLs including GM3 due to deficient in ceramide glycosyltransferase (GlcT-1), the first step of glucosylceramide (GlcCer) synthesis[29]. As GM3 is the most abundant GSLs in B16 cells, we focus on the function of GM3 on DENV replication in the present study.
Methods
Ethics Statements- All animal experiments were conducted according to the Guide for the Care and Use of Medical Laboratory Animals (Ministry of Health, People's Republic of China, 1998) and approved by the Animal Care and Use Committee as well as the Ethical Committee of Soochow University.
Virus, cells and replicons- DENV type 2 (DENV-2) New Guinea C (NGC) strain was propagated in mosquito C6/36 cells (from ATCC® CRL-1660). B16 and GM95 cells were initially from Dr. Steven B Levery (University of New Hampshire) and maintained in our lab in DMEM supplemented with 10 %( v/v) FBS and 1 %( v/v) penicillin-streptomycin. Cells were infected with DENV-2 at a multiplicity of infection (MOI) of 1, unless otherwise stated. DNA-based replicons (for DENV type 1) expressing secreted Gaussia luciferase (DGL2 and DGL2-mut) [30], were generously provided by Dr. Takayuki Hishiki (Kyoto University, Kyoto, Japan).
Inhibitors and antibodies- Pharmacological inhibitors 1-phenyl-2-palmitoylamino-3-morpholino-1-propanol (PPMP) and Soyasaponin-I were purchased from Sigma Aldrich. Primary antibodies Mouse anti-DENV (Santa Cruz), mouse anti-GM3 (NeuAc) mAb M2590 (Cosmo Bio Co. Ltd, Tokyo, Japan) and secondary antibodies FITC labeled Donkey anti-mouse IgG (Jackson ImmunoResearch), Goat anti-Mouse IgM Secondary Antibody, FITC conjugate (Pierce) were used in this study.
TCID50 assay and viral growth kinetics- B16 and GM95 cells were seeded in 24-well plate and infected with DENV for 12, 24 and 48 hrs, respectively. The cell-free supernatants were collected and the DENV titers were assayed with a TCID50 assay according to standard protocols on Vero cells (ATCC® CCL-81). The viral replication levels inside cells, in terms of the transcript levels of the DENV-2 envelop gene (E), were quantified by quantitative reverse transcription PCR (qRT-PCR) and normalized to mouse beta actin gene.
Preparation of Rhodamine-labeled DENV- Viruses were concentrated by ultracentrifugation and then labelled with Rhodamine dye using a NHS-Rhodamine antibody labelling kit (Pierce) according to manufacturer's protocol. Briefly, purified viruses were dialyzed in 0.67M borate buffer and incubate in the dark at room temperature with NHS-Rhodamine Reagent. After 1 hr, the unincorporated dye was removed by resin filter. Rhodamine-Labelled viruses were used immediately.
Virus binding and endocytosis assay- For virus binding assay, B16 and GM95 cells were incubated with DENV (MOI=10) at 4 ℃ for 1 hr. The cells were washed three times with pre-cold PBS to remove unbound viruses. Two approaches were used to determine the virus binding efficiency: (a) cells were lysed directly for qRT-PCR analysis of virus E gene copy; (b) cells were lysed and the number of bound virus was measured by TCID50 assay.
To assess viral internalization efficiency (virus endocytosis assay), B16 and GM95 cells were incubated with DENV (MOI=10) at 37 ℃ for 30 min. Cells were washed with PBS and treated with 0.5% trypsin for 10 min to remove the viruses that not internalized. Finally, cells were washed with PBS and harvested by low-speed centrifugation. Intracellular viral RNA was quantified by qRT-PCR.
For a visualized DENV binding and endocytosis assay, the same experimental procedure was performed using a Rhodamine-labeled DENV as described above. Cells were incubated with labelled virus for 1 hr at 4 ℃ (for virus binding) or incubated at 37 ℃ for 30 min (for virus internalization). Cells were fixed in 4% formaldehyde, permeabilized with 0.1% Triton X-100 and then examined by inverted fluorescence microscope (cell nucleus were stained with DAPI).
RT-PCR of virus negative strand RNA (-ssRNA)- To generate the cDNA template specific for DENV negative strand RNA, the total RNA was extracted from DENV-infected cells at 6, 12 and 24 hr postinfection. Forward PCR primer for DENV E gene was used to transcribe the cDNA for negative strand RNA using First Strand cDNA Synthesis Kit (Thermo). DENV E gene and mouse beta actin were amplified by PCR with rTaq DNA polymerase (TaKaRa Biotechnology), and detected by electrophoresis on a 2% agarose gel.
DENV replicon Gaussia luciferase reporter assay- In 48-well plates, 50% confluent B16 and GM95 cells were transfected with 100 ng of DGL2-WT or DGL2-mut replicon plasmids. For the Gaussia luciferase assay, culture supernatant were collected at different time points ( 0, 24, 48, 72 and 96 hr post transfection) and luciferase was measured using BioLux™ Gaussia Luciferase Assay Kit (New England Biolabs) according to the manufacturer's instructions.
RNA interference and qRT-PCR- siRNA targeting mouse T3gal5 gene and negative controls were ordered from Ribotask (Guangzhou, China). B16 cells were seeded in 24-well plates prior to transfection and the siRNA were transfected into cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. After 24 hrs, siRNA-transfected cells were then infected with DENV. Viral replication were measured by amplifying the DENV-2 E gene and normalized to mouse beta actin. (siRNA and oligo-primer sequences for this study were shown in Table S1 in the supplemental materials).
