ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2016; 12(10):1181-1190. doi:10.7150/ijbs.16158 This issue Cite
Research Paper
Changes in the Expression of FUS/TLS in Spinal Cords of SOD1 G93A Transgenic Mice and Correlation with Motor-Neuron Degeneration
Jiao Li1*, Yi Lu1*, Huiting Liang1*, Chunyan Tang1*, Lei Zhu1*, Jie Zhang1,2, Renshi Xu1 ![]()
1. Department of Neurology, the First Affiliated Hospital of Nanchang University, Nanchang 330006, Jiangxi, China.
2. Department of Biochemistry and Molecular Biology, College of Basic Medical Science, Nanchang University, Nanchang 330006, Jiangxi, China.
* indicates equal contributors.
Abstract

In order to searching the possible pathogenesis of amyotrophic lateral sclerosis (ALS), we examined the expression and distribution of FUS/TLS protein in the different anatomic regions, segments and neural cells of adult spinal cord at the different stages of the SOD1 wild-type and G93A transgenic mice using the fluorescent immunohistochemistry. Result revealed that, in the SOD1 wild-type mice, the FUS/TLS expression almost wasn't detected. However, in the SOD1 G93A mice, the FUS/TLS expression in the white matter was significantly more than that in the gray matter. In the white matter, the FUS/TLS expression in the anterior funiculus was more than that in the lateral funiculus more than that in the posterior funiculus. In the gray matter, the FUS/TLS expression in the ventral horn was more than that surrounding the central canal more than that in the dorsal horn. The FUS/TLS expression in the thoracic segment was more than that in the cervical segment more than that in the lumbar segment. Almost all FUS/TLS expressed in the nuclear of the GFAP positive cell at the onset stage, but it expressed in both the nuclear and the cytoplasm of the GFAP positive cell at the progression stage, almost didn't detected FUS/TLS expression in the NeuN and Oligo positive cells. The FUS/TLS expression was positively correlated with the neuron death. Our data suggested that the expressive increase and mislocalization of FUS/TLS in the astrocyte cell might cause the motor neuron degenerative death in the SOD1 G93A transgenic mice.
Keywords: FUS/TLS, astrocyte cell, amyotrophic lateral sclerosis, spinal cord, pathogenesis.
Introduction
Amyotrophic lateral sclerosis (ALS) also is known as Lou Gehrig's disease or Charcot disease, is a relentlessly progressive neurodegenerative disorder that selectively damages motor neuron [1]. In the United Kingdom, the term of motor neuron disease (MND) is commonly used [2]. MND usually is the term of a disease group including four different subtypes, among them, ALS is the most common [3, 4]. The clinical manifestations of ALS are mainly characterized by the damage of up and lower motor neurons, show the progressive muscle powerlessness, stiffness, twitch and atrophy of limbs, and gradually spread to the muscle of whole body including the respiratory and laryngopharyngeal muscles, and the partial patients appear the hyperactivity of tendon reflexes and the positive of pathologic sign. At the late stage of ALS, patients often occur the difficulty of speaking, swallowing and breathing, ultimately die from the respiratory failure [1-4]. The pathogenesis of 90-95% ALS has not been known now [1-4]. Approximate 5-10% of ALS inherit from their ancestry [5]. Approximate half of these genetic ALS are associated with one of two specific genes, one is the mutation of Cu/Zn-superoxide dismutase (SOD1) on chromosome 21, another is a genetic abnormality of a hexanucleotide repeat in a region of C9orf72, they all cause the neuron death in ALS.
At present, the diagnosis of ALS mainly according to the clinical signs and symptoms, some clinical accessory examinations only are used to rule out the other similar diseases [1-4]. There have not been any effectively cured and preventive methods for ALS in the worldwide up to now [1-4], and there have not been any objective diagnostic technologies in the clinical accessory examinations. Still to now, the sole drug of riluzole is produced, and several clinical trials reported that it might extend the life expectancy of approximate two to three months [6]. The sporadic ALS usually occurs around the age of 60 years; the age of the inherited ALS generally is around 50 years [5]. The average survival time from onset to death is about 3-5 years [1]. No more than 10% ALS survive longer than 10 years [1-4]. In the most regions of the worldwide, the rate of ALS has not been unknown at present [5]. In the European and the United States, the morbidity of ALS is about 2 people per 100,000 per year [5, 7]. Although the prevalence rate of ALS is very low, the disease seriously damages the health of the middle and elderly populations, and largely shortens their life-span because of its significant disability and mortality. And up to now, a lot of questions about ALS including the etiology, pathogenesis, treatment, prevention, pathogens and so on are waiting for clarifying, especially in the pathogenesis of ALS, because the studied results about pathogenesis will provide the important evidence for the prevention and treatment of ALS. Therefore, the study about the pathogenesis of ALS is necessary to preventing and treating of ALS.
The pathologic features of ALS are the progressive death of motor neurons in the motor cortex of the cerebrum, the brain stem and/or the spinal cord. Prior to the destruction of motor neurons, the protein-rich inclusions generate in their cell bodies and axons of the motor neurons, which may be partly due to defect in the protein degradation [8]. These inclusions often contain ubiquitin, and generally also consist of one of the ALS-associated proteins like SOD1, TAR DNA binding protein (TDP-43, or TARDBP), and/or fused in sarcoma/translocated in liposarcoma (FUS, or FUS/TLS) [9-14]. Thus, may investigators suggest that the pathogenesis of ASL is closely associated with the abnormal expression, redistribution and dislocation of some special proteins.
