ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Abbreviation
Acknowledgements
References

Int J Biol Sci 2016; 12(12):1488-1499. doi:10.7150/ijbs.15583 This issue Cite
Research Paper
LIS1 Regulates Osteoclastogenesis through Modulation of M-SCF and RANKL Signaling Pathways and CDC42
Shiqiao Ye1*, Toshifumi Fujiwara1*, Jian Zhou1,3, Kottayil I Varughese2, Haibo Zhao1,2 ![]()
1. Center for Osteoporosis and Metabolic Bone Diseases, Division of Endocrinology and Metabolism, University of Arkansas for Medical Sciences, Little Rock, Arkansas, USA;
2. Department of Physiology and Biophysics, University of Arkansas for Medical Sciences, Little Rock, Arkansas, USA.
3. Department of Orthopedics, First Affiliated Hospital of Anhui Medical University, Hefei, Anhui, P. R. China.
*Ye, S and Fujiwara, T contributed equally to this work.
Abstract

We have previously reported that depletion of LIS1, a key regulator of microtubules and cytoplasmic dynein motor complex, in osteoclast precursor cells by shRNAs attenuates osteoclastogenesis in vitro. However, the underlying mechanisms remain unclear. In this study, we show that conditional deletion of LIS1 in osteoclast progenitors in mice led to increased bone mass and decreased osteoclast number on trabecular bone. In vitro mechanistic studies revealed that loss of LIS1 had little effects on cell cycle progression but accelerated apoptosis of osteoclast precursor cells. Furthermore, deletion of LIS1 prevented prolonged activation of ERK by M-CSF and aberrantly enhanced prolonged JNK activation stimulated by RANKL. Finally, lack of LIS1 abrogated M-CSF and RANKL induced CDC42 activation and retroviral transduction of a constitutively active form of CDC42 partially rescued osteoclastogenesis in LIS1-deficient macrophages. Therefore, these data identify a key role of LIS1 in regulation of cell survival of osteoclast progenitors by modulating M-CSF and RANKL induced signaling pathways and CDC42 activation.
Keywords: LIS1, M-CSF, RANKL, MAPK, Cdc42, osteoclast.
Introduction
Osteoclasts are multinucleated cells capable of resorbing calcified cartilage and bone matrix during skeletal development, homeostasis, and repair [1]. Osteoclastic bone resorption coupling with bone formation by osteoblasts remodel the adult skeleton and help to maintain bone mass [2]. However, excessive bone resorption, either caused by increased osteoclast number or enhanced activity under pathological conditions, leads to bone loss in metabolic bone diseases such as postmenopausal osteoporosis, rheumatoid arthritis, Paget's disease of bone, periodontal disease and lytic tumor bone metastasis [3]. Thus, identification of the molecular mechanisms governing osteoclast differentiation and function will not only improve our understanding of the pathogenesis of human skeletal diseases but provide new therapeutic targets for treatment of these diseases as well.
Mature osteoclasts are formed by fusion of mononuclear precursors of the monocyte/macrophage lineage of hematopoietic origin. Macrophage colony-stimulating factor (M-CSF) and the receptor activator of nuclear factor-κB (NF-κB) ligand (RANKL) are two indispensable cytokines for osteoclastogenesis in vitro and in vivo [4]. While M-CSF stimulates the proliferation of macrophages and the survival of osteoclasts by activating extracellular signal-regulated kinase (ERK) and phosphoinositide-3-kinase/Akt (PI3K/AKT) pathways, RANKL is a major osteoclast differentiation factor. RANKL activates mitogen-activated protein kinase (MAPK), NF-κB, and PI3K/AKT pathways and induces calcium oscillation that also requires co-stimulating signals from immunoglobulin-like receptors and their associated adapter proteins. These pathways converge to induce and activate nuclear factor of activated T cells 1 (NFATc1), a master transcription factor of osteoclast differentiation [4, 5]. Systemic hormones and other cytokines/growth factors in the bone marrow microenvironment regulate osteoclast number or activities through controlling the expression of M-CSF and RANKL in other cell types of bone marrow or modulating the downstream signaling pathways of these two cytokines in osteoclast lineage cells [6].