Pharmaceutical inhibitors treatment and cell viability assay- Two inhibitors were used for inhibiting ganglioside biosynthesis. PPMP, an inhibitor of glucosylceramide synthase that catalyses the first step of ganglioside biosynthesis; Soyasaponin-I, a sialyltransferase inhibitor, inhibits the last step of synthesis of GM3. B16 cells were cultured in 48-well plates, 1μM PPMP (or 100 μM Soyasaponin-I) was added to cells. At 48 hr post treatment, cells were washed three times with PBS and then infected with DENV. The viral replication efficiency was measured as described above.
For cell viability assay, B16 cells were cultivated in 96-well plates to 90% confluency and treated with the inhibitor compounds as described concentration above. Following treatment at 37 °C for 48 hrs, cell viability was evaluated using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega) according to the manufacturer's instructions.
Addition of exogenous GM3 to GM95 cells- To evaluate the effect of exogenous ganglioside GM3 on GM95 cells during DENV infection, GM3 (bovine, purity >98%, from Matreya LLC.) was added to the GM95 culture medium at a final concentration of 100 μg/μl. Unincorporated gangliosides were removed by washing twice with 20% FBS-DMEM prior to DENV infection. Then, the cells were infected with DENV-2 and viral replication efficiency was measured as described above.
Immunofluorescence staining and confocal microscopy- B16 and GM95 cells grown on 48-well plate were infected with DENV-2. At 24 hr postinfection, the cells were fixed with 4% paraformaldehyde (PFA) for 10 min, permeabilized with 0.1% Triton X-100 in PBS, and then blocked with 5% FBS in PBS. Plates were incubated with a DENV envelope monoclonal antibody overnight at 4 °C, washed, and then incubated with FITC goat anti-mouse secondary antibody for 1 hr at room temperature. Samples were analyzed by an inverted fluorescence microscope.
To analyze of the changes of GM3 in B16 cells upon DENV infection, DENV-2 infected or non-infected B16 cells were fixed and immunostained with antibody against GM3. Then images were collected using confocal fluorescence microscopy.
For co-localization study, B16 cells were transfected with YFP-KDEL (an ER lumen protein) and infected with DENV-2. 48 hr postinfection, cells were fixed with 4% PFA. DENV NS4A protein and GM3 were probed with primary antibodies (rabbit IgG anti-DENV NS4A and mouse anti-GM3 mAb ) and stained with Dy light 649 donkey anti-rabbit IgG and TRITC labeled anti-mouse IgM, respectively. The localizations of DENV NS4A, GM3 and ER inside cells were then examined by confocal microscope.
GSL extraction, Mass Spectrometry, and thin-layer chromatography (TLC)- GSLs extraction was performed as described previously[31]. Briefly, cell pellets (1x108) were extracted by sonication with each of the following different solvents: (i) chloroform/methanol (1:1, v/v); (ii) isopropanol/hexane/water (55:25:20, v/v/v). Supernatants were dried and whole lipids were dissolved with chloroform/methanol/water (30:60:8, v/v/v) and then applied to a DEAE Sephadex A-25 (Sigma, St. Luis, MO, USA) on a glass column (Internal diameter×Length, 5×80 mm) for separation of neutral and acidic lipid fraction. The acidic lipid fraction was dried, dialyzed and lyophilized.
GSLs were completely methylated as described previously[31, 32]. Methylated GSLs were analyzed by LTQ-XL mass spectrometer (Thermo Fischer Scientific, Waltham) in the positive ion mode ( ion spray voltage 3.5 kV, capillary voltage 35 V, capillary temperature 350 ℃, injection time 100 ms, activation time 30 min, and isolation width m/z 1.5).
Acid GSLs were further analyzed by thin-layer chromatography (TLC) as described [29]. The purified acid GSLs were developed on TLC plates of silica gel 60 (Merck) with chloroform/methanol/0.25% CaCl2˙H2O (50:40:10, v/v/v). The plate was sprayed with a resorcinol/H2SO4 reagent and then heated at 110 ℃ for 30 min. The purified ganglioside GM3 (Matreya LLC, Pleasant Gap, PA, USA) was used as standard.
DENV infection of suckling mice- Breeder BALB/c mice were purchased from Shanghai SLAC Laboratory animal CO. LTD. Each 3- or 4-day-old suckling mouse was inoculated intracerebrally with 100 PFU of DENV-2 as previously described[33], together with or without 15 μg of GM3 synthetase inhibitor soyasaponin-I. Survival of mice were monitored daily and the DENV replication level in cerebrum at day 2 and day 4 postinfection were measured by qRT-PCR method described above.
Statistical analysis- Statistical significances were calculated with an unpaired two tailed Student's t- test and Log-rank (Mantel-Cox) Test (for survival data only) using Prism 5 software (GraphPad).
Results
GM95 cells are resistant to DENV infection
GM95 cells, a mutant B16 melanoma cell line, are incapable of synthesizing GSLs due to a defective GlcT-1 gene. As shown in Fig. 1A, GSL GM3 was highly expressed in wild type cells, but not in GM95 cells.
GM95 cells were resistant to DENV infection. (A) Immunofluorescence analysis of GM3 in B16 and GM95 cells. B16 and GM95 cells were fixed and immunostained with GM3 monoclonal antibody. The images were collected using confocal fluorescence microscopy. (B) Growth curves of DENV-2 in B16 and GM95 cells. The viral burdens were analyzed by measuring the virus E gene copy using qRT-PCR, and normalized to mouse beta actin. (C) B16 and GM95 cells were seeded in 24-well plate and infected with DENV for 12, 24 and 48hrs, respectively. The cell-free supernatants were collected and the DENV titers were assayed with a TCID50 assay on Vero cells. Results were expressed as the mean + the SEM. * p < 0.05 and ** p < 0.01 (t-test). Representative results from at least 3 independent experiments. (D) The supernatants from Fig. 1C were used to test cell pathogenic effect (CPE) on Vero cells. Supernatants from infected B16 cells, but not from infected GM95 cells, had significantly cell pathogenic effect on Vero cells.
