ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2016; 12(8):990-999. doi:10.7150/ijbs.14077 This issue Cite
Research Paper
Fibroblast Growth Factor Receptor 3 Deficiency Does Not Impair the Osteoanabolic Action of Parathyroid Hormone on Mice
Yangli Xie1*, Lingxian Yi1,3*, Tujun Weng 1, Junlan Huang 1, Fengtao Luo 1, Wanling Jiang1, Cory J Xian2, Xiaolan Du1 ![]() , Lin Chen1
, Lin Chen1 ![]()
1. Center of Bone Metabolism and Repair, Department of Rehabilitation Medicine, State Key Laboratory of Trauma, Burns and Combined injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing 400042, China;
2. Sansom Institute for Health Research, School of Pharmacy and Medical Sciences, University of South Australia, Adelaide, SA 5001, Australia.
3. Intensive Care Unit, The 306th hospital of PLA, Beijing 100101, China
* These authors contributed equally to this work.
Abstract
Summary: PTH stimulates bone formation in Fgfr3 knockout mice through promotion of proliferation and differentiation in osteoblasts.
Introduction: Previous studies showed that endogenous fibroblast growth factor 2 (FGF-2) is required for parathyroid hormone (PTH)-stimulated bone anabolic effects, however, the exact mechanisms by which PTH stimulate bone formation and the function of FGF receptors in mediating these actions are not fully defined. FGF receptor 3 (FGFR3) has been characterized as an important regulator of bone metabolism and is confirmed to cross-talk with PTH/PTHrP signal in cartilage and bone development.
Methods: Fgfr3 knockout and wild-type mice at 2-month-old and 4-month-old were intraperitoneally injected with PTH intermittently for 4 weeks and then the skeletal responses to PTH were assessed by dual energy X-ray absorptiometry (DEXA), micro-computed tomography (μCT) and bone histomorphometry.
Results: Intermittent PTH treatment improved bone mineral density (BMD) and femoral mechanical properties in both Fgfr3-/- and wild-type mice. Histomorphometric analysis showed that bone formation and bone resorption were increased in both genotypes following PTH treatment. PTH treatment increased trabecular bone volume (BV/TV) in WT and Fgfr3-deficient mice. The anabolic response in Fgfr3-deficient and wild-type bone is characterized by an increase of both bone formation and resorption-related genes following PTH treatment. In addition, we found that Fgfr3 null osteoblasts (compared to wild-type controls) maintained normal abilities to response to PTH-stimulated increase of proliferation, differentiation, expression of osteoblastic marker genes (Cbfa1, Osteopontin and Osteocalcin), and phosphorylation of Erk1/2.
Conclusions: Bone anabolic effects of PTH were not impaired by the absence of FGFR3, suggesting that the FGFR3 signaling may not be required for osteoanabolic effects of PTH activities.
Keywords: PTH, FGFR3, Knockout mice, Bone remodeling, Osteoblast
Introduction
Parathyroid hormone (PTH) is a critical regulator of bone remodeling and anabolic skeletal benefits of intermittent PTH1-34 treatment are well recognized [1, 2]. Intermittent PTH treatment increases bone mass in a variety of animal models and decreases fracture risk, which has been approved to treat osteoporotic patients for a decade [3, 4]. At the cellular level, osteoblastic lineages are the principal target cells for PTH action on the bone, and osteoclast-mediated bone resorption coupled by osteoblastic bone formation is also involved in the effects of PTH on skeletal homeostasis [5].
PTH stimulates bone anabolic activity by prolonging osteoblast lifespan and increasing osteoblast number [6]. With the advances in gene targeting technology, genetically modified mice have been utilized to uncover the anabolic effects of PTH on skeleton. A number of molecules have been identified as important for the anabolic actions of PTH on bone, including insulin-like growth factor-1 (IGF-I) [7], bone sialoprotein [8], sclerostin [9], cox2 [10] and β-arrestin 2 [11] et al. Several mechanisms have been postulated to be responsible for the bone anabolic effects of PTH, including increasing osteo-differentiation from stem cell / differentiation of osteoblast progenitors[12, 13], activating osteoblastic lining cells [3], extending life span of mature osteoblasts via suppressing apoptosis [1], and indirect effects by promoting production of local osteotrophic growth factors [13-16]. Recently, Qiu T et.al found that transforming growth factor-β type II receptor (Tgfbr2) directly phosphorylates the PTH1R cytoplasmic domain and mice lacking Tgfbr2 in osteoblasts have increased bone mass due to the augment of PTH signaling [17]. However, the underlying mechanism responsible for bone anabolic action of PTH is yet to be fully elucidated. A better understanding of these mechanisms will help to develop more effective approaches to manage patients with dysregulated bone remodeling and bone loss.
A growing number of evidences suggest that PTH signaling cross-talks with fibroblast growth factor (FGF) pathway in the bone development and maintaining of bone homeostasis. Previous studies have shown that PTH stimulates the production of FGF-23 and FGF-23 signaling does not seem to be involved in the anabolic functions of PTH [18, 19]. In addition, Hurley et.al showed that bone anabolic action of PTH in human was associated with an increased serum level of FGF-2 [20]. FGF-2 is another important regulator of osteoblast differentiation and bone anabolic metabolism. Intermittent PTH treatment increased FGF-2 production in osteoblasts, furthermore, the bone anabolic action of PTH was blunted in Fgf2-/- null mice [21]. These observations suggest that FGF-2 signaling is essential for the bone anabolic actions of PTH. FGFR3 as one of the major receptor binding with FGF-2 in osteoblasts, both gain- or loss-of-function of which lead to abnormal bone metabolism, indicating that FGFR3 may play critical role in bone remodeling [22, 23]. Amizuka et al. reported that signaling by FGFR3 and PTHrP coordinate to affect cartilage and bone development [24]. Hence, we hypothesized that FGFR3 may play a role in regulating the anabolic actions of PTH on bone.
Until now, there is no direct genetic evidence about the function of FGFR3 signaling in the anabolic effects of PTH on skeleton. Whether or not and to what extent the bone anabolic action of intermittent PTH depends on FGFR3 signaling have not been reported. To test our hypothesis, we use wild-type and Fgfr3-deficient mice in this study to evaluate bone microstructure and cellular response upon intermittent PTH treatment.