Heterozygous disruption of the Lis1 (lissencephaly-1) gene, resulting in haploinsufficiency of LIS1, causes classical lissencephaly, a severe human developmental brain disorder manifested by a smooth cerebral surface and disorganized cortical layers due to defects in neuronal migration [7, 8]. LIS1 interacts with NDE1 (nudE neurodevelopment protein 1)/NDEL1 (nudE neurodevelopment protein 1 like 1) and regulates microtubule organization and the function of minus-end oriented microtubule motor, the cytoplasmic dynein [9, 10]. LIS1 is also known as PAFAH 1b1 (platelet-activating factor (PAF) acetylhydrolase 1b complex subunit 1, a regulatory subunit of PAFAH 1b complex that inactivates PAF [11]. PAF is one of the most potent lipid messengers and is involved in a variety of physiological and pathological events [12]. PAF binds to a unique lipid G-protein coupled PAF receptor (PAFR) that initiates intracellular signals leading to mobilization of intracellular Ca2+ and activation of MAPK [13]. Osteoclasts express high levels of PAF biosynthetic enzymes and PAFR [14]. More importantly, PAFR-deficient mice have lower osteoclast survival rate and bone resorption [14]. Both LIS1 and its binding protein NDEL1 regulate the activity of CDC42, a member of small GTPase Rho family, by direct interaction with CDC42 and its endogenous inactivator CDC42GAP, respectively [15, 16]. We have previously reported that CDC42 is critical for osteoclast formation and bone homeostasis through modulation of M-CSF and RANKL signaling [17].
Given that LIS1-depletion in macrophages by short-hairpin RNAs inhibits osteoclastogenesis in vitro [18] and that PAF and CDC42 play an important role in regulation of osteoclast formation in vitro and in vivo [14, 17], we hypothesize that LIS1 may play an important role in osteoclastogenesis via PAF and/or CDC42. To elucidate the role of LIS1 in osteoclasts, we generated LIS1 conditional knockout mice in which LIS1 is specifically deleted in osteoclast precursor cells. We here provide in vivo and in vitro evidence indicating that LIS1 plays an important role in regulation of cell survival of osteoclast progenitors by modulating signaling pathways and CDC42 activated by M-CSF and RANKL.
Materials and Methods
Mice
The LIS1-floxed mice (stock number 008002) and LysM-Cre mice (stock number 004781), both in C57BL6/129 mixed background, were obtained from The Jackson Laboratory. The sequences of genotyping primers and PCR protocols followed those provided by The Jackson Laboratory. All animal procedures used in this study were approved by the Institutional Animal Care and Use Committees of the University of Arkansas for Medical Sciences.
Micro-CT
The left femurs and L4 vertebrae of 5-month old control (+/+; LysM-Cre) and LIS1 conditional knockout mice (LIS1 flox/flox;LysM-Cre) were cleaned of soft tissues and fixed in 10% Millonig's formalin with 0.5% sucrose for 24 hours. The bone samples were gradually dehydrated into 100% ethanol. The bones were loaded into a 12.3-mm diameter scanning tube and were imaged in a μCT (model μCT40, Scanco Medical). We integrated the scans into 3-D voxel images (1024 x 1024 pixel matrices for each individual planar stack) and used a Gaussian filter (sigma = 0.8, support = 1) to reduce signal noise. A threshold of 200 was applied to all scans, at medium resolution (E = 55 kVp, I = 145 μA, integration time = 200ms).
Histology and bone histomorphometry
The histology and histomorphometry measurements were performed as previously described [19]. In brief, the right femurs were fixed in 10% Millonig's formalin with 0.5% sucrose for 24 h and were gradually dehydrated into 100% ethanol. The femurs were embedded undecalcified in methyl methacrylate and stained for Goldner trichrome staining. The histomorphometric examination of trabecular bone, osteoblast number, and osteoid volume, was done on 5 μm longitudinal sections with a digitizer tablet (OsteoMetrics, Inc., Decatur, GA, USA) interfaced to a Zeiss Axioscope (Carl Zeiss, Thornwood, NY, USA) with a drawing tube attachment. The right tibia was fixed in 10% Millonig's formalin for 24 h and were decalcified in 14% EDTA for 7-10 days. The bones were embedded in paraffin before obtaining 5-μm longitudinal sections. After removal of paraffin and rehydration, sections were stained for TRAP activity and counter-stained with haematoxylin and osteoclasts were enumerated on the trabecular bone surface.
Bone marrow monocyte and osteoclast cultures
Cell culture media and L-Glutamine-penicillin-streptomycin (GPS) solution were purchased from Invitrogen and Sigma-Aldrich, respectively. Fetal bovine serum was purchased from Hyclone. Bone marrow monocyte (BMM) and osteoclast (OC) cultures were conducted as before [20]. Whole bone marrow was extracted from tibia and femurs of one or two 8-10-week-old mice. Red blood cells were lysed in buffer (150 mM NH4Cl, 10 mM KNCO3, 0.1 mM EDTA, pH 7.4) for 5 minutes at room temperature. 5 × 106 bone marrow cells were plated onto a 100mm petri-dish and cultured in α-10 medium (α-MEM, 10% heat-inactivated FBS, 1 × GPS) containing 1/10 volume of CMG 14-12 (conditioned medium supernatant containing recombinant M-CSF at 1µg/ml) for 4 to 5 days. Pre-osteoclasts and osteoclasts were generated by culturing BMMs (at density of 1.5 × 104/well of a 48-well tissue culture plate) with 1/100 vol of CMG 14-12 culture supernatant and 100 ng/ml of recombinant RANKL to 2 and 4 days, respectively.