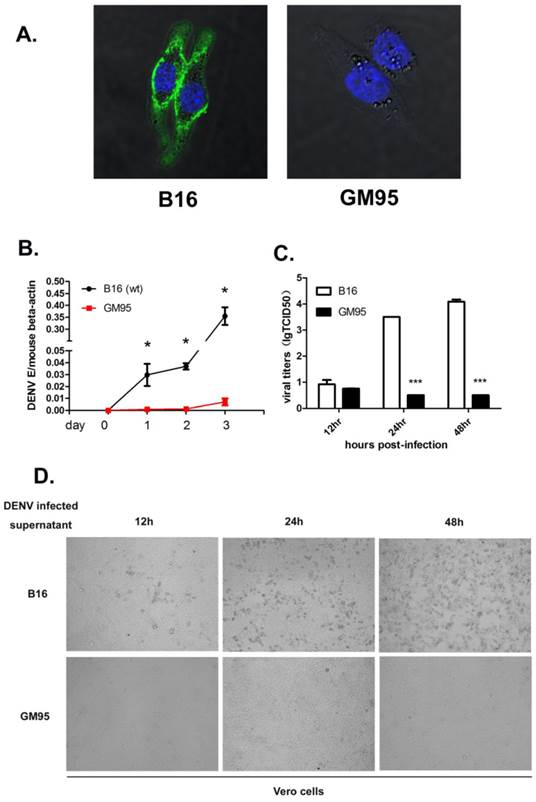
To determine the roles of GSLs on DENV infection, we exposed the B16 and GM95 cells to DENV-2. Viral infection level was measured by amplifying the viral envelope gene (E) copies using qRT-PCR at 0, 24, 48 and 72 hr postinfection and normalized to mouse beta-actin gene. As shown in Fig. 1B, B16 cells were highly susceptible to DENV infection, while GM95 cells were resistant to DENV infection. To further test whether GSLs impact production of infectious virus particles, the titers of virus in the supernatant of infected B16 and GM95 cells were measured by TCID50 assay. The numbers of DENV virions in cell supernatants from GM95 cells were much lower than that from B16 cells (Fig. 1C). As shown in Fig. 1D, supernatants from infected B16 cells caused significantly cell pathogenic effect (CPE) on Vero cells. However, supernatants from infected GM95 cells did not cause noticeable cell death. Taken together, these data suggested that GM95 cells were resistant to DENV infection.
Inhibition of GM3 synthesis impedes DENV infection
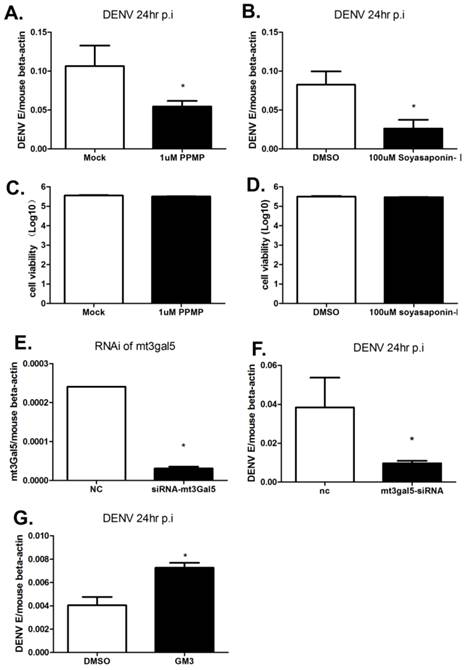
To inhibit GSLs metabolism, inhibitors PPMP and Soyasaponin-I were used to treat B16 cells. PPMP is a glucosylceramide synthase inhibitor, and deplete GSLs production by inhibiting GlcT-1 activity. Soyasaponin-I, a specific sialytransferase inhibitor, inhibits the last step of synthesis of GM3. As shown in Fig. 2A and B, DENV RNA levels were reduced more than 50% following the treatment of PPMP or Soyasaponin-I. We have verified that the inhibitor treatment has a negligible impact on cell viability as determined using the CellTiter-Glo Luminescent Cell Viability Assay (Promega) (Fig. 2C and D). To further confirm the role of GM3 on DENV infection, GM3 synthetase was silenced by siRNA targeting mouse T3gal5 gene (mt3gal5) (Fig. 2E). Consistent with the data shown in Fig. 2B, the intracellular viral loads were decreased in GM3 synthetase knockdown cells (Fig. 2F). Moreover, exogenous ganglioside GM3 added to GM95 cells partially restored the DENV infection (Fig. 2G). These data suggested that GM3 played important roles in DENV infection.
Inhibition of GM3 synthesis impeded DENV infection. (A) A glucosylceramide synthase inhibitor, PPMP, inhibited DENV replication in B16 cells. (B) Soyasaponin-I, the GM3 synthetase inhibitor, inhibited DENV replication in B16 cells. (C, D) Viability of B16 cells treated with PPMP (C) or Soyasaponin-I (D). (E) The RNAi efficiency of mt3gal5 siRNA was tested in B16 cells. (F) DENV replication was inhibited in mt3gal5 knockdown B16 cells. (G) Exogenous ganglioside GM3 added to GM95 cells partially restored the DENV infection. Results were expressed as the mean + the SEM. * p < 0.05 and ** p < 0.01 (t-test). Representative results from at least 3 independent experiments.