Materials and Methods
Animals and PTH treatment
Fgfr3 knock out (Fgfr3-/-) mice were maintained on C3H background and the genotyping was conducted as described previously [25]. All experiments were performed in accordance with protocols approved by the Institutional Animals Care and Use Committee of Daping Hospital (Chongqing, China). Two-month-old or 4-month-old male Fgfr3-/- and wide-type mice from the same litters were intraperitoneally injected with rhPTH1-34 (80μg/kg, Anaspec, Fremont, CA) [21] or vehicle (0.9% saline, 0.01 mM β-mercaptoethanol, 0.1% acetic acid) once a day for 4 weeks. Mouse body weight was measured every seven days. For dynamic bone formation analysis, calcein (20 mg/kg body weight, Sigma, St. Louis, MO) was intraperitoneally injected at day 10 and day 3 before tissue collection. At the end of experiment, femora and tibiae were dissected for BMD assessment, micro-computed tomography (μCT) analysis and histological examination.
Measurement of bone mineral density
Right femora were dissected free of soft tissues and fixed in 90% ethanol. High-resolution X-ray images of these femora were obtained using a Faxitron MX20 [23]. BMD was measured by dual energy X-ray absorptiometry (DEXA; PIXimus Mouse 11 densitometer, GE Medical System, Madison, WI).
μCT scanning and analysis
Femora from Fgfr3-/- and WT mice were scanned with a Skyscan 1072 μCT instrument and analyzed for bone structure. Briefly, serial 9-μm tomographic images were acquired at the condition of 70 kV and 113 mA. Constant thresholds were performed in binary images to segment bone from marrow and soft tissues. Lower and upper grey thresholds were 66 and 170 for trabecular compartment whereas those of cortical compartment were 66 and 123, respectively. Region of interest (ROI) in trabecular bone was located at 30 slides (270 μm) below the lowest point of growth plate and extended for 900 μm width. Trabecular morphometry parameters analyzed included the bone volume fraction (BV/TV), trabecular thickness (Tb.Th), trabecular number (Tb.N), trabecular spacing (Tb.Sp), and structure model index (SMI). Cortical ROI was selected from femoral midshaft and extended for 200 slides (1800 μm) thickness. Average cortical area and cortical thickness were used to analyze cortical morphometry.
Bone histology and histomorphometry
Tibiae and femora were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer at 4°C overnight, decalcified in 0.5M EDTA-PBS for 10 days and embedded with paraffin. Five μm thick sections were stained with tartrate-resistant acid phosphatase (TRAP), which was performed to evaluate the osteoclast activity and bone resorption. For tissues to be used for dynamic bone formation analysis, after dehydration, the undecalcified tibiae were embedded in methylmethacrylate and 10 μm and 5 μm sections were cut and stained with Toluidine blue for histomorphometric analyses by OsteoMeasure (OsteoMetrics, Inc.) bone histomorphometric analysis software. The regions of interest for trabecular bone data collection were measured in an area 1.0 mm in length from 0.25 mm below the growth plate of the proximal tibiae.
Femoral biomechanical testing
Mechanical properties of femora were examined by a three-point bending test according to the protocol described previously [26]. Briefly, femoral specimens from 4-month-old mice were rehydrated at room temperature in phosphate buffered saline (PBS) and analyzed at the right femur midshaft using a mechanical testing machine (Instron Model 5865, Instron, Melbourne, Australia) with a displacement rate of 6 mm/min (span length = 8 mm) until broken. Whole femur mechanical parameters of maximum load, stiffness, Yang's elastic modulus were recorded by load-deflection diagrams.
Primary cell cultures and PTH exposure
Bone marrow cells were flushed from tibiae and femora from 6-8 week-old mice and cultured at a density of 106 cells/well in 6-well plates up to 21 days for osteoblast differentiation assay [23]. In addition, primary calvarial osteoblasts from newborn mice were digested serially with 0.1% collagenase I (Gibco, Life Technologies, Carlsbad, CA) as previously described [26]. Cells were cultured with α-MEM/10% FBS until confluent and then re-plated for experiments. Osteogenic culture medium was α-MEM with 10% FBS, 50mg/ml ascorbic acid, 10mM β-glycerophosphate, 10-8M dexamethasone (Sigma), 100U/ml penicillin, and 50mg/ml streptomycin. To mimic intermittent exposure to PTH, cells were treated with 10nM PTH1-34 or vehicle during the first 6 hours and then treated with vehicle in the subsequent 42 hours in a 48-hour cycle [27].
ALP staining and Alizarin red staining
Alkaline phosphatase (ALP) staining was used to examine osteoblastic differentiation as previously described [26]. Briefly, primary cells were fixed with 4% paraformaldehyde for 15min and stained with ALP detection solution (Sigma) for 30min at 37°C. For mineralization analysis, cells were induced for osteogenesis as above up to 21 days, then washed with PBS, fixed in 75% ethanol for 15min at 4°C, and stained with 1% alizarin red in 2% ethanol for 30min. Cells were then washed with water, and dried on 37°C.
mRNA expression analysis by Real-time PCR
Primary calvarial osteoblasts from Fgfr3-/- or WT mice were plated in 6-well plates and grown in osteogenic culture medium for 7 days. Cells were serum deprived for 24 h, followed by being treated with 10nM PTH1-34 or vehicle for 6 hours. After wash, osteoblasts were processed for total RNA extraction using the TRizol reagent (Invitrogen, Life Technologies, Carlsbad, CA) according to the manufacturer's instructions. Total RNA was reverse transcribed to cDNA using PrimerScript RT reagent kit (TaKaRa, Dalian, China). Quantitative PCR was performed to analyze relative gene abundance in a Mx3000P PCR machine (Stratagene, Agilent Technologies, Santa Clara, CA) with the Two-Step QuantiTect SYBR Green RT-PCR Kit (Takara). The relative abundance of osteogenic transcription factor Cbfa1, bone matrix proteins osteocalcin (OC) and osteopontin (OP), and IGF-1 as well as internal control Cyclophillin A were analyzed using primers published previously [23]. The data were analyzed with 2-△△CT method [28], where threshold cycle (CT) values of target genes were calculated and averaged from triplicate runs, and calibrated in relation to CT values of Cyclophillin A. Levels of gene expression (fold changes) are presented in relation to expression levels of vehicle-treated control of WT osteoblasts.