Tartrate-resistant acid phosphatase (TRAP) staining TRAP5b ELISA
BMMs were cultured on 48-well tissue culture plate in α-10 medium with M-CSF and RANKL for 4-5 days. The cells were fixed with 4% paraformaldehyde/phosphate buffered saline (PBS) and TRAP was stained with NaK Tartrate and Napthol AS-BI phosphoric acid (Sigma-Aldrich). The level of TRAP5b in the culture medium was measured by an ELISA kit (SB-TR103, Immunodiagnosticsystems) following the manufacture's instruction.
RNA isolation and quantitative real time RT-PCR
Total RNA was purified using RNeasy mini kit (Qiagen) according to the manufacture's protocol. First-strand cDNAs were synthesized from 1 μg of total RNA using the High Capacity cDNA Reverse Transcription kits (Thermo-Fisher Scientific) following the manufacturer's instructions. TaqMan quantitative real-time PCR was performed using the following primers from Life Technologies: Acp5 (Mm00475698_m1); CalcR (Mm00432282_m1); Ctsk (Mm00484039_m1). Samples were amplified using the StepOnePlus real-time PCR system (Thermo-Fisher Scientific) with an initial denaturation at 95 ºC for 10 min, followed by 40 cycles of 95 ºC for 15 s and 60 ºC for 1 min. The relative cDNA amount was calculated by normalizing to that of the mitochondrial gene Mrps2 (mitochondrial ribosomal protein S2), which is steadily expressed in both BMMs and osteoclasts, using the ΔCt method [21, 22].
Antibodies and Western blotting
The antibodies were obtained from the following resources: mouse anti-LIS1 monoclonal antibody was a generous gift from Dr. O Reiner (The Weizmann Institute of Science, Rehovot, Israel); mouse monoclonal anti-Cathepsin K (CTSK) (clone 182-12G5) (Millipore); rabbit polyclonal anti-ERK1/2; mouse monoclonal anti-phospho-ERK1/2 (Thr202/Tyr204); mouse monoclonal anti-AKT (pan) (clone 40D4); rabbit monoclonal anti-phosphor-AKT (Ser473) (clone 193H12); mouse monoclonal anti-phospho-JNK (Thr183/Tyr185) (clone G9;, rabbit polyclonal anti-IKB-α; mouse monoclonal anti-phospho-IKB-α (Ser32/36) (clone 5A5) (Cell Signaling); mouse monoclonal anti-Dynein intermediate chain (74.1) (Millipore). Cultured cells were washed with ice-cold PBS twice and lysed in 1 x RIPA buffer (catalog number R-0278, Sigma) containing 1 mM DTT and Complete Mini EDTA-free protease inhibitor cocktail (catalog number 04693159001, Roche), as described previously [23]. After incubation on ice for 30 min, the cell lysates were clarified by centrifugation at 14,000 rpm for 15 min at 4°C. 10 to 30 µg of total protein were subjected to 8% or 10% SDS-PAGE gels and transferred electrophoretically onto polyvinylidene difluoride membrane (catalog number IPVH00010, EMD Millipore) by a semi-dry blotting system (Bio-Rad). The membrane was blocked in 5% fat-free milk/Tris-buffered saline for 1 hour and incubated with primary antibodies at 4°C overnight followed by secondary antibodies conjugated with horseradish peroxidase (Santa Cruz Biotechnology). After rinsing 3 times with Tris-buffered saline containing 0.1% Tween 20, the membrane was subjected to western blot analysis with enhanced chemiluminescent detection reagents (catalog number WBKLS0100, EMD Millipore).
Flow cytometric analysis of cell cycle progression
BMMs were lifted by 1 × Trypsin/EDTA (Thermo-Fisher Scientific) and were suspended in cold PBS at 1 × 106/ml. Cells were incubated with 25ug/ml of 7-aminoactinomycin-D (7'AAD) (BD Biosciences) for 30 minutes on ice. The cell cycle analysis was performed by flow cytometry at Flow-cytometry Core Facility of University of Arkansas for Medical Sciences.