GM3 synthesis is upregulated in DENV infected cells
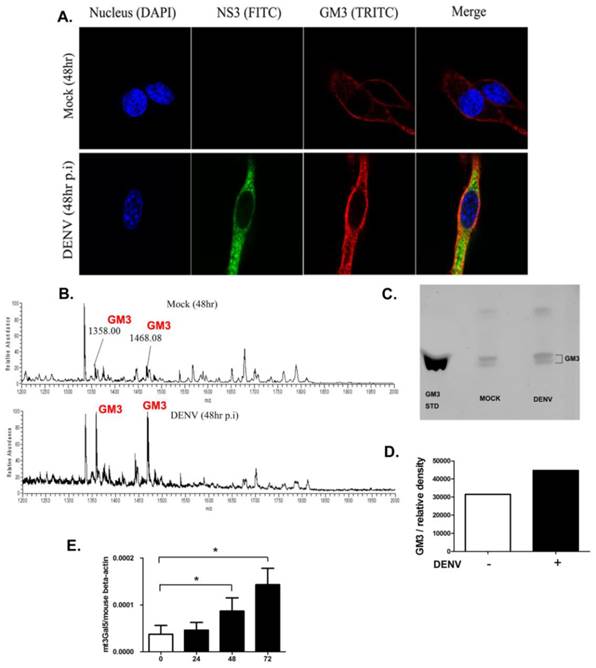
To explore whether the expression of GM3 changed upon DENV infection, immunofluorescence assay was performed with immunofluorescence staining using GM3 mAbs in B16 cells. As shown in Fig. 3A, the production of GM3 was significantly higher in DENV infected B16 cells. The result was further confirmed by mass spectrometry and thin layer chromatography analysis. As shown in Fig. 3B, the GM3 specific m/z peaks (1358.00 and 1468.08) were significantly higher in infected B16 cells compared with non-infected cells. In a TLC analysis, the fraction of GM3 was increased in the infected cells compared with non-infected cells (Fig. 3 C and D). In line with these results, the mRNA level of GM3 synthetase gene mT3gal5 in B16 cells was significantly increased post DENV infection as determined by qRT-PCR and normalized to mouse beta-actin gene (Fig. 3E). These data suggested that GM3 production was increased in DENV infected B16 cells, further indicating that GM3 is important in DENV infection.
GM3 does not influence binding and endocytosis of DENV
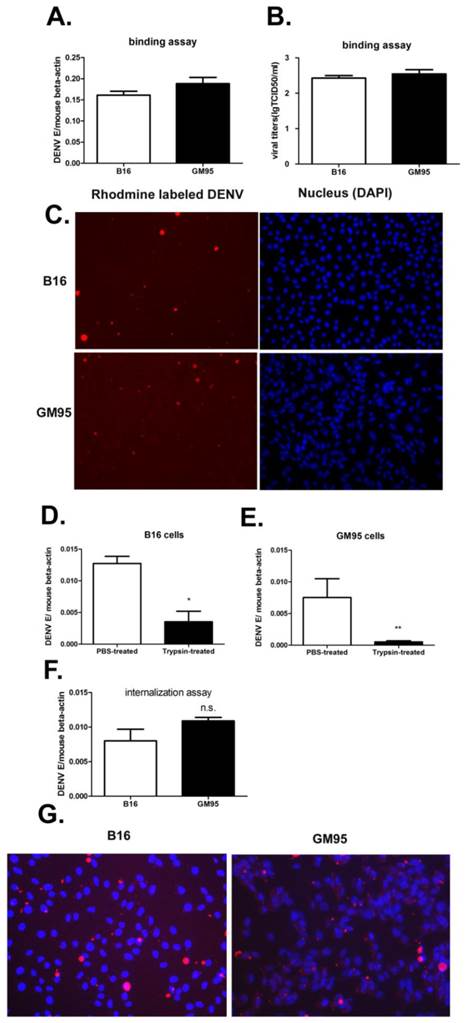
To assess whether the GM3 was necessary in early stage of viral infection, the amount of membrane-bound and internalized viruses were measured by qRT-PCR of virus E gene and Rhodamine-labeled virus imaging. The amount of viruses attached to B16 and GM95 cells was also analyzed by TCID50 assay on Vero cells. For virus binding assay, B16 and GM95 cells were incubated with DENV (MOI=10) at 4 ℃ for 1 hr. The results suggested that no significant differences in the efficiency of virus binding between B16 and GM95 cells (Fig. 4, A-C). To quantity the viruses that entered into the cells, the viruses were incubated with cells at 37 ℃ for 30 min and then the cells were treated with trypsin. As shown in Fig. 4D and 4E, trypsin, but not PBS, could effectively remove the virions which just binding to the cell membrane. After trypsin treatment, the viral RNA could still be detected in GM95 cells, with a comparable level of those in B16 cells (Fig. 4F). The Rhodamine-labeled DENV were also obviously detected inside the GM95 cells in the virus endocytosis assay (Fig. 4G). These results suggested that GM3 was not involved in the virus binding and internalization.
GM3 synthesis was upregulated in DENV infected cells. (A) Immunofluorescence analysis of GM3 in DENV infected and non-infected B16 cells. GM3 mouse mAb and DENV NS3 mAb were used to perform the experiment. (B) Mass spectrometry analysis of GSLs in DENV infected and non-infected B16 cells. Both m/z peaks (1358.0 and 1468.08) represented GM3. (C) The GM3 fractions from DENV-infected and noninfected B16 cells were detected on TLC plates. Purified GM3 were served as a standard. (D) Relative amount of GM3 fractions from Fig. 3C by density analysis. (E) The mRNA expression level of GM3 synthetase mt3gal5 at different time points after DENV infection, normalized to mouse beta actin gene. Results were expressed as the mean + the SEM. * p < 0.05 and ** p < 0.01 (t-test). Representative results from at least 3 independent experiments.