Western blot analysis
Primary osteoblasts were plated on 60-mm plates (1×106 per plate) and cultured in osteogenic induction medium. After fully confluence, osteoblasts were starved overnight with serum free medium and then stimulated with 10nM PTH1-34 or vehicle for 6 hours. Total cell proteins were extracted with RIPA buffer containing a protease inhibitors cocktail (Roche). Protein samples (30 μg each sample) were loaded on 10% SDS-PAGE gels and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore). Primary antibodies used were p-Erk1/2 and Total Erk1/2 (Cell Signaling).
Statistical analysis
Results are expressed as mean ± SD and analysed by 2-way analysis of variance (ANOVA) using PASW 17.0, assessing the individual effects of PTH treatment and of the Fgfr3 deficiency alleles, as well as their 2-way interaction. When significant levels (P<0.05) were achieved, Tukey's Post Hoc test was performed.
Results
Effects of PTH1-34 treatment on bone mineral density and bone microstructure in WT and Fgfr3-deficient mice
To determine whether FGFR3 signaling modulates PTH function in vivo, male WT and Fgfr3-/- mice were intraperitoneally injected with vehicle or PTH1-34once a day over 4 weeks. Since the phenotype of bone abnormalities of Fgfr3-/- mice was evident as early as 2-month-old and worsened by 4-month-old and bone remodeling is dominant at 4 months of age [22], we simultaneously administrated with intermittent PTH in these two age groups over 4 weeks period. No significant increases in body weight gain and femur length were observed in both WT and Fgfr3-/- mice and in these two age groups between PTH1-34-treated and vehicle control (data not shown).
Effects of intermittent PTH treatment on femoral trabecular and cortical bone mineral density (BMD) from Fgfr3-/- and wild-type (WT) mice. (A) Radiographic images of femurs in WT and Fgfr3-/- mice treated with intermittent PTH (+) or vehicle (-) at the age of 4-month-old group (A). (B-D) BMDs of femurs from 4-month-old group were assessed by dual-energy X-ray absorptiometry (DEXA) in the whole bone (B), and quantified separately in trabecular bone (C) and cortical bone (D). Data are means ± SD of 7-9 animals/group. Results of 2-way ANOVA are shown above the graphs.
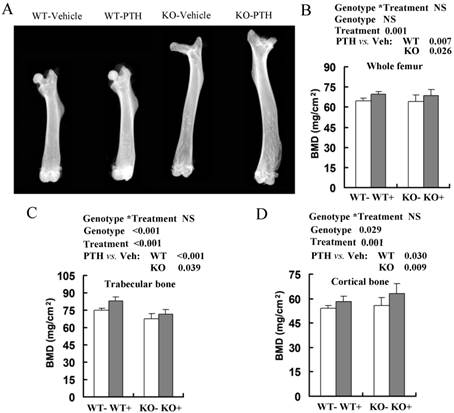
We then analyzed anabolic response in bone by comparing the femoral bone mineral density (BMD) between PTH-treated and vehicle-treated mice by DEXA (Fig 1A). Total BMD, trabecular BMD and cortical BMD were higher in both WT and Fgfr3-/- mice injected with PTH at 2-month-old (data not shown) and 4-month-old groups (Fig 1B-D). Intermittent PTH treatment induced similar percentage increases in total femoral BMD in WT mice (69.5 ± 3.0 mg/cm2 versus 64.8 ± 2.9 mg/cm2, 7% increase), and Fgfr3-/- mice (68.7 ± 3.5 mg/cm2 versus 64.1 ± 3.5 mg/cm2, 7% increase) at 4 month old group (Fig 1B). PTH stimulation also produced a substantial increase in trabecular BMD and cortical BMD in WT and Fgfr3-/- mice compared with vehicle-treated control (Fig 1C, D). The only difference between PTH-treated WT and Fgfr3-/- mice was a slightly more pronounced increase in femoral cortical BMD in Fgfr3 KO compared with WT mice (Fig 1D).
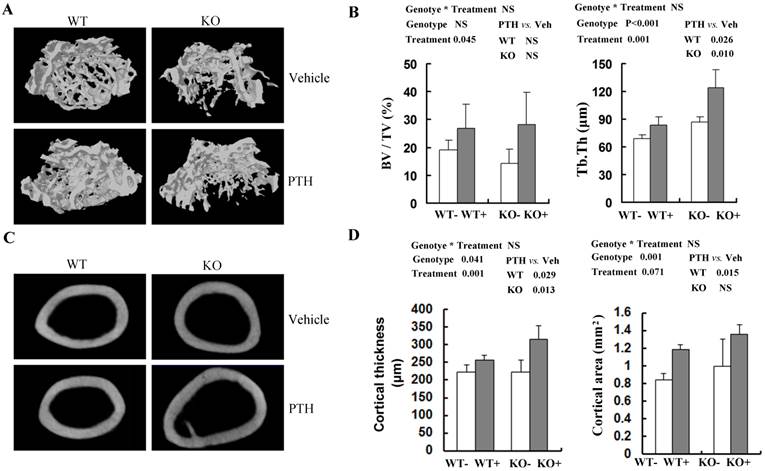
To obtain more accurate information about bone architecture, we further performed 3D μCT analysis on cancellous bone and cortical bone in femora (Fig 2A, C). In trabecular bone, bone volume (BV/TV) tended (P=0.045) to be higher in PTH-treated mice compared with vehicle-treated mice at 4 month old group, irrespective of genotype (Fig 2B). However, no significant increase of trabecular BV/TV with PTH treatment was detected in distal femur in either WT or Fgfr3-/- mice respectively (Fig 2B). In both genotypes, trabecular thickness (Tb.Th) was higher (P=0.001) in PTH-treated than that in vehicle-treated animals (Fig 2B). At 4-month-old group, trabecular separation (Tb.Sp) and structure model index (SMI) were decreased after PTH treatment, again irrespective of genotype (Table S2). In addtion, PTH treatment increased trabecular number (Th.N) significantly at 2-month-old group in both genotypes (Table S1). Increased trabecular bone volume (BV/TV) in femora from PTH treated mice was also observed at the 2-month-old group, of both genotype (P<0.05, Tables S1). In cortical bone, PTH produced a substantial increase in cortical thickness at femoral mid-shaft in Fgfr3-/- mice (223.5 ± 15.7 μm in untreated versus 315.1 ± 39.0 μm in treated) and WT mice (223.2 ± 18.9 μm in untreated versus 256.1 ± 13.3 μm in treated) at 4-month-old group (Fig 2D). However, no remarkable change in cortical bone area with PTH treatment was detected at femoral mid-shaft irrespective genotypes (Fig 2D). These results suggest that the absence of FGFR3 signaling does not attenuate the skeletal response to the anabolic effects of PTH on cancellous and cortical bone.