Cell Death ELISA
For the detection of osteoclast apoptosis, BMMs were cultured with M-CSF and RANKL for 2 days. Cell death of pre-osteoclasts was analyzed using cell death detection ELISA PLUS kit (catalog number 11774425001, Roche), which detects cytoplasmic histone-associated DNA fragmentation.
CDC42 activation assay
Pre-osteoclasts were serum and cytokine starved for three hours and were stimulated with 50ng/ml of M-CSF or 100ng/ml of RANKL for the indicated time. The cells were lysed and CDC42-GTP was examined by an effector domain, GST-fusion pulldown protocol using EZ-detect CDC42 activation kit (catalog number 16119, Thermo-Fisher Scientific).
Retroviral Transduction
The pMX retroviral empty and recombinant CDC42Q61L vectors were transfected into Plat E retroviral packing cells using TransIT-LT1 transfection reagent (catalog number MIR2300, Mirus Bio LLC). Virus supernatants were collected at 48 h after transfection. BMMs were transduced with viruses for 24 h in α-10 medium containing M-CSF and 20 μg/ml of protamine. Cells were then selected in α-10 medium containing M-CSF and 1.5 μg/ml of blasticidin (catalog number 203350, EMD Chemicals) for 3 days.
Statistics
For all graphs, data are represented as the mean ± standard deviation. Data of 2-group comparisons were analyzed using a 2-tailed Student's t test. For comparison of more than 2 groups, data were analyzed using one way Analysis of Variance (ANOVA) and the Bonferroni procedure was used for Tukey comparison.
Results
Conditional deletion of LIS1 gene in osteoclast precursor cells in mice leads to increased bone mass and decreased osteoclasts on trabecular bone
To elucidate the role of LIS1 in osteoclastogenesis in vivo, we generated conditional knockout mice with specific deletion of LIS1 gene in osteoclast precursor cells by crossing LIS1-floxed mice with LysM-Cre mice in which the Cre recombinase is expressed from the endogenous Lysozyme B locus in myeloid cells, including monocytes, mature macrophages, and granulocytes. The LIS1 flox/flox;Cre/+ mice were designated as conditional knockout mice (cKO). The initial examination of bone mineral density of wild type, LIS1 flox/flox; +/+, and +/+;Cre/+ mice showed no difference among these three mouse lines. We used +/+; Cre/+ mice as littermate controls (con) for further studies. The LIS1 cKO mice were viable, fertile, and normal in size and body weight. They did not display any overt abnormalities.
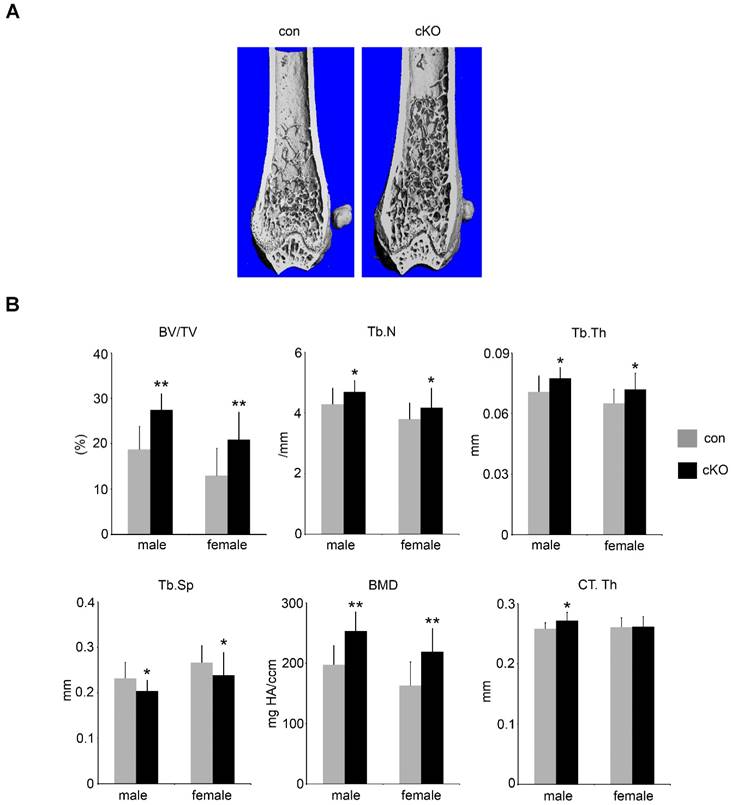
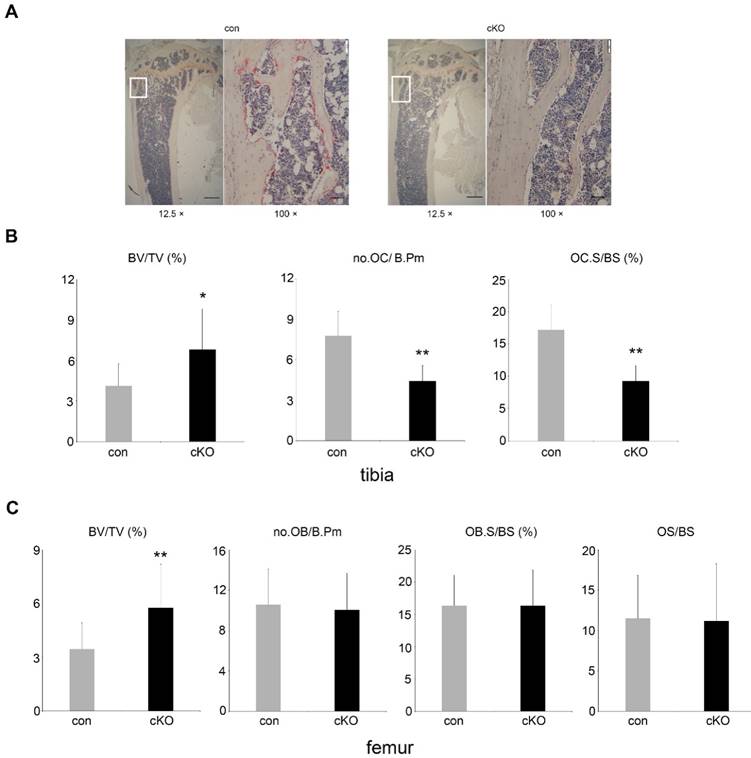