GM3 was not necessary for DNEV binding and endocytosis. (A, B) B16 and GM95 cells were incubated with DENV (MOI=10) at 4 ℃ for 1 hr. The cells were washed three times with pre-cold PBS to remove unbound viruses. The amount of membrane-bound DENV as measured by qRT-PCR of DENV E gene RNA (A) and TCID50 assay (B). (C) 4 ℃ binding assay with Rhodamine-labeled DENV in B16 and GM95 cells (cell nucleus were stained with DAPI). (D, E) The amount of viral RNA copies in B16 (D) and GM95 (E) cells with or without trypsin treatment. The results confirmed that trypsin treatment could efficiently remove DENV virions that not been internalized. (F) The amount of internalized DENV in B16 cells and GM95 cells as measured by qRT-PCR of DENV E gene RNA. (G) The amount of internalized Rhodamine-labeled DENV in B16 cells and GM95 cells as analyzed via an inverted fluorescence microscope.
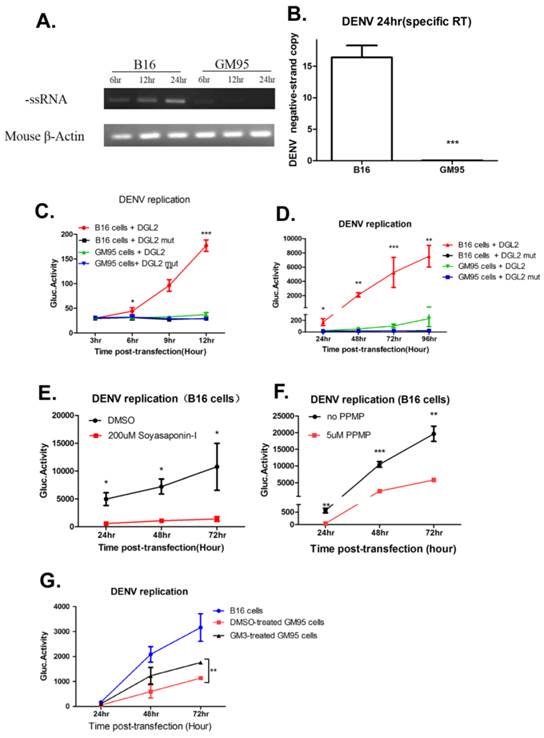
GM3 is indispensable for DENV genome replication
The replication of the positive-strand RNA of DENV is semi-conservative, and viral (-) strand RNA serves as a template for the synthesis of new (+) strand RNA. Therefore, the detection of viral (-) strand RNA is indicative for DENV replication. Total RNA was isolated from infected B16 and GM95 cells for indicated time points and transcribed into (-) cDNA with forward primer of DENV E gene. During 6 to 24 hr postinfection, the amount of negative strands in infected B16 cells was significantly increased, but not in infected GM95 cells (Fig. 5A). In addition, qRT-PCR quantitation of negative strand RNA in B16 and GM95 cells at 24 hr postinfection also suggested that there was no detectable (-) strand subgenomic RNA in GM95 cells (Fig. 5B). In order to better understand the kinetics of DENV RNA replication, a wild-type DENV DNA-based replicon (DGL2) or a NS5 mutant replicon (DGL2 mut)[30] was transfected into B16 and GM95 cells. As shown in Fig. 5C and 5D, the DENV replicon replication was severely impaired in GM95 cells. In line with this, when Soyasaponin-1 (Fig. 5E) and PPMP (Fig. 5F) was added to B16 cells, the DENV replicon replications were obviously decreased. Meanwhile, exogenous ganglioside GM3 added to GM95 cells partially restored the DENV replicon replication (Fig. 5G). These results demonstrated that GM3 was necessary for DENV genome replication.
GM3 was indispensable for DENV genome replication. (A, B) Semi-quantified PCR (A) and qRT-PCR (B) analysis of negative strand RNA (-ssRNA) of DENV in infected B16 and GM95 cells. (C, D) The replication kinetics of DENV replicon (DGL2 or DGL2 mut) in B16 and GM95 cells. Cells supernatant was harvested at different time points and Gaussia luciferase was measured. (E, F) DGL2 replicon replication was inhibited by Soyasaponin-I (E) and PPMP (F) in B16 cells, individually. (G) Exogenous ganglioside GM3 added to GM95 cells partially restored the DGL2 replicon replication in GM95 cells. Results were expressed as the mean + the SEM. * p < 0.05 and ** p < 0.01 (t-test). Representative results from at least 3 independent experiments.
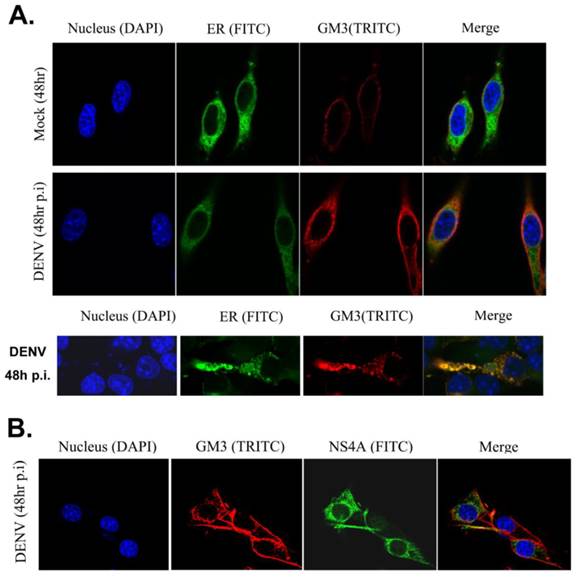
GM3 is associated with DENV replication complex
Then we examined whether GM3 co-localized with DENV replication complex. Studies have been reported that DENV RNA replication takes place in replication complex (RC) on ER, which comprise the viral RNA template with the nonstructural proteins and presumably associated with virus-induced intracellular membrane structures. GM3 was stained in B16 cells with or without DENV infection and the ER structure were indicated by a YFP-labeled ER marker. As shown in Fig. 6A, GM3 is mainly located on the cell membrane. After DENV infection, the amount of GM3 significantly increased and some of them were localized on ER. Immunofluorescence staining also suggested that GM3 was partially colocalized with DENV NS4A, the non-structural protein that anchored the RC to ERs, in B16 cells (Fig. 6B). These data suggested that GM3 colocalized with DENV replication complex in ER.