Effects of intermittent PTH treatment on bone biomechanical properties in WT and FGFR3-deficient mice
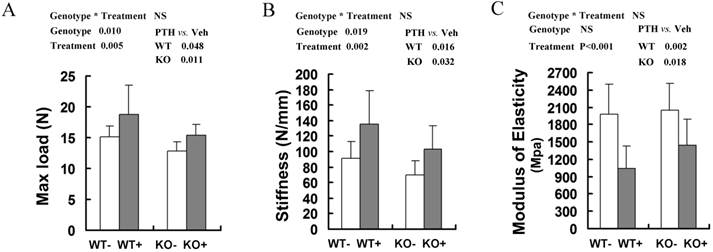
The bone mechanical properties are largely determined by bone mass and architecture. To investigate changes in the biomechanical strength and resilience of long bones in the WT and Fgfr3-/- mice following intermittent PTH treatment, right femora from 4-month-old group mice were performed on three-point bending test, in which the femoral mid-shaft was deflected to failure by a loaded bar from above. There were a lower maximum load to failure and a lower stiffness in the vehicle-treated femora from Fgfr3-/- mice compared to the vehicle-treated WT controls (P<0.05 for both, Fig 3A, B). All of these data were consistent with the results reported previously [22]. After PTH treatment, there were significantly increased maximum load and stiffness in WT and Fgfr3-deficient bone (P<0.05). In addition, elasticity of femora in both genotypes was decreased following PTH treatment (both at P<0.05) (Fig 3C), which was consistent with a previous report that high dose (40μg/kg) PTH treatment reduced elasticity of femurs [29]. These results demonstrated that PTH administration improved bone strength in WT and Fgfr3-/- mice, suggesting FGFR3 signaling deficiency does not impair the bone mechanical properties enhanced by PTH treatment.
Effects of PTH treatment on femoral bone microstructure, cortical bone and bone parameters analyzed by μCT in the WT and Fgfr3-/- mice. (A) Representative μCT 3-dimensional images of femoral trabeculae from 4-month-old group after 4 weeks' intermittent administration of PTH or vehicle. (B) Bone volume (BV/TV) and trabecular thickness (Tb.Th) in distal femoral cancellous bone. (C) Two-dimensional cortical images of the mid-shaft of femora from control and Fgfr3-deficient mice of the 4-month-old treatment group. (D) Cortical thickness and cortical bone area of the distal femoral metaphysis upon PTH treatment. Data are means ± SD of 5 animals / group. Results of 2-way ANOVA are shown above the graphs.
Effects of intermittent PTH treatment on biomechanical properties of femoral bones in WT and Fgfr3-/- mice. Whole bone mechanical properties of femoral diaphyses were assessed by three-point bending test with test parameters including max load (A), stiffness (B), and modulus of elasticity (C). Data are means ± SD of 7-9 animals/group. Results of 2-way ANOVA are shown above the graphs.
Effects of PTH1-34 treatment on bone histomorphometric parameters and expression of osteogenesis-related genes in WT and Fgfr3-deficient mice
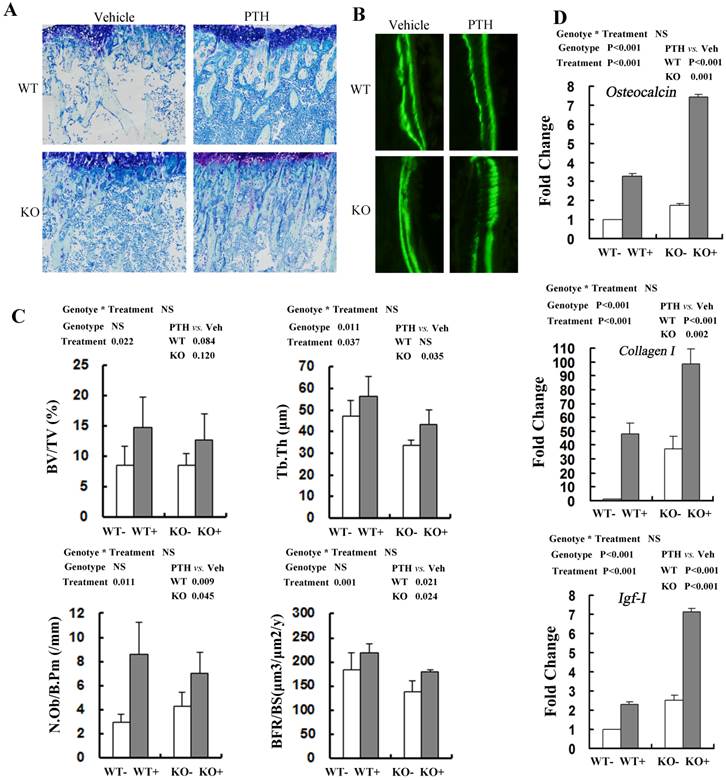
Consistent with the PTH-induced increase in distal femoral BV/TV by μCT, histomorphometric analysis of undecalcified sections stained with toluidine blue demonstrated that trabecular bone volume (BV/TV) in tibia treated with PTH was increased significantly compared with that of vehicle control, irrespective of genotypes (Fig 4A, C). PTH treatment increased trabecular BV/TV in both WT and Fgfr3-/- mice, although the effect did not reach the statistical level (Fig 4C). Trabecular thickness (Tb.Th) was also higher in mice treated with PTH than that in vehicle-treated animals, again irrespective of genotype (Fig 4C). Based on the double calcein labeling, PTH treatment resulted in increased trabecular bone formation (BFR/BS) in WT and Fgfr3-/- mice (Fig 4B, C). Administration of PTH also caused increased the number of osteoblasts/bone perimeter (N.Ob/B.Pm) (P=0.011) in WT and Fgfr3-/- mice (Fig 4C). We next explored the effects of FGFR3 disruption on PTH induction of osteobalst function-associated genes in vivo by real-time PCR assay. PTH treatment significantly increased the expressions of these genes including differentiation marker Osteocalcin (P<0.001, Fig 4D), extracellular matrix Collagen-I (P<0.05, Fig 4D), and local growth factor IGF-I (P<0.001, Fig 4D) in WT and Fgfr3-/- bone. These results further indicate that FGFR3 functions are not required for osteoanabolic actions of PTH.