5-month-old male and female LIS1 cKO mice displayed an increase in trabecular bone mass (BV/TV) in long bones and vertebrae (Figure 1-2 and supplemental Figure 1). Micro-CT analysis demonstrated an elevation in trabecular number (Tb.N), trabecular thickness (Tb. Th), bone mineral density (BMD), and decreased trabecular separation (Tb. Sp) in LIS1 cKO mice as compared to their littermate controls. A slight increase of cortical bone thickness was observed only in male cKO mice (Figure 1B). Histomorphometry analysis of paraffin-embedded, TRAP-staining sections of proximal tibias and plastic-embedded, Goldner-staining sections of distal femurs from 5-month old female LIS1 cKO and con mice revealed a similar increase in BV/TV in LIS1 cKO mice (Figure 2). In accordance with our previous in vitro findings [18], loss of LIS1 in osteoclast progenitor cells led to decreased osteoclast number and surface on trabecular bone in vivo (Figure 2A-B). The osteoblast differentiation, as shown by osteoblast number and surface, and the deposition of new bone, as indexed by the volume of osteoid (uncalcified new bone matrix), remained normal in LIS1 cKO mice (Figure 2C). These data indicate that LIS1 plays an important role in bone homeostasis by regulating osteoclast formation and bone resorption.
LIS1 regulates osteoclastogenesis through modulation of M-CSF/RANKL signaling pathways and survival/differentiation of osteoclast precursors
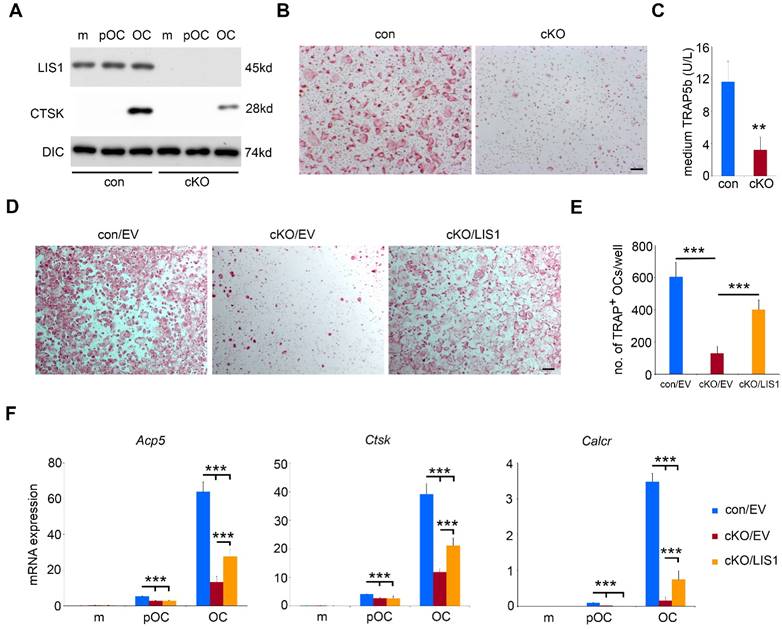
To uncover the cellular and molecular mechanisms by which LIS1 regulates osteoclastogenesis, we conducted in vitro osteoclast cultures using bone marrow cells isolated from control and LIS1 cKO mice. As shown in Figure 3A, LIS1 protein expression was completely diminished in monocytes, pre-osteoclasts and mature osteoclasts generated from bone marrow cells of LIS1 cKO mice. Loss of LIS1 attenuated the formation of multinucleated osteoclasts, stained positively for TRAP (Figure 3B). This finding was further confirmed and quantified by decreased protein expression of Cathepsin K, a lysosomal acidic hydrolase highly expressed in osteoclasts (Figure 3A), and reduced level of medium TRAP5b, a specific marker of osteoclasts (Figure 3C), in LIS1-deficient mature osteoclasts. Furthermore, the mRNA expression of osteoclast marker genes, such as Acp5 (encoding TRAP), Ctsk (encoding cathepsin K), and Calcr (encoding calcitonin receptor), in LIS1-null osteoclast lineage cells was greatly decreased, as measured by quantitative real-time PCR (Figure 3F). More importantly, the observed osteoclastogenic defect seemed to be intrinsic to osteoclast precursor cells because retroviral transduction of LIS1 in LIS1-null monocytes partially restored the formation of TRAP+, multinucleated osteoclasts (Figure 3D and 3E) and mRNA expression of osteoclast marker genes (Figure 3F). The exogenous expression of murine LIS1 in control monocytes had no effects on osteoclastogenesis (data not shown).