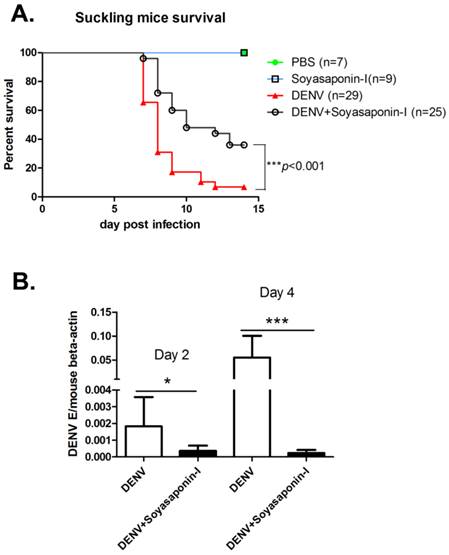
Soyasaponin-I reduces the mortality rate of DENV infected suckling mice
To test whether GM3 synthetase inhibitor soyasaponin-I could protect mice against DENV infection, 3- or 4-day-old suckling mice were inoculated intracerebrally with 100 PFU of DENV-2 together with or without 15 μg soyasaponin-I, respectively. As shown in Fig. 7A, soyasaponin-I significantly reduced the mortality rate of suckling mice suffered with DENV infection. To test whether soyasaponin-I protect mice against DENV infection via inhibiting the viral replication, the DENV replication level in cerebral tissues were analyzed at day 2 and day 4 postinfection. As shown in Fig. 7B, the DENV replications in mouse brain were significantly impaired in soyasaponin-I treated mice. These data further suggested that GM3 was indispensable for DENV replication.
GM3 was associated with DENV replication complex. (A) GM3 was co-localized with ER marker (YFP-KDEL) in DENV infected B16 cells. B16 cells were transfected with YFP-KDEL (an ER lumen protein as ER marker) and infected with DENV-2. After 48 hr, the cells were fixed. GM3 were stained mouse mAb followed by a TRITC labeled goat anti mouse IgM secondary Ab. The lower panel showed a detailed co-localization of GM3 and ER marker. (B) GM3 was colocalized with DENV NS4A in DENV infected B16 cells. B16 cells were infected with DENV-2 for 48 hrs. Then the cells were fixed and labeled with GM3 mouse mAb and NS4A rabbit Ab followed by fluorescence labeled secondary Abs.
Soyasaponin-I reduced the mortality rate of DENV infected suckling mice. (A) Survival rate of the suckling mice received intracerebral injection of 100 PFU of DENV-2 with or without 15 μg soyasaponin-I. Injected suckling mice were monitored daily for signs of illness until day 15. PBS or Soyasaponin-I was injected into suckling mice separately as the controls. Results were analyzed by Log-rank (Mantel-Cox) Test. ***p =0.0004. (B) Soyasaponin-I impaired the DENV replication in mouse brain. RNA was isolated from cerebral tissues of DENV-2 infected suckling mice with or without 15 μg soyasaponin-I at day 2 and day 4 postinfection. And the qRT-PCR was performed as described before. Results were expressed as the mean + the SEM. * p < 0.05 and ** p < 0.01 (t-test).Representative results from at least 3 independent experiments.
Discussion
GSLs are amphipathic lipids consisting of oligosaccharide and ceramide moieties. Ceramide, the common precursor of GSLs, is synthesized at the membranes of the ER. Newly synthesized ceramide transports from ER to the trans-Golgi regions, where it is converted to the most GSLs by glycosyltransferases. Finally, the de novo biosynthesis of GSLs reaches the plasma membrane via vesicular exocytotic membrane flow[19]. Viruses are not capable of replicating themselves independently, and must utilize host cell cofactors to facilitate its entry, replication, assembly and budding of virions. Among the host factors required by viruses, lipids like GSLs, play important roles in viral infection [20, 21]. The vast majority of articles showed that viruses exploit GSLs for viral binding, entry, assembly or release. For example, HIV-1 entry to CD4+ T cells via its envelope protein gp120 recognizes of CD4 on target cells, this interaction gives rise to a conformational change in gp120 third variable loop (V3) that allow for binding to GalCer, globotriaosylceramide (Gb3) and GM3. In addition, GalCer can support HIV entry into non-CD4 expressing cells through binding to gp120 [22, 34-36]. Several members of the polyomavirus (Py), such as simian virus 40 (SV40), BK virus and Py viruses, also bind to specific gangliosides (sialic acid-containing GSLs) on cell membrane to cause infection[23]. In the case of virus budding, HIV-1 preferentially buds from GSL-enriched lipid microdomains on the plasma membrane. The HIV-1 virions contain higher levels of GM3 and boosts virus capture by mature dendritic cells (MDCs)[37]. However, the distinct role of GSLs in the DENV life cycle is still unclear.