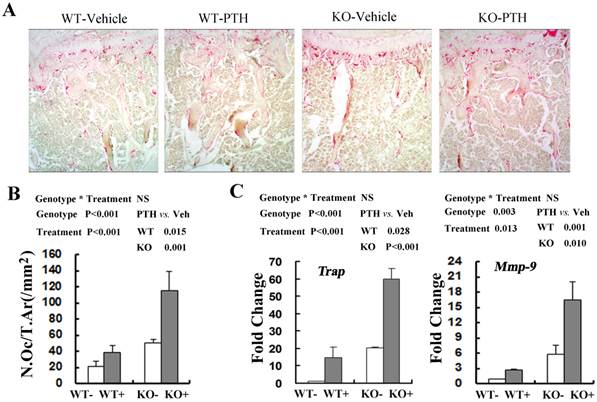
Since bone homeostasis was determined by osteoblast-mediated bone formation and osteoclast-mediated bone resorption, we then explored whether osteoclast activity was changed following PTH treatment by performing tartrate-resistant acid phosphatase (TRAP) staining in the tibiae from WT and Fgfr3-/- mice at the 4-month old group(Fig 5A). The number of TRAP staining-positive osteoclasts (N.Oc/T.Ar) was significantly elevated in treated WT and Fgfr3-/- mice when compared with vehicle control (Fig 5A, B). Real-time PCR quantification showed that basal expression of TRAP (20.3-fold) and MMP-9 (5.3 fold) in the bone tissue was higher in Fgfr3-/- mice than that in WT mice not receiving PTH treatment (Fig 5C). Consistent with histomorphormetric analysis on osteoclasts, intermittent PTH treatment induced a substantial increase in expression of Trap and Mmp-9 in WT and Fgfr3 KO bone (Fig 5C). Our results revealed that intermittent PTH treatment stimulates bone formation and bone resorption in WT and Fgfr3 KO bone.
Effects of intermittent PTH treatment on osteoblast differentiation are maintained in the absence of FGFR3 signaling
Our finding of nearly unchanged skeletal response to PTH in Fgfr3-/- mice led us to investigate the response of primary cultures from Fgfr3-/- mice upon PTH exposure in vitro. We isolated calvarial osteoblasts and bone marrow stromal cells (BMSCs) from Fgfr3-/- and WT mice and analysed the changes of their proliferation and differentiation following PTH1-34 treatment. In order to mimic intermittent exposure to PTH, calvarial osteoblasts and BMSCs were treated with 10nM PTH1-34 or vehicle during the first 6 hours and then treated with vehicle in the subsequent 42 hours in a 48-hour cycle as described [27]. MTT assay showed that PTH treatment increased the proliferation of osteoblasts from WT and Fgfr3-/- mice, and the extents of increase in both genotypes were comparable (data not shown).
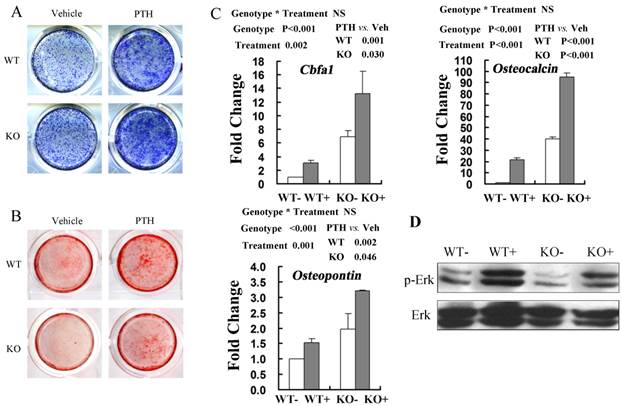
ALP staining was performed in the cultures of MSCs after osteogenic induction for 6 days with PTH or vehicle treatment. These results showed that PTH treatment stimulated osteogenic differentiation of BMSCs in both WT and FGFR3 KO culture (Fig 6A). Alizarin Red staining showed that the impaired mineralization in Fgfr3-deficient cells was partially restored by PTH1-34 treatment and exposure of PTH increased numbers of mineralized bone nodules in both WT and FGFR3 KO culture (Fig 6B). Real-time PCR analysis also showed that PTH exposure increased expressions of osteoblastic differentiation markers Cbfa1, Osteopontin and Osteocalcin in primary Fgfr3-deficient and WT osteoblasts over 6-day's osteogenic induction (Fig 6C). Western blot showed that decreased basal level of phosphorylation of extracellular signal-regulated kinase 1/2 (p-Erk1/2) in osteobalsts from Fgfr3-deficient mice as compared with that in WT. Interestingly, treatment of Fgfr3-deficient osteoblasts with PTH induced a clear activation of Erk1/2 that was similar with that observed in control osteobalsts when compared with basal levels (Fig 6D). These results suggest that primary Fgfr3-deficient culture maintained normal response to osteoanabolic actions of PTH.
Effects of PTH (+) or vehicle (-) treatment on trabecular bone formation in WT and Fgfr3-/- tibiae. (A) Representative images of undecalcified sections stained with Toluidine blue in the tibiae from WT and Fgfr3-/- mice treated with PTH or vehicle at the 4-month-old group (Original magnification, 100×). (B) Representative images of double calcein labeling in the trabecular bone at the 4-month-old group by fluorescence microscopy. Original magnification, 400×. (C) Quantification of trabecular bone volume fraction (BV/TV), trabecular thickness (Tb.Th), osteoblast number per bone perimeter (N.Ob/B.Pm) and bone formation rate (BFR/BS) in the proximal tibias in WT and KO mice was determined by OsteoMeasure image-analysis software. (D) Real-time RT-PCR mRNA expression analyses were performed for evaluating expression of Osteocalcin Collagen-I, and IGF-I in bone tissue of WT and Fgfr3-/- mice after PTH (+) or vehicle (-) treatment at the 4-month-old group and resultes were expressed as fold changes relative to expression level of vehicle-treated WT control (WT-). The real-time RT-PCR analysis was repeated for three times. Results of 2-way ANOVA are shown above the graphs.