The FUS/TLS gene was initially found to be a component of fusion pro-oncogene, which causes a chromosomal translocation in liposarcomas. FUS/TLS belongs to a member of RNA binding protein family, it consists of an N-terminal serine-tyrosine-glycine-glutamine (SYGQ) region and the RNA recognition motif flanked by the glycine rich regions, a cysteine (2)/cysteine (2) zinc finger motif and the multiple RGG repeats. The FUS/TLS interacts with RNA, the single and double stranded DNA, and exerts some special roles in the procession and transport of mRNA, the regulation of transcription and the maintenance of genomic stability. Recently, the mutations of FUS/TLS were found in some patients of ALS, and suggested that the altered metabolism of FUS/TLS RNA might play a role in the pathogenesis of ALS. Several mutations in the FUS/TLS gene have been detected in the patients of ALS yet. The mutant forms of FUS/TLS gene show the similar pathology with other ALS causative genes, including the aberrant cytoplasmic inclusions and an increased ratio between cytoplasm and nuclear of FUS/TLS [15-19].
FUS/TLS is a multiple functional protein participated in a wide range of cellular processes including the processing and transport of mRNA, the regulation of transcription and the maintenance of genomic stability. Although FUS/TLS is currently suggested to be related to the pathogenesis of ALS, its mechanisms exerted the role in the pathogenesis of ALS has not been clearly known, they need to further be studied. Here, we used the commonly used ALS animal model of SOD1 G93A transgenic mouse to identify the possible roles in the motor neuron degeneration potentially induced by the alteration of FUS/TLS expression, redistribution and dislocation in the pathogenesis of ALS. In this study, we confirmed that the expressive increase and mislocalization of FUS/TLS in the astrocyte cell might cause the progressive motor neuron degeneration in the SOD1 G93A transgenic mouse model of ALS.
Materials and methods
Study approval
All animal studies were conducted in accordance with the guide for the care and use of laboratory animals of China. All experiments involving animal were reviewed and approved by the ethics committee for animal care and use of the First Affiliated Hospital of Nanchang University, China.
Animals
The C57BL/6J SOD1 G93A transgenic mice [20] (Jackson laboratory, Bar Harbour, Maine) were maintained through mating the transgenic males with the C57BL/6J SOD1 wild-type mice in the neurological laboratory of the First Affiliated Hospital of Nanchang University. The SOD1 G93A transgenic mice were detected by the PCR of the genomic DNA derived from tail to identify whether or not the positive SOD1 G93A transgenic mice. The following primer sets were used: The reward-primer of hmSOD1 5'-CAT CAG CCC TAA TCC ATC TGA-3', the reverse-primer of hmSOD1 5'-CGC GAC TAA CAA TCA AAG TGA-3'; The forward-primer of IL-2 5'-CTA GGC CAC AGA ATT GAA AGA TCT-3', the reverse-primer of IL-2 5'-GTA GGT GGA AAT TCT AGC ATC ATC C-3'. The amplification conditions were: the degeneration of 94°C for 3 seconds, the annealing of 60°C for 1minute and the extension of 72°C for 1minute, the cycles for 35 times.
The disease stages were divided into three stages based on the days of disease duration, the pre-onset (60-70days), onset (90-100days) and progression stages (120-130days) [20, 21]. Animals were sacrificed at the time points of the pre-onset, onset and progression stages. The gastrocnemius muscles of abnormal limbs were taken to perform HE staining at the different stages, observed the changes of muscle structures in the light microscope, aimed to identify the paralytic scale of limb muscle and further decided the pre-onset, onset and progression stages of the disease courses. The above described experimental methods were the same with that in our previous published paper [22].
Fluorescent immunohistochemical staining of spinal cord
The SOD1 wild-type and G93A transgenic mice were anesthetized and perfused with 20 ml of 0.9% saline and 40 ml of 4% paraformaldehyde (PFA) in 1xPBS (pH 7.5) at room temperature (RT). The spinal cord was separated and put in 4% PFA buffer in 1xPBS (pH 7.5) overnight, then incubated in 20% sucrose in 1xPBS (pH 7.5) for 3 days, embedded using the optimum cutting temperature compound (OCT, Water soluble mixture of polyethylene glycol and polyvinyl alcohol). The tissues of spinal cord were coronally and successively cut the sections of 12 μm on a Leica cryostat and collected on the Superfrost Plus slides.
On the processes of the immunohistochemical staining, sections were permeabilized by 0.2% TritonX-100 and blocked by 10% goat serum in 1xPBS after rehydrated in 1xPBS (pH 7.4), followed with incubating with the following antibodies: FUS/TLS 1:100 (Santa cruz biotechnology Inc), NeuN 1:250, GFAP 1:1000 and Oligo-2 1:100 (Abcam Hong Kong Ltd.) at 4 °C overnight, then washed using 0.2% Triton X-100 in 1xPBS for 6 times each 5 minutes, incubated using the secondary antibody (Donkey anti goat, 1:250, donkey anti rabbit, 1:200) conjugated to fluorescein (Green) or/and rhodamine (Red) for 2 hours at RT, and DAPI stained (Blue), extensively washed 6 times each 5 minutes, mounted by the antifade medium, and observed under a Nikon E800 fluorescent microscope with a spot digital camera (Diagnostic Instruments, Sterling Heights, MI, USA) and Photoshop software (Adobe Systems, San Jose, CA, USA). The representative staining sections were taken photos. Multiple labeling fluorescent immunohistochemistry conjugated to anti-FUS/TLS, NeuN, GFAP and Oligo-2 antibodies was used to observe and analyze the alteration of FUS/TLS distribution and dislocation in the spinal cord of the SOD1 G93A transgenic mouse model of ALS.