Conditional deletion of LIS1 gene in osteoclast precursor cells in mice leads to increased bone mass in femurs. (A) µCT images of distal femur of control (con) and LIS1 conditional knockout (cKO) mice. (B) µCT analysis of distal femurs of 5-month old male (con = 8, cKO = 11) and female (con = 20, cKO = 14) in C57BL6/129 mixed background. * p < 0.05, ** p < 0.01 vs con by student t-test.
Loss of LIS1 in osteoclast precursors results in decreased number of osteoclasts on trabecular bone. (A) Lower (12.5 ×) and higher (100 ×) magnification microscopic images of TRAP-stained paraffin-embedded sections of distal tibia from 5-month old control (con) and LIS1 conditional knockout (cKO) female mice. White rectangles in lower magnification images indicate the areas shown in higher magnification images. Scale bars = 500 µm and 50 µm, respectively. (B) Histomorphometric analysis of TRAP-stained sections of distal tibia from 5-month old female control (n= 20) and cKO (n= 14) mice. (C) Histomorphometric analysis of Goldner-trichrome staining of distal femurs of 5-month old control and cKO female mice. * p < 0.05, ** p < 0.01 vs control by Student's t-test.
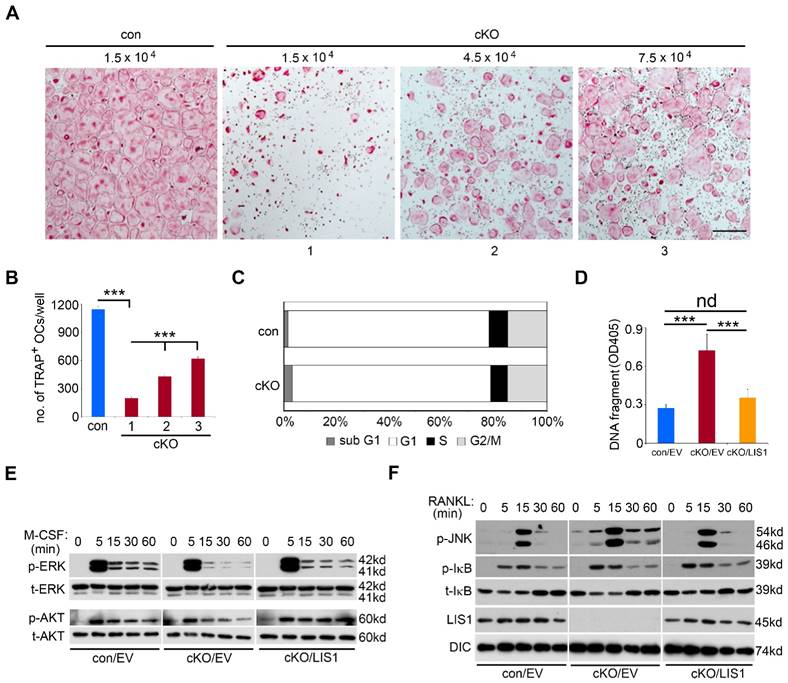
The magnitude of osteoclast formation in vivo and in vitro is regulated by proliferation, differentiation, and survival of osteoclast precursor and mature cells. To dissect which of these cellular processes was affected by LIS1 deficiency, we first plated increasing number of LIS1-null macrophages in osteoclast cultures. While increased cell density in control cultures resulted in earlier osteoclast formation and cell death after 4 days' cultures (data not shown), three and five times more LIS1-deficient monocytes in the cultures significantly increased the formation of TRAP-positive, multinucleated osteoclasts after 4 days' culture with M-CSF and RANKL (Figure 4A-B), suggesting that decreased number of osteoclast precursor cells led to defective osteoclast formation. Since LIS1 has been reported to regulate cell cycle progression in hematopoietic stem cells [24], we next tried to determine if such is a case in osteoclast precursor cells. To this end, control and LIS1-null macrophages were labelled with the fluorescent dye 7-aad (7-aminoactinomycin D) and the percentage of cell number in each stage of cell cycles was examined by flow-cytometry. Loss of LIS1 had little effect on cell cycle progression except a slight increase in sub G1 cells, an index of apoptotic cells (Figure 4C). Increased apoptosis in LIS1-deficient pre-osteoclasts, which was rescued by LIS1 reconstitution, was further confirmed by a Cell-Death ELISA (Figure 4D).