Effects of PTH (+) or vehicle (-) treatment on trabecular bone resorption in WT and Fgfr3-/- mice. (A) Representative images of TRAP staining in the tibia from WT and Fgfr3-/- mice upon PTH treatment at the 4-month-old group, Original magnification, 100×. (B) Bone resorption parameters N.Oc/T.Ar were quantified by OsteoMeasure image-analysis software. (C) Real-time RT-PCR mRNA expression analyses were conducted for osteoclasts-related genes Trap and Mmp-9 in tibia from WT and Fgfr3-/- mice treated with PTH (+) or vehicle (-) at the 4-month-old group and data are expressed as fold changes relative to expression level of vehicle-treated WT control (WT-). Results of 2-way ANOVA are shown above the graphs.
Anabolic response to intermittent PTH treatment in primary calvarial osteoblasts and bone marrow stromal cells from WT and Fgfr3-/- mice. (A) Alkaline phosphatase (ALP) staining performed on primary bone marrow stromal cells isolated from WT and Fgfr3-/- mice induced for osteogenesis with PTH or Vehicle for 6 days. (B) Alizarin Red-stained mineralization potential of primary bone marrow cultures induced for osteogenesis with PTH or not for 21 days. (C) Effects of intermittent PTH treatment on Cbfa1, Osteopontin and Osteocalcin mRNA levels in primary calvarial osteoblasts from WT and Fgfr3-/- mice after being cultured under osteogenic condition and treated with intermittent PTH (10nM) or vehicle for 6 days. Expression was expressed as fold changes normalized to the vehicle-treated WT control. (D) Western blot analysis showing the levels of phosphor-Erk1/2 and total Erk1/2 after PTH treatment in WT and FGFR3-deficient calvarial osteobalsts. Results of 2-way ANOVA are shown above the graphs.
Discussion
Previous studies have shown that FGF-2 signaling contributes to the bone anabolic effects of intermittent PTH treatment; however, the underlying mechanisms have remained unclear. Both FGFR3 deficient mice and mice mimicking ACH show defective bone mineralization and osteopenia, which suggests that FGFR3 plays important role in bone homeostasis [22, 23]. While FGFR3 as one of FGF-2 receptor known to be a negative regulator for bone growth and an important regulator in bone remodeling, its roles in mediating PTH anabolic effects are unknown. In the current study, by examining the effects of Fgfr3 gene knockout and PTH treatment on skeleton, we found that intermittent PTH treatment improved the BMD, cortical thickness, bone formation and bone resorption in both Fgfr3-/- and wild-type mice, suggesting that mice lacking FGFR3 maintained normal abilities to response to osteoanabolic actions of intermittent PTH on skeleton.
Previous studies showed that FGF signal crosstalked with PTH signals. PTH treatment leads to increased expression of FGF-2, FGFR1 and FGFR2 in pre-osteoblast cell lines and primary cells [16], and endogenous FGF-2 is essential for the anabolic effects of PTH on bone homeostasis [30]. We and others have found that mice with FGFR3 activating mutations exhibit enhanced expressions of osteogenic differentiation makers decreased expression of PTHrP receptor, and that overexpression of FGFR3 in chondrocytes down-regulates PTH receptor and PTHrP expression [31-33]. FGFR3 inhibits expression of PTH/PTHrP receptor via JAK/STAT signaling in chondrocytes and FGFR3 appears to be an inhibitor in osteoid synthesis and early osteoblastic differentiation [22, 31]. However, whether FGFR3 signaling plays a role in PTH's anabolic effects on bone formation is not reported before. Thus, the major issue we addressed in this work was to evaluate the role, if any, of FGFR3 signaling in the bone anabolic actions of PTH. However, no significant difference in the skeleton response to intermittent PTH treatment was observed between WT and Fgfr3-/- mice. In addition, the stimulatory effects of PTH treatment on osteoblastic proliferation, differentiation and expression of osteogenesis-related genes were maintained in the Fgfr3 deficient mice. Furthermore, we also found that PTH stimulated bone anabolic action in the adult mice with FGFR3 gain-of-function mutation and wild-type mice[34]. Thus, FGFR3 signaling seems to be dispensable for the anabolic effects of PTH on bone.
Our results revealed that skeletal response to intermittent PTH treatment was characterized with increased osteoblastic bone formation and osteocalstic bone resorption in both Fgfr3-/- and WT mice. Similarly, it has been reported that intermittent PTH treatment increases bone formation and bone resorption in both wild-type and Lrp5-/- or BSP-/- mice[8, 35]. Although PTH-stimulated bone formation is partially antagonized by increased bone resorption, the net effects of intermittent PTH treatment in both WT and Fgfr3-/- mice is an improvement in BMD and bone strength. PTH treatment improves bone quality of femur in an osteoporosis rat model and enhancement of femoral bone strength stimulated by PTH in mice is associated with increased BMD [36, 37]. In our study, PTH induced increased BMD and cortical thickness may contribute to bone mechanical property of resistance to fracture. Our results demonstrated that the bone strength following intermittent PTH was improved significantly in WT and Fgfr3-/- mice.