Analysis of immunohistochemical positive cells
The fluorescent immunohistochemical positive cells was analyzed by counting the amounts of the positive cells in the different anatomic regions, segments and neural cells of spinal cord under 200 magnifications. 10 slices each mouse, 3 sections each slices, 3 mice per group. Calculated total amounts of positive cells of all sections, then total amounts of positive cells were divided by total number of section, the averaged amount was used for quantitatively analyzing.
Statistical Analysis
All experimental data were expressed using mean ± SD. Specific comparison between the control and the experimental individual was analyzed by ANOVA. The correlation between the FUS/TLS positive cells and the neuronal cells was analyzed by the Pearson Correlation of SPSS17. P<0.05 was considered as statistically significant.
Results
Identification of C57BL/6J G93A SOD1 transgenic mice
In our bred SOD1 G93A transgenic mice, we observed the characteristics of the typical ALS-like symptoms at the different time points of ALS pre-onset (60-70d), onset (90-100d) and progression (120-130d) stages, usually suffered from the strong gait impairment, the muscle atrophy and the reduced motor function which were consistent with the description of Gurney et al [20]. The pathological examination of abnormal limb muscle was the same with the description in our previous published paper [22].
Alteration of FUS/TLS expression in the adult spinal cord of the SOD1 wild-type and G93A transgenic mice
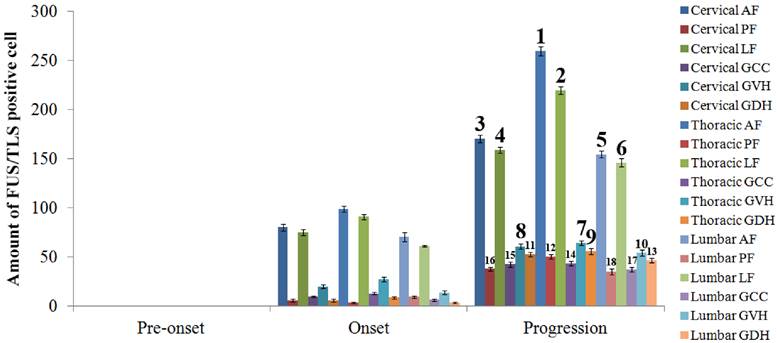
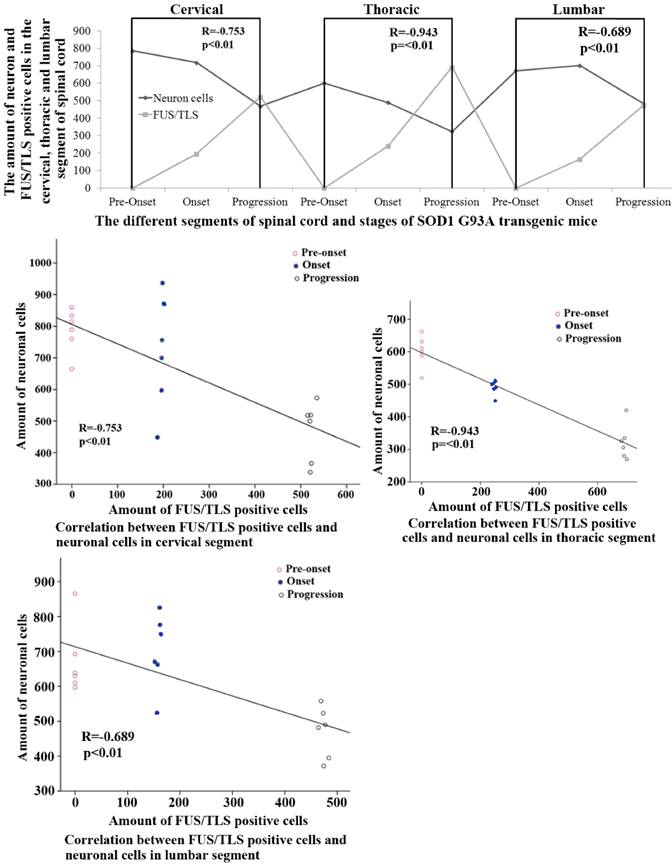
The FUS/TLS positive cell was observed and analyzed in the different anatomic regions, segments and neural cells in the entire adult spinal cord of the SOD1 wild-type and G93A transgenic mice. The FUS/TLS positive cell almost wasn't detected in the different segments and anatomic regions of spinal cord in the SOD1 wild-type transgenic mice. The distribution of FUS/TLS positive cell in the different segments and anatomic regions of spinal cord was significantly different during the different stages in the SOD1 G93A mice. The FUS/TLS positive cell almost wasn't detected in the cervical, thoracic and lumbar segments at the pre-onset stage in the SOD1 G93A mice (Figure 1-3). The FUS/TLS positive cell significantly increased in the cervical, thoracic and lumbar segments at the onset and progression stages, particularly at the progression stage (Figure 1-3, 2A). During the onset and progression of the SOD1 G93A mice, the most significantly increased segment of spinal cord was in the thoracic segment, the secondary was in the cervical segment, the third was in the lumbar segment (Figure 1-3, 2B). The FUS/TLS positive cell almost distributed in the entire anatomic regions of spinal cord including the anterior funiculus, the posterior funiculus, the lateral funiculus, the gray matter surrounding the central canal, the gray matter of ventral horn and dorsal horn in the G93A SOD1 mice. The increase rank order of total FUS/TLS positive cell amount in the cervical, thoracic and lumbar segments of integral spinal cord was the thoracic greater than the cervical greater than the lumbar segment during the onset and progression stages of SOD1 G93A mice (i.e. thoracic>cervical>lumbar segment) (Figure 1-3, 2B). The increase rank order of total FUS/TLS positive cell amount in the different anatomic regions was the anterior funiculus>the lateral funiculus>the gray matter of ventral horn>the gray matter surrounding the central canal>the gray matter of dorsal horn>the posterior funiculus (Figure 1-3, 2C). Among the different segments and anatomic regions of the entire spinal cord during the onset and progression stages, the increase rank order of FUS/TLS positive cell amount was the thoracic AF>the thoracic LF>the cervical AF>the cervical LF>the lumbar AF>the lumbar LF>the thoracic GVH>the cervical GVH>the thoracic GDH>the lumbar GVH>the cervical GDH>the thoracic PF>the lumbar GDH> the thoracic GCC>the cervical GCC>the cervical PF>the lumbar GCC>the lumbar PF (Figure 3). The white matter was more than the gray matter. The amount of FUS/TLS positive cell in the other anatomic regions and segments of integral spinal cord wasn't significantly different.