Intracellular signaling pathways activated by both M-CSF and RANKL play a critical role in osteoclastogenesis and cell survival in vivo and in vitro [25]. We then determined whether LIS1 modulates downstream signalings of M-CSF and RANKL in bone marrow monocytes. As shown in Figure 4E, the peak activation of ERK and AKT pathways, as demonstrated by their phosphorylation after 5-minute stimulation of M-CSF, was indistinguishable between control and LIS1-null monocytes. However, the prolonged activation of ERK induced by M-CSF was diminished in LIS1-deficient cells as compared to control monocytes. The maximum activation of JNK pathway by RANKL at 15-minute stimulation was intact in LIS1-deficient macrophages. In contrast, RANKL induced a prolonged JNK activation in LIS1-deficient cells that was not observed in control monocytes (Figure 4F). RANKL-activated NF-κB pathway, as detected by either phosphorylation or degradation of IκB, was not affected by LIS1-depletion. More importantly, the aberrant changes in M-CSF and RANKL signalings in LIS1-deficient monocytes were rescued by LIS1 re-expression in these cells, suggesting that these defects were caused intrinsically by the lack of LIS1 (right panels in Figure 4E and 4F). Since prolonged ERK activation has been reported to promote osteoclast survival whereas prolonged JNK induces osteoclast apoptosis [26, 27], these data indicate that LIS1 regulates survival of osteoclast lineage cells by fine-tuning modulation of ERK and JNK pathways activated by M-CSF and RANKL, respectively.
Loss of LIS1 in osteoclast precursor cells attenuates osteoclastogenesis in vitro. (A) Western blots of protein expression of LIS1, cathepsin K (CTSK) in control (con) and LIS1 conditional knockout (cKO) bone marrow monocytes (m), pre-osteoclasts (pOC), and mature osteoclasts (OC). Dynein intermediate chain (DIC) served as a loading control. (B) Microscopic images of TRAP-stained osteoclasts. The original magnification: × 20. Scale bar = 200µm. (C) medium level of TRAP5b detected by an ELISA kit. The data are presented as mean ± s.d., n = 6. ** p < 0.01 vs con by Student's t-test. (D) Microscopic images of TRAP-stained osteoclasts cultured from empty vector (EV) transduced con (con/EV), cKO cKO/EV), and cKO/LIS1-reconstituted bone marrow monocytes. The original magnification: × 20. Scale bar = 200µm. (E) The number of osteoclasts with more than three nuclei per well of a 48-well plate of cultures in (D). The data are presented as mean ± s.d., n = 6, *** p < 0.001 by one-way ANOVA. (F) Real-time quantitative PCR analysis of mRNA expression of osteoclast marker genes, Trap (encoded by Acp5), Cathepsin K (encoded by Ctsk) , and Calcitonin receptor (encoded by Calcr). The data are presented as mean ± s.d., n = 3, *** p < 0.001 by one-way ANOVA.