In the present study, we discovered that DENV infection severely impaired in GSL deficient GM95 cells when compared with wild type B16 cells. Interestingly, DENV binding and entry were not affected in GM95 cells, suggesting that GSLs were not necessary for virus binding and endocytosis. However, Chie Aoki and colleagues reported that DENV-2 could recognize the neutral GSLs nLc4Cer (Galβ1-4GlcNAcβ1-3Galβ1-4Glcβ1-1'Cer) and L-3 (GlcNAcβ1-3Manβ1-4Glcβ1-1'Cer) expressed on the cell surface of DENV-susceptible cells[38]. Whether those specific GSLs were involved in DENV infection are still unknown. In our study, we focused on the role of GM3, the most dominant GSL in B16 cells. Actually, we did not detect any Gb3 and LC3 in B16 cells by mass spectrometry. We found that GM3 was not required for DENV binding and endocytosis.
To test whether GM3 play a role in DENV genome replication, DNA-based DENV replicon DGL-2 and DGL-2 mut[30] were used to assess the dynamic replication of DENV in B16 and GM95 cells. Without any binding or entry issue, transfected DENV replicon replicated much more efficiently in B16 cells compared to GM95 cells. Replication of DENV RNA occurs in ER membrane-associated replication complex (RC) composed of viral replicase proteins, viral RNA and other host factors [10, 39]. Subcellular colocalization analysis showed that GM3 colocalized with the DENV NS4A, NS3 and ER marker in DENV infected B16 cells, suggesting that GSLs may participate in organization of DENV RCs. Recently, GSLs were reported to play a key role in Hepatitis C virus (HCV) replication. Knockdown of four-phosphate adaptor protein 2 (FAPP2) could attenuate HCV infectivity and impede HCV RNA replication. In addition, HCV increased LacCer and Gb3 levels, and addition of these lipids to FAPP2-depleted cells partially rescued replication[14]. The results were in line with our findings that GM3, a major subtype of GSLs, was indispensable for DENV genome replication. Although both HCV and DENV belong to the family Flaviviridae, the replication of DENV shows significant difference from that of HCV[10]. DENV's NS4A induced rearrangements of the ER membranes after infection [40]. Electron tomography and three-dimensional reconstructions analyzes showed that DENV-induced ER-derived structure are termed vesicle packets and convoluted membranes, while HCV infection induces double-membrane vesicles[10].
Up to date, the roles of GSLs during DENV infection are largely unknown. Our findings showed that GM3 was not required for DENV's binding and entry, but indispensable for its replication. GM3 synthetase inhibitor significantly reduced the mortality rate of DENV infected mice. Therefore, inhibiting GM3 biosynthesis could be a new strategy for control of DENV infection.
Supplementary Material
Table S1.
Acknowledgements
We thank Guanghao Li and Xiangyang Zuo for assistance with the experiments. This work was supported by A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT), National Natural Science Foundation of China (NSFC) (31400737, 81471571, 81271792, 31300714 and 31500700) and Jiangsu Natural Science Foundation (BK20140322). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Authors Contribution
P.W., Y.L., and J.D. designed the experiments and prepared the manuscript. K.W., J.W., T.S., G. B., W.P., and T. F. performed and analyzed the experiments. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Guzman MG, Halstead SB, Artsob H, Buchy P, Farrar J, Gubler DJ. et al. Dengue: a continuing global threat. Nat Rev Microbiol. 2010;8:S7-16
2. Horstick O, Tozan Y, Wilder-Smith A. Reviewing dengue: still a neglected tropical disease?. PLoS Negl Trop Dis. 2015;9:e0003632
3. Murrell S, Wu SC, Butler M. Review of dengue virus and the development of a vaccine. Biotechnol Adv. 2011;29:239-47
4. da Costa VG, Marques-Silva AC, Floriano VG, Moreli ML. Safety, immunogenicity and efficacy of a recombinant tetravalent dengue vaccine: A meta-analysis of randomized trials. Vaccine. 2014
5. Hidari KI, Suzuki T. Dengue virus receptor. Trop Med Health. 2011;39:37-43
6. Fischl W, Bartenschlager R. Exploitation of cellular pathways by Dengue virus. Curr Opin Microbiol. 2011;14:470-5
7. Jain B, Chaturvedi UC, Jain A. Role of intracellular events in the pathogenesis of dengue; an overview. Microb Pathog. 2014;69-70:45-52
8. Rodenhuis-Zybert IA, Wilschut J, Smit JM. Dengue virus life cycle: viral and host factors modulating infectivity. Cell Mol Life Sci. 2010;67:2773-86
9. den Boon JA, Diaz A, Ahlquist P. Cytoplasmic viral replication complexes. Cell Host Microbe. 2010;8:77-85
10. Chatel-Chaix L, Bartenschlager R. Dengue virus- and hepatitis C virus-induced replication and assembly compartments: the enemy inside-caught in the web. J Virol. 2014;88:5907-11
11. Paranjape SM, Harris E. Control of dengue virus translation and replication. Curr Top Microbiol Immunol. 2010;338:15-34
12. Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P. et al. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe. 2009;5:365-75
13. Alvisi G, Madan V, Bartenschlager R. Hepatitis c virus and host cell lipids: An intimate connection. RNA Biology. 2014;8:258-69