The expression of FUS/TLS in the different anatomic regions of the adult spinal cord at the pre-onset (60-70d), onset (90-100d) and progression (120-130d) stages of ALS-like G93A transgenic mice. The expression of FUS/TLS in all anatomic regions of the adult spinal cord including the anterior funiculus (AF), the posterior funiculus (PF), the lateral funiculus (LF), the gray matter surrounding the central canal (GCC), the gray matter of the ventral horn (GVH) and the dorsal horn (GDH) almost wasn't detected at the pre-onset stages (A). The expression of FUS/TLS in almost all anatomic regions including AF, PF, LF, GCC, GVH and GDH significantly increased at the pre-onset (B) and the progression (C) stages, and that at the progression stage was more significant than that at the onset stage (B, C). There was significant difference in the different anatomic regions, among them. The increased rank order of total FUS/TLS positive cell amount in the different anatomic regions was AF>LF>GVH>GCC>GDH>PF (D). Comment: The number indicated the rank order from more to less. The white arrow marked the positive cells.
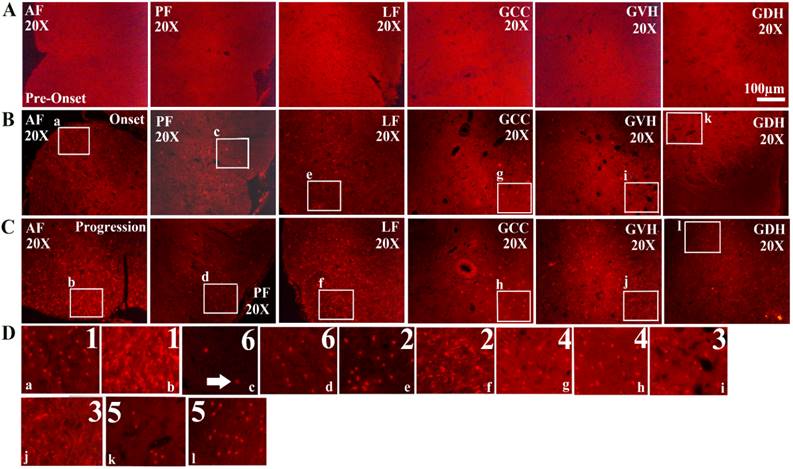
The analysis on the positive cell amount of FUS/TLS expression in the different segments and anatomic regions of spinal cord at the pre-onset, onset and progression stages of the ALS-like G93A transgenic mice. The expression of FUS/TLS in all segments of spinal cord including the cervical, thoracic and lumbar segments almost wasn't detected at the pre-onset stage (A, B, C). The FUS/TLS expression in the cervical, thoracic and lumber segments significantly increased at the onset and progression stages, among them, the increased FUS/TLS expression at the progression stage was significantly more than that at the onset stage (A). The FUS/TLS expression at the onset and progression stages significantly increased in the cervical, thoracic and lumber segments, among them, the increase of FUS/TLS expression in the thoracic segment was especially significant, and the increased rank order of FUS/TLS expression in the thoracic was more than that in the cervical more than that in the lumbar segment (i.e. thoracic>cervical>lumbar segment) (B). The expression of FUS/TLS expression in the different anatomic regions of the cervical, thoracic and lumbar segment significantly increased at the onset and progression stages, the increase rank order of FUS/TLS expression was AF>LF>GVH>GCC> GDH>PF (C).