LIS1 regulates survival of osteoclast precursors by modulating M-CSF and RANKL signaling pathways. (A) Microscopic images of TRAP-stained osteoclasts cultured from control (con) and increasing number of conditional knockout (cKO) macrophages. The original magnification: × 20. Scale bar = 200µm. (B) quantification of TRAP-positive mature osteoclasts with more than 3 nuclei. Data are presented as mean ± s.d., n = 6, *** p < 0.001 by one-way ANOVA. (C) Flow-cytometry analysis of cell cycle progression in con and cKO macrophages. n = 4. (D) Quantification of apoptosis by a Cell-Death ELISA in empty vector (EV) transduced con (con/EV), cKO (cKO/EV), and cKO/LIS1-reconstituted pre-osteoclasts. Data are presented as mean ± s.d., n = 6, *** p < 0.001 by one-way ANOVA. nd, no statistical difference. (E) and (F) western blots of 50ng/ml M-CSF-induced activation of ERK and AKT pathways and 100ng/ml RANKL-stimulated JNK and NF-κB pathways in EV transduced con, cKO and cKO/LIS1-reconstituted bone marrow monocytes after an overnight serum and cytokine starvation. p-ERK, phosphorylated ERK; t-ERK, total ERK; p-AKT, phosphorylated AKT; t-AKT, total AKT; p-JNK, phosphorylated JNK; p-IκB, phosphorylated IκB; t- IκB, total IκB; DIC, Dynein intermediate chain.
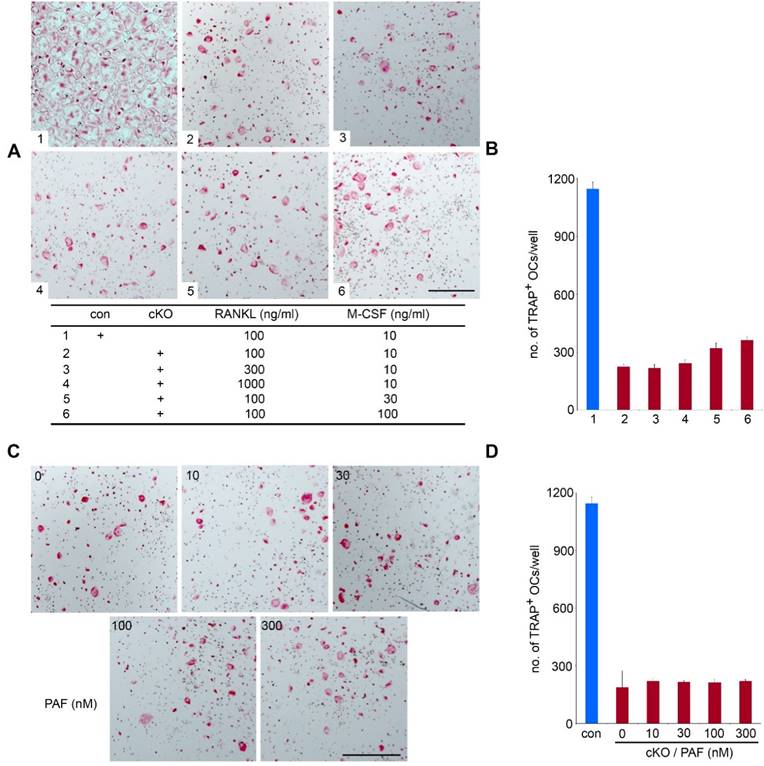
High doses of M-CSF, RANKL and PAF fail to rescue the osteoclastogenic defect in LIS1-null monocytes
It has been reported that high-dose of M-CSF and/or RANKL can overcome defects in osteoclast formation in certain in vitro experimental models [28, 29]. To test if such is case in LIS1-deficiency, we increased concentrations of M-CSF and/or RANKL in the culture medium. While causing earlier osteoclast formation and cell death by high doses of M-CSF and RANKL in control cultures (data not shown), 3- and 10-fold more RANKL failed to normalize osteoclast formation in LIS1-null cultures (Figure 5A-B). High-dose M-CSF slightly increased number of TRAP positive, multinucleated osteoclasts. LIS1 was originally identified as the regulatory subunit of PAFAH 1b complex which inactivates PAF by removing the acetyl group form the glycerol backbone of PAF [11]. Since PAF has been implicated as an important autocrine lipid messanger regulating osteoclast survival and function [14], we next determined whether addition of exogenous PAF in LIS1-deficient cultures could rescue osteoclastogenic defect. Figure 5 C-D showed that PAF, up to 300nM of concentration, could not stimulate osteoclast formation in LIS1-deficient osteoclast precursor cells. Thus, LIS1 regulates osteoclast survival and formation through a PAF-independent mechanism.
High doses of M-CSF, RANKL, and PAF fail to rescue osteoclastogenic defects in LIS1-deficient osteoclast precursors. (A) and (C) Microscopic images of TRAP-stained
Loss of LIS1 attenuates M-CSF and RANKL induced CDC42 activation and a constitutively active form of CDC42 partially rescues osteoclastogenesis in LIS1-deficient osteoclast precursors