14. Khan I, Katikaneni DS, Han Q, Sanchez-Felipe L, Hanada K, Ambrose RL. et al. Modulation of hepatitis C virus genome replication by glycosphingolipids and four-phosphate adaptor protein 2. J Virol. 2014;88:12276-95
15. Heaton NS, Perera R, Berger KL, Khadka S, Lacount DJ, Kuhn RJ. et al. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:17345-50
16. Mackenzie JM, Khromykh AA, Parton RG. Cholesterol manipulation by West Nile virus perturbs the cellular immune response. Cell Host Microbe. 2007;2:229-39
17. Lingwood CA. Glycosphingolipid functions. Cold Spring Harbor perspectives in biology. 2011:3
18. Kolter T. Ganglioside Biochemistry. ISRN Biochemistry. 2012;2012:1-36
19. D'Angelo G, Capasso S, Sticco L, Russo D. Glycosphingolipids: synthesis and functions. FEBS J. 2013;280:6338-53
20. Schneider-Schaulies J, Schneider-Schaulies S. Sphingolipids in viral infection. Biol Chem. 2015;396:585-95
21. Schneider-Schaulies J, Schneider-Schaulies S. Viral infections and sphingolipids. Handb Exp Pharmacol. 2013:321-40
22. Hammache D, Yahi N, Pieroni G, Ariasi F, Tamalet C, Fantini J. Sequential interaction of CD4 and HIV-1 gp120 with a reconstituted membrane patch of ganglioside GM3: implications for the role of glycolipids as potential HIV-1 fusion cofactors. Biochem Biophys Res Commun. 1998;246:117-22
23. Gilbert J, Dahl J, Riney C, You J, Cui C, Holmes R. et al. Ganglioside GD1a restores infectibility to mouse cells lacking functional receptors for polyomavirus. J Virol. 2005;79:615-8
24. Han L, Tan M, Xia M, Kitova EN, Jiang X, Klassen JS. Gangliosides are ligands for human noroviruses. J Am Chem Soc. 2014;136:12631-7
25. Martinez MA, Lopez S, Arias CF, Isa P. Gangliosides Have a Functional Role during Rotavirus Cell Entry. J Virol. 2013;87:1115-22
26. Bonsch C, Zuercher C, Lieby P, Kempf C, Ros C. The globoside receptor triggers structural changes in the B19 virus capsid that facilitate virus internalization. J Virol. 2010;84:11737-46
27. Tani H, Shiokawa M, Kaname Y, Kambara H, Mori Y, Abe T. et al. Involvement of ceramide in the propagation of Japanese encephalitis virus. J Virol. 2010;84:2798-807
28. Yu X, Feizpour A, Ramirez NG, Wu L, Akiyama H, Xu F. et al. Glycosphingolipid-functionalized nanoparticles recapitulate CD169-dependent HIV-1 uptake and trafficking in dendritic cells. Nat Commun. 2014;5:4136
29. Ichikawa S, Nakajo N, Sakiyama H, Hirabayashi Y. A mouse B16 melanoma mutant deficient in glycolipids. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:2703-7
30. Kato F, Kobayashi T, Tajima S, Takasaki T, Miura T, Igarashi T. et al. Development of a novel Dengue-1 virus replicon system expressing secretory Gaussia luciferase for analysis of viral replication and discovery of antiviral drugs. Japanese journal of infectious diseases. 2014;67:209-12
31. Wang Z, Wen L, Ma X, Chen Z, Yu Y, Zhu J. et al. High expression of lactotriaosylceramide, a differentiation-associated glycosphingolipid, in the bone marrow of acute myeloid leukemia patients. Glycobiology. 2012;22:930-8
32. Yu RK, Ledeen RW. Gangliosides of human, bovine, and rabbit plasma. Journal of lipid research. 1972;13:680-6
33. Lee YR, Huang KJ, Lei HY, Chen SH, Lin YS, Yeh TM. et al. Suckling mice were used to detect infectious dengue-2 viruses by intracerebral injection of the full-length RNA transcript. Intervirology. 2005;48:161-6
34. Hammache D, Pieroni G, Yahi N, Delezay O, Koch N, Lafont H. et al. Specific interaction of HIV-1 and HIV-2 surface envelope glycoproteins with monolayers of galactosylceramide and ganglioside GM3. The Journal of biological chemistry. 1998;273:7967-71
35. Puri A, Rawat SS, Lin HM, Finnegan CM, Mikovits J, Ruscetti FW. et al. An inhibitor of glycosphingolipid metabolism blocks HIV-1 infection of primary T-cells. AIDS. 2004;18:849-58
36. Lingwood CA, Branch DR. The role of glycosphingolipids in HIV/AIDS. Discovery medicine. 2011;11:303-13
37. Puryear WB, Yu XW, Ramirez NP, Reinhard BM, Gummuluru S. HIV-1 incorporation of host-cell-derived glycosphingolipid GM3 allows for capture by mature dendritic cells. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:7475-80
38. Aoki C, Hidari KI, Itonori S, Yamada A, Takahashi N, Kasama T. et al. Identification and characterization of carbohydrate molecules in mammalian cells recognized by dengue virus type 2. J Biochem. 2006;139:607-14
39. Bartenschlager R, Miller S. Molecular aspects of Dengue virus replication. Future microbiology. 2008;3:155-65
40. Miller S, Kastner S, Krijnse-Locker J, Buhler S, Bartenschlager R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. The Journal of biological chemistry. 2007;282:8873-82
Author contact
![]() Corresponding authors: Jianfeng Dai, Ph.D., daijianfengedu.cn and Yunsen Li, Ph.D., yunsenliedu.cn; Institutes of Biology and Medical Sciences, Soochow University, Building 703, 199 Ren-ai Road, Suzhou 215123, P.R. China; Tel/Fax: (86-512) 6588 2472
Corresponding authors: Jianfeng Dai, Ph.D., daijianfengedu.cn and Yunsen Li, Ph.D., yunsenliedu.cn; Institutes of Biology and Medical Sciences, Soochow University, Building 703, 199 Ren-ai Road, Suzhou 215123, P.R. China; Tel/Fax: (86-512) 6588 2472
Received 2016-3-24
Accepted 2016-5-3
Published 2016-5-25