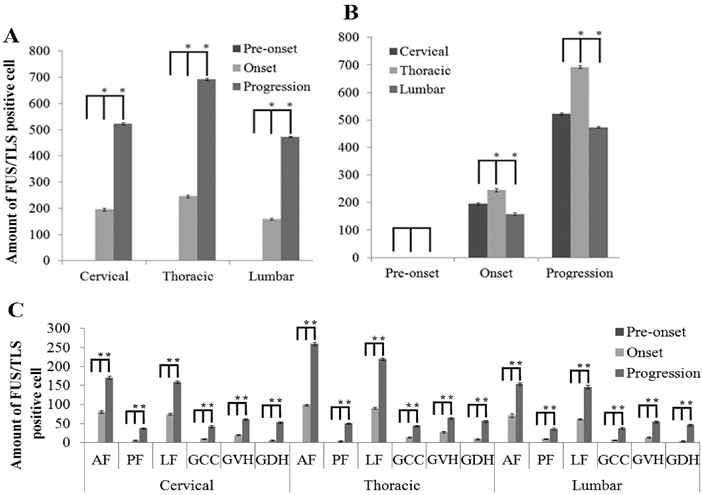
The analysis on the positive cell amount of FUS/TLS expression at the pre-onset, onset and progression stages of ALS-like G93A transgenic mice in the different anatomic regions of spinal cord. The FUS/TLS positive cell at the pre-onset stage of ALS-like G93A transgenic mice in the different anatomic regions of spinal cord almost wasn't detected. The increase of FUS/TLS positive cell amount showed a significant difference among the different anatomic regions of spinal cord at the onset and progression stages, the increase rank order of FUS/TLS expression was the thoracic AF> the thoracic LF> the cervical AF> the cervical LF> the lumbar AF> the lumbar LF> the thoracic GVH> the cervical GVH> the thoracic GDH> the lumbar GVH> the cervical GDH> the thoracic PF> the lumbar GDH> the thoracic GCC> the cervical GCC> the cervical PF> the lumbar GCC> the lumbar PF, that at both the onset and progression stages was the same.
The expression of FUS/TLS in the astrocyte cell in the different anatomic regions and segments of spinal cord at the pre-onset, onset and progression stages of the ALS-like G93A transgenic mice. The expression of FUS/TLS in the astrocyte cell (GFAP positive cell) in the different anatomic regions and segments of spinal cord almost wasn't detected at the pre-onset stage of the ALS-like G93A transgenic mice. The FUS/TLS expressed in the astrocyte cell in almost all anatomic regions and segments of spinal cord at the onset and progression stages. Comment: The Figure 4 was the representative images of the FUS/TLS expression in the astrocyte cell in the different anatomic regions and segments of spinal cord at the pre-onset, onset and progression stages of the ALS-like G93A transgenic mice.
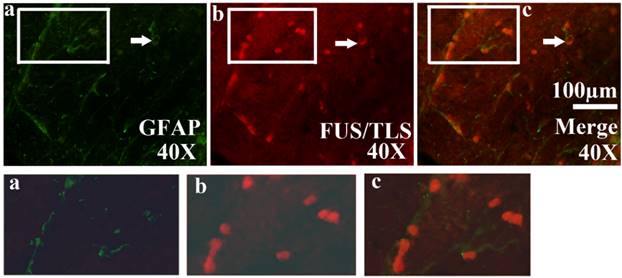
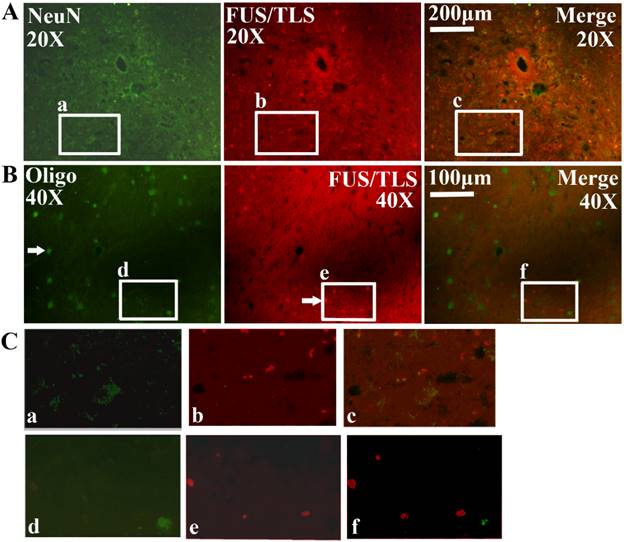
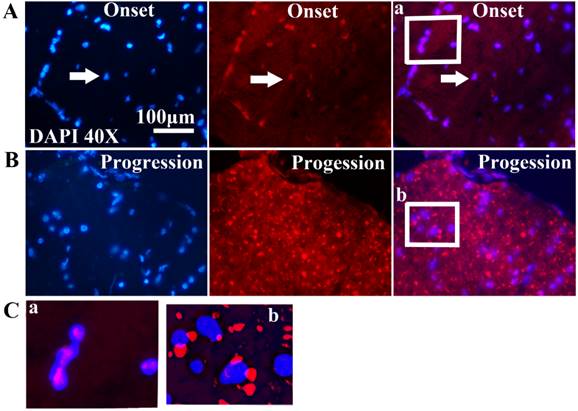
All FUS/TLS positive cells were the GFAP positive cell (Astrocyte cell) (Figure 4), the expression of FUS/TLS wasn't detected in the NeuN positive cell (Neuron cell) and the Oligo positive cell (Oligodendrocyte cell) (Figure 5), which indicated that the expressive increase of FUS/TLS mainly occurred in the astrocyte cell during the onset and progression stages of the SOD1 G93A mice. The FUS/TLS positive cell mainly expressed in the nuclear of the astrocyte cell at the onset stage of the SOD1 G93A mice, but it mainly expressed in the cytoplasm of the astrocyte cell, only fewer expressed in the nuclear at the progression stage (Figure 6), which indicated that FUS/TLS emerged the redistribution or translocation (Mislocalisation) from the nuclear to the cytoplasm of the astrocyte cell during from the onset to the progression stage in the SOD1 G93A mice.