Previous studies have showed that a number of genes play an important role in the osteoanabolic action of PTH with the help of geneticly modified mice. Intermittent PTH treatment stimulated bone anabolic action was impaired in IGF-1, FGF-2, or ATF4 null mice, while Cox-2 deficiency enhanced bone anabolic response to PTH stimulation [7, 10, 21, 38]. However, there are some genes are not required for the PTH anabolic action on bone. Dkk-1 overexpression mice, Waved-5 mice (carring a point mutation in the kinase domain of the epidermal growth factor receptor gene), low-density lipoprotein receptor-related protein 5 (lrp5), FGF-23 and Klotho deficient mice are reported to have normal skeletal response to the anabolic effects of intermittent PTH [19, 35, 39, 40]. To our surprise, mice lacking Fgfr3 maintained nearly normal response to the anabolic effects of PTH. In contrast, earlier studies indicated that FGF signal is a potentially important mediator of the bone stimulatory effects of PTH [16, 21]. The explanation for this disagreement may be that 1) FGFR3 signaling actually is not involved in the bone anabolic action of PTH. 2) Other components of FGF receptor family might compensate for the absence of FGFR3 activity. There are four members of FGF receptor (FGFR1-4), and these components may be redundant to some degree and thus functionally compensate for the absence of FGFR3. Therefore, to clarify the function of FGF signal in the bone anabolic actions of PTH, the role of other member of FGFs/FGFRs in mediating intermittent PTH action on bone need to be further investigated.
In summary, our study revealed that the anabolic effects of intermittent PTH treatment were not impaired in Fgfr3-/- mice compared with that observed in WT mice. The results obtained from our in vivo and in vitro studies definitely showed that bone anabolic effects of PTH were not blocked by the absence of FGFR3, suggesting that the FGFR3 signaling may not be required for the stimulatory effects of PTH on bone, and PTH can be used to improve the bone quality of patients with mutations of FGFR3, such as ACH.
Abbreviations
FGFR, fibroblast growth factor receptor; BMD, bone mineral density; μCT, micro-computed tomography; PTH, parathyroid hormone; WT, wild-type; TRAP, tartrate-resistant acid phosphatase.
Supplementary Material
Tables S1-S2.
Acknowledgements
The authors thank Professor Xizheng Zhang and associate professor Ruixin Li of the Institute of Medical Equipment, Academy of Tianjin Medical Science for kindly providing femoral three-point bending test service. This project was funded in part by grants from Major State Basic Research Program of China (973 Program, grant numbers: 2014CB 942904), National Natural Science Foundation of China (grant numbers: 81301593, 81170809), and Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT) (IRT1216).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jilka RL, Weinstein RS, Bellido T, Roberson P, Parfitt AM, Manolagas SC. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J Clin Invest. 1999;104:439-46
2. Karaplis AC, Goltzman D. PTH and PTHrP effects on the skeleton. Reviews in endocrine & metabolic disorders. 2000;1:331-41
3. Dobnig H, Turner RT. Evidence that intermittent treatment with parathyroid hormone increases bone formation in adult rats by activation of bone lining cells. Endocrinology. 1995;136:3632-8
4. Orwoll ES, Scheele WH, Paul S, Adami S, Syversen U, Diez-Perez A. et al. The effect of teriparatide [human parathyroid hormone (1-34)] therapy on bone density in men with osteoporosis. J Bone Miner Res. 2003;18:9-17
5. Wu X, Pang L, Lei W, Lu W, Li J, Li Z. et al. Inhibition of Sca-1-positive skeletal stem cell recruitment by alendronate blunts the anabolic effects of parathyroid hormone on bone remodeling. Cell Stem Cell. 2010;7:571-80
6. Jobert AS, Zhang P, Couvineau A, Bonaventure J, Roume J, Le Merrer M. et al. Absence of functional receptors for parathyroid hormone and parathyroid hormone-related peptide in Blomstrand chondrodysplasia. J Clin Invest. 1998;102:34-40
7. Miyakoshi N, Kasukawa Y, Linkhart TA, Baylink DJ, Mohan S. Evidence that anabolic effects of PTH on bone require IGF-I in growing mice. Endocrinology. 2001;142:4349-56
8. Wade-Gueye NM, Boudiffa M, Laroche N, Vanden-Bossche A, Fournier C, Aubin JE. et al. Mice lacking bone sialoprotein (BSP) lose bone after ovariectomy and display skeletal site-specific response to intermittent PTH treatment. Endocrinology. 2010;151:5103-13
9. Kramer I, Loots GG, Studer A, Keller H, Kneissel M. Parathyroid hormone (PTH)-induced bone gain is blunted in SOST overexpressing and deficient mice. J Bone Miner Res. 2010;25:178-89
10. Xu M, Choudhary S, Voznesensky O, Gao Q, Adams D, Diaz-Doran V. et al. Basal bone phenotype and increased anabolic responses to intermittent parathyroid hormone in healthy male COX-2 knockout mice. Bone. 2010;47:341-52
11. Bianchi EN, Ferrari SL. Beta-arrestin2 regulates parathyroid hormone effects on a p38 MAPK and NFkappaB gene expression network in osteoblasts. Bone. 2009;45:716-25
12. Ishizuya T, Yokose S, Hori M, Noda T, Suda T, Yoshiki S. et al. Parathyroid hormone exerts disparate effects on osteoblast differentiation depending on exposure time in rat osteoblastic cells. J Clin Invest. 1997;99:2961-70
13. Yu B, Zhao X, Yang C, Crane J, Xian L, Lu W. et al. Parathyroid hormone induces differentiation of mesenchymal stromal/stem cells by enhancing bone morphogenetic protein signaling. J Bone Miner Res. 2012;27:2001-14
14. Canalis E, Centrella M, Burch W, McCarthy TL. Insulin-like growth factor I mediates selective anabolic effects of parathyroid hormone in bone cultures. J Clin Invest. 1989;83:60-5
15. Pfeilschifter J, Laukhuf F, Muller-Beckmann B, Blum WF, Pfister T, Ziegler R. Parathyroid hormone increases the concentration of insulin-like growth factor-I and transforming growth factor beta 1 in rat bone. J Clin Invest. 1995;96:767-74