The relationship of between the expression of FUS/TLS and the neuron death in the adult spinal cord of the SOD1 wild-type and G93A transgenic mice
The systematic analysis based on the alteration of FUS/TLS in the different anatomic regions, segments and neural cells of spinal cord during the different stages in the SOD1 wild-type and G93A transgenic mice demonstrated that the increase of FUS/TLS positive cells amount was negatively correlated with the decrease of neuron cell amount in the cervical, thoracic and lumbar segments of the adult spinal cord at the onset and progression stages in the SOD1 G93A transgenic mice, the death of neuron cell was closely related to the increase of FUS/TLS expression in the astrocyte cell, which implied the more expression of FUS/TLS and the more death of neuron cell (Figure 7). Notably, almost all FUS/TLS positive astrocyte cell showed the significant translocation from nuclear to cytoplasm (an increased FUS/TLS cytoplasmic to nuclear ratio) during the progression stages of the ALS-like G93A SOD1 transgenic mice (Figure 6). The data indicated that the overexpression and mislocalization of FUS/TLS participated in the progressive motor neuron degeneration in the SOD1 G93A transgenic mouse model of ALS.
Discussion
Our study demonstrated the following major findings: 1). The FUS/TLS almost didn't express in the adult spinal cord in the SOD1 wild-type transgenic mice and at the pre-onset stage of the SOD1 G93A transgenic mice, but the FUS/TLS extensively expressed in almost all anatomic regions and segments of the adult spinal cord at the onset and progression stages, and mainly distributed in the astrocyte cell, which indicated that the expressive increase of FUS/TLS in the astrocyte cell participated in the disease development of the SOD1 G93A transgenic mice. 2). The FUS/TLS mainly expressed in the nuclear of astrocyte cell in the adult spinal cord at the onset stage of the SOD1 G93A transgenic mice, but almost all FUS/TLS expressed in the cytoplasm of astrocyte cell at the progression stage, which indicated that the mislocalization of FUS/TLS expression from nuclear to cytoplasm of astrocyte cell from onset to progression stages. 3). A significant increase of FUS/TLS followed with a significant increase of neuron cell death at the onset and progression stages in the SOD1 G93A transgenic mice, which indicated that the expressive increase of FUS/TLS caused the neuron death in the SOD1 G93A transgenic mice. Our results suggested that the expressive increase and mislocalization in the astrocyte cell might cause the progressive motor neuron degeneration in the SOD1 G93A transgenic mouse model of ALS.
The expression of FUS/TLS in the neuron and oligodendrocyte cell in the different anatomic regions and segments of spinal cord at the pre-onset, onset and progression stages of ALS-like G93A transgenic mice. The expression of FUS/TLS in the neuron (NeuN positive cell) (A, C) and oligodendrocyte cell (Oligo positive cell) (B, C) in the different anatomic regions and segments of spinal cord almost wasn't detected at the pre-onset, onset and progression stage. Comment: The Figure 5 was the representative images of the FUS/TLS expression in the neuron and oligodendrocyte cell in the different anatomic regions and segments of spinal cord at the pre-onset, onset and progression stages of ALS-like G93A transgenic mice.
The observation and analysis on the mislocalization of FUS/TLS expression in neural cells in the different anatomic regions and segments of spinal cord at the pre-onset, onset and progression stages of ALS-like G93A transgenic mice. Almost all FUS/TLS expressed in the nuclear of astrocyte cell in the different anatomic regions and segments of spinal cord at the onset stage (A, C), but the vast majority of FUS/TLS expressed in the cytoplasm of astrocyte cell, only fewer expressed in the nuclear at the progression stage (B, C).
The correlation between the FUS/TLS expression and the neuron cell death in the different anatomic regions and segments of spinal cord at the pre-onset, onset and progression stages of the ALS-like G93A transgenic mice. The FUS/TLS expression was positively correlated with the neuron death in the different anatomic regions and segments of spinal cord at the onset and progression stages of the ALS-like G93A transgenic mice, the significant increase of FUS/TLS expression followed by the significant increase of neuron death.
This study performed a partially observation and analysis on the alteration of FUS/TLS expression, redistribution and dislocation in the spinal cord of SOD1 wild-type and G93A transgenic mice in order to further investigate its mechanism in the pathogenesis of ALS. We found some evidences in the association between the alteration of FUS/TLS and the pathogenesis of ALS. Firstly, our results revealed that the FUS/TLS almost didn't expressed in the adult spinal cord in the SOD1 wild-type transgenic mice and the pre-onset stages of SOD1 G93A transgenic mice, only expressed in the adult spinal cord at the onset and progression stages of the SOD1 G93A transgenic mice, which suggested that the expressive increase of FUS/TLS at the onset and progression stages was one of the pathogenesis of ALS.
Secondly, our results showed that the expressive increase of FUS/TLS mainly occurred in the astrocyte cell, almost wasn't detected the expressive increase in the other neural cells including neuron and oligodendrocyte cells. This suggested that the expressive increase of FUS/TLS firstly generated the toxicity to the astrocyte cell, which contributed to the dysfunction of the astrocyte cell, subsequently caused the death of motor neuron in ALS. Meanwhile, our results also provided a further evidence for the mechanism of motor neuron death in ALS, which was that the pathological alteration didn't only occur in the motor neuron, but also occurred in the glial cells like astrocyte cell. The degeneration of motor neuron in ALS might be a secondary pathological result of the glial cell lesion like the astrocyte cell.
Thirdly, the FUS/TLS mainly expressed in the nuclear of astrocyte cell, the other cell structures almost weren't detected the FUS/TLS expression at the onset stages, but almost all FUS/TLS expressed in the cytoplasm of astrocyte cell at the progression stages. The result inferred that the mislocalization of FUS/TLS didn't occur in all neural cells during the whole disease course of ALS, but only in the partial cells and the late stage of ALS.
Fourthly, our results found the increase of FUS/TLS expression was significantly different in the different anatomic regions of spinal cord at the onset and progression stages. The most significant increase of FUS/TLS expression was in the lateral funiculus and the gray matter of the ventral horn, which was consistent with the pathological damaged regions in ALS, which further suggested that the increase of FUS/TLS expression was closely related to the pathogenesis of ALS.
Fifthly, we analyzed the correlation between the FUS/TLS expression and the neuron death, the results showed that the significant increase of neuron death followed with the significant increase of FUS/TLS expression at the onset and progression stages of the ALS-like G93A transgenic mice, which indicated that the expressive increase and mislocalization of FUS/TLS were closely associated with the neuron death in the ALS disease courses. Our results demonstrated that the expressive increase and mislocalization of FUS/TLS in the astrocyte cell causes the progressive motor neuron degenerative death in ALS.
FUS/TLS protein predominantly expresses and distributes in the nuclear of cells [23]. Although FUS/TLS protein mainly expresses and distributes in the nuclear of cells under the physiological conditions, it accumulates in the inclusions of the cytoplasm of the lower motor neurons in the spinal cord of the patients with the pathogenic mutations FUS/TLS [24, 25]. Major mutations of FUS/TLS protein happen in the extreme C-terminus containing a nuclear localized signal, therefore, FUS/TLS protein has been suggested that either protein losses from the cellular nucleus, or the toxicity of protein aggregation accumulated in the cellular cytoplasm causes the neuron degeneration [26]. The overexpression of the human ALS mutant FUS/TLS in the adult transgenic rat causes the progressive paralysis produced by the degeneration of motor axons and the substantial loss of cortical and hippocampal neurons [27, 28], and the overexpression of the wild-type FUS/TLS protein also causes the cognitive deficits in the older animals due to a loss of cortical and hippocampal neurons [29, 30]. Furthermore, it is reported that that the mutant FUS/TLS is more toxic to the motor neurons than the normal FUS/TLS [31]. There is the evidence showing that the increased expression of wild-type FUS/TLS is sufficient to result in neuron degeneration [32]. In our study, we found that the expressive increase and mislocalization of FUS/TLS almost occurred in the astrocyte cell at the onset and progression stages of SOD1 G93A mice. Therefore, we suggested that the expressive increase and mislocalization of FUS/TLS firstly might produce the toxic to the astrocyte cell, but not to the neuron cells, subsequently induce the degeneration of neuron cells due to the dysfunction of the glial cells like the astrocyte cell, ultimately develop ALS.
At present, although the SOD1 G93A transgenic mouse model might not accurately reflect the pathogenesis of ALS found in the vast majority of human cases, it still is the most commonly used mouse models of ALS in studying the pathogenesis of ALS now [33]. Thus, in this study we still applied the SOD1 G93A transgenic mouse model to observe and analyze the role of the alteration of FUS/TLS expression, redistribution and dislocation in the development of ALS. Our result provided some novel evidences and clues for further studying the role of FUS/TLS in the developmental of ALS. Because of the deficiency of the SOD1 G93A transgenic mouse model, there might exist some differences with the result of different mouse models and the clinical case study. Our data might exist some error, they need further validated in other animal models and the vast majority of human ALS cases.
In summary, our study about the alteration of expression, redistribution and dislocation of FUS/TLS in the adult spinal cord of the SOD1 G93A transgenic mouse model preliminary clarified that the expressive increase and mislocalization in the astrocyte cell might cause the progressive motor neuron degeneration in the pathogenesis of ALS. In addition, this study also identified the lesion of astrocyte cell might play an important role in the development of motor neuron death during the ALS disease course, suggested that the pathogenesis of motor neuron death in ALS wasn't only occur in the motor neuron, but also was involved in the dysfunction of the glial cells such as astrocyte cell. The mechanism might be that the expressive increase and mislocalization of FUS/TLS in the astrocyte cell generated toxicity to the astrocyte cell, leaded to the damage of its functions protecting the motor neurons, subsequently contributed to the neuron degenerative death in ALS.
Abbreviations
FUS/TLS, fused in sarcoma/translocated in liposarcoma; GFAP, glial fibrillary acidic protein; NeuN, hexaribonucleotide Binding Protein-3; Oligo-2, anti-oligodendrocyte antibody-2; DAPI, 4',6-diamidino-2- phenylindole.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (30560042, 81160161, 81360198), Education Department of Jiangxi Province (GJJ13198) and Jiangxi provincial department of science and technology ([2014]-47).