16. Hurley MM, Tetradis S, Huang YF, Hock J, Kream BE, Raisz LG. et al. Parathyroid hormone regulates the expression of fibroblast growth factor-2 mRNA and fibroblast growth factor receptor mRNA in osteoblastic cells. J Bone Miner Res. 1999;14:776-83
17. Qiu T, Wu X, Zhang F, Clemens TL, Wan M, Cao X. TGF-beta type II receptor phosphorylates PTH receptor to integrate bone remodelling signalling. Nature cell biology. 2010;12:224-34
18. Burnett-Bowie SM, Henao MP, Dere ME, Lee H, Leder BZ. Effects of hPTH(1-34) infusion on circulating serum phosphate, 1,25-dihydroxyvitamin D, and FGF23 levels in healthy men. J Bone Miner Res. 2009;24:1681-5
19. Yuan Q, Sato T, Densmore M, Saito H, Schuler C, Erben RG. et al. FGF-23/Klotho signaling is not essential for the phosphaturic and anabolic functions of PTH. J Bone Miner Res. 2011;26:2026-35
20. Hurley M, Yao W, Lane NE. Changes in serum fibroblast growth factor 2 in patients with glucocorticoid-induced osteoporosis treated with human parathyroid hormone (1-34). Osteoporos Int. 2005;16:2080-4
21. Hurley MM, Okada Y, Xiao L, Tanaka Y, Ito M, Okimoto N. et al. Impaired bone anabolic response to parathyroid hormone in Fgf2-/- and Fgf2+/- mice. Biochem Biophys Res Commun. 2006;341:989-94
22. Valverde-Franco G, Liu H, Davidson D, Chai S, Valderrama-Carvajal H, Goltzman D. et al. Defective bone mineralization and osteopenia in young adult FGFR3-/- mice. Human molecular genetics. 2004;13:271-84
23. Su N, Sun Q, Li C, Lu X, Qi H, Chen S. et al. Gain-of-function mutation in FGFR3 in mice leads to decreased bone mass by affecting both osteoblastogenesis and osteoclastogenesis. Human molecular genetics. 2010;19:1199-210
24. Amizuka N, Davidson D, Liu H, Valverde-Franco G, Chai S, Maeda T. et al. Signalling by fibroblast growth factor receptor 3 and parathyroid hormone-related peptide coordinate cartilage and bone development. Bone. 2004;34:13-25
25. Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911-21
26. Weng T, Mao F, Wang Y, Sun Q, Li R, Yang G. et al. Osteoblastic molecular scaffold Gab1 is required for maintaining bone homeostasis. Journal of cell science. 2010;123:682-9
27. Sun ZF, Jiang H, Ye ZQ, Jia B, Zhang XL, Zhang KQ. Expression of Rho GDIalpha in rat osteoblasts intermittently exposed to parathyroid hormone in vitro and in vivo. Acta Pharmacol Sin. 2009;30:1001-7
28. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402-8
29. Hirano T, Burr DB, Turner CH, Sato M, Cain RL, Hock JM. Anabolic effects of human biosynthetic parathyroid hormone fragment (1-34), LY333334, on remodeling and mechanical properties of cortical bone in rabbits. J Bone Miner Res. 1999;14:536-45
30. Sabbieti MG, Agas D, Xiao L, Marchetti L, Coffin JD, Doetschman T. et al. Endogenous FGF-2 is critically important in PTH anabolic effects on bone. J Cell Physiol. 2009;219:143-51
31. Li M, Seki Y, Freitas PH, Nagata M, Kojima T, Sultana S. et al. FGFR3 down-regulates PTH/PTHrP receptor gene expression by mediating JAK/STAT signaling in chondrocytic cell line. Journal of electron microscopy. 2010;59:227-36
32. Chen L, Li C, Qiao W, Xu X, Deng C. A Ser(365)->Cys mutation of fibroblast growth factor receptor 3 in mouse downregulates Ihh/PTHrP signals and causes severe achondroplasia. Hum Mol Genet. 2001;10:457-65
33. Chen L, Adar R, Yang X, Monsonego EO, Li C, Hauschka PV. et al. Gly369Cys mutation in mouse FGFR3 causes achondroplasia by affecting both chondrogenesis and osteogenesis. The Journal of clinical investigation. 1999;104:1517-25
34. Xie Y, Su N, Jin M, Qi H, Yang J, Li C. et al. Intermittent PTH (1-34) injection rescues the retarded skeletal development and postnatal lethality of mice mimicking human achondroplasia and thanatophoric dysplasia. Hum Mol Genet. 2012;21:3941-55
35. Iwaniec UT, Wronski TJ, Liu J, Rivera MF, Arzaga RR, Hansen G. et al. PTH stimulates bone formation in mice deficient in Lrp5. J Bone Miner Res. 2007;22:394-402
36. Shahnazari M, Yao W, Wang B, Panganiban B, Ritchie RO, Hagar Y. et al. Differential maintenance of cortical and cancellous bone strength following discontinuation of bone-active agents. J Bone Miner Res. 2011;26:569-81
37. Iida-Klein A, Hughes C, Lu SS, Moreno A, Shen V, Dempster DW. et al. Effects of cyclic versus daily hPTH(1-34) regimens on bone strength in association with BMD, biochemical markers, and bone structure in mice. J Bone Miner Res. 2006;21:274-82
38. Yu S, Franceschi RT, Luo M, Fan J, Jiang D, Cao H. et al. Critical role of activating transcription factor 4 in the anabolic actions of parathyroid hormone in bone. PloS one. 2009;4:e7583
39. Yao GQ, Wu JJ, Troiano N, Insogna K. Targeted overexpression of Dkk1 in osteoblasts reduces bone mass but does not impair the anabolic response to intermittent PTH treatment in mice. J Bone Miner Metab. 2011;29:141-8
40. Schneider MR, Dahlhoff M, Andrukhova O, Grill J, Glosmann M, Schuler C. et al. Normal epidermal growth factor receptor signaling is dispensable for bone anabolic effects of parathyroid hormone. Bone. 2012;50:237-4
Author contact
![]() Corresponding authors: Lin Chen, PhD & MD, Center of Bone Metabolism and Repair, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing 400042, China. Tel: 86-23-68702991; Fax: 86-23-68702991; E-mail: linchen70com or Xiaolan Du, Center of Bone Metabolism and Repair, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing 400042, China. Tel: 86-23-68702991; Fax: 86-23-68702991; E-mail: DXL_xiaolancom
Corresponding authors: Lin Chen, PhD & MD, Center of Bone Metabolism and Repair, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing 400042, China. Tel: 86-23-68702991; Fax: 86-23-68702991; E-mail: linchen70com or Xiaolan Du, Center of Bone Metabolism and Repair, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing 400042, China. Tel: 86-23-68702991; Fax: 86-23-68702991; E-mail: DXL_xiaolancom
Received 2015-10-8
Accepted 2016-4-18
Published 2016-7-17