Authors' contributions
R.X. and J.L. designed the experiments and analyzed the data. J.L. Y.L. H.L. C.T. L.Z. and J.Z. conducted the experiments. R.X. and J.L. wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ludolph AC, Brettschneider J, Weishaupt JH. Amyotrophic lateral sclerosis. Current Opinion in Neurology. 2012;25:530-5
2. Bäumer D, Talbot K, Turner MR. Advances in motor neurone disease. Journal of the Royal Society of Medicine. 2014;107:14-21
3. Arbab M, Baars S, Geijsen N. Modeling motor neuron disease: the matter of time. Trends in Neurosciences. 2014;37:642-652
4. Sendtner M. Motoneuron disease. Handbook of Experimental Pharmacology. 2014;220:411-41
5. Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O. et al. Amyotrophic lateral sclerosis. Lancet. 2011;377:942-55
6. Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database of Systematic Reviews. 2012;3:CD001447
7. Chiò A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA. et al. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology. 2013;41:118-30
8. Kawaja MD. A proteomic approach to assess intraneuronal inclusions associated with neurodegenerative disorders. Current Opinion in Molecular Therapeutics. 2005;7:565-8
9. Perry JJ, Shin DS, Tainer JA. Amyotrophic lateral sclerosis. Advances in Experimental Medicine and Biology. 2010;685:9-20
10. Fiesel FC, Kahle PJ. TDP-43 and FUS/TLS: cellular functions and implications for neurodegeneration. The FEBS Journal. 2011;278:3550-68
11. Traub R, Mitsumoto H, Rowland LP. Research advances in amyotrophic lateral sclerosis, 2009 to 2010. Current Neurology and Neuroscience Reports. 2011;11:67-77
12. Wang XN, Cui LY. TAR DNA binding protein-43 and fused in sarcoma/translocated in liposarcoma protein in two neurodegenerative diseases. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2012;34:286-92
13. Baralle M, Buratti E, Baralle FE. The role of TDP-43 in the pathogenesis of ALS and FTLD. Biochemical Society Transactions. 2013;41:1536-40
14. Bunton-Stasyshyn RK, Saccon RA, Fratta P, Fisher EM. SOD1 Function and Its Implications for Amyotrophic Lateral Sclerosis Pathology: New and Renascent Themes. Neuroscientist. 2015;21:519-29
15. Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136:1001-4
16. Valdmanis PN, Daoud H, Dion PA, Rouleau GA. Recent advances in the genetics of amyotrophic lateral sclerosis. Current Neurology and Neuroscience Reports. 2009;9:198-205
17. Da Cruz S, Cleveland DW. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Current Opinion in Neurobiology. 2011;21:904-19
18. Liscic RM, Breljak D. Molecular basis of amyotrophic lateral sclerosis. Progress in Neuro-psychopharmacology & Biological Psychiatry. 2011;35:370-2
19. Sama RR, Ward CL, Bosco DA. Functions of FUS/TLS from DNA repair to stress response: implications for ALS. ASN Neuro. 2014;6:pii 1759091414544472
20. Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD. et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772-5
21. Henriques A, Pitzer C, Schneider A. Characterization of a novel SOD-1(G93A) transgenic mouse line with very decelerated disease development. PLoS One. 2010;5:e15445
22. Zhou Y, Lu Y, Fang X, Zhang J, Li J, Li S. et al. An astrocyte regenerative response from vimentin-containing cells in the spinal cord of amyotrophic lateral sclerosis's disease-like transgenic (G93A SOD1) mice. Neurodegenerative Disease. 2015;15:1-12
23. Romano M, Buratti E. Targeting RNA binding proteins involved in neurodegeneration. Journal of Biomolecular Screening. 2013;18:967-83
24. Daigle JG, Lanson NA Jr, Smith RB, Casci I, Maltare A, Monaghan J. et al. RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations. Human Molecular Genetics. 2013;22:1193-205
25. Shelkovnikova TA, Peters OM, Deykin AV, Connor-Robson N, Robinson H, Ustyugov AA. et al. Fused in sarcoma (FUS) protein lacking nuclear localization signal (NLS) and major RNA binding motifs triggers proteinopathy and severe motor phenotype in transgenic mice. Journal of Biological Chemistry. 2013;288:25266-74
26. Masuda A, Takeda JI, Ohno K. FUS-mediated regulation of alternative RNA processing in neurons: insights from global transcriptome analysis. Wiley Interdisciplinary Reviewers. RNA. 2016;7:330-40
27. Huang C, Tong J, Bi F, Wu Q, Huang B, Zhou H. et al. Entorhinal cortical neurons are the primary targets of FUS mislocalization and ubiquitin aggregation in FUS transgenic rats. Human Molecular Genetics. 2012;21:4602-14
28. Huang C, Zhou H, Tong J, Chen H, Liu YJ, Wang D. et al. FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genetics. 2011;7:e1002011
29. Belly A, Moreau-Gachelin F, Sadoul R, Goldberg Y. Delocalization of the multifunctional RNA splicing factor TLS/FUS in hippocampal neurones: exclusion from the nucleus and accumulation in dendritic granules and spine heads. Neuroscience Letters. 2005;379:152-7
30. Schoen M, Reichel JM, Demestre M, Putz S, Deshpande D, Proepper C. et al. Super-Resolution Microscopy Reveals Presynaptic Localization of the ALS/FTD Related Protein FUS in Hippocampal Neurons. Frontiers in Cellular Neuroscience. 2016;9:496
31. Sharma A, Lyashchenko AK, Lu L, Nasrabady SE, Elmaleh M, Mendelsohn M. et al. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nature Communications. 2016;7:10465
32. Mitchell JC, McGoldrick P, Vance C, Hortobagyi T, Sreedharan J, Rogelj B. et al. Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathologica. 2013;125:273-88
33. Turner BJ, Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Progress in Neurobiology. 2008;85:94-134
Author contact
![]() Corresponding author: Prof. Renshi Xu. Department of Neurology, the First Affiliated Hospital of Nanchang University, Nanchang 330006, Jiangxi, China. Email address: 13767015770com.
Corresponding author: Prof. Renshi Xu. Department of Neurology, the First Affiliated Hospital of Nanchang University, Nanchang 330006, Jiangxi, China. Email address: 13767015770com.
Received 2016-5-13
Accepted 2016-7-14
Published 2016-9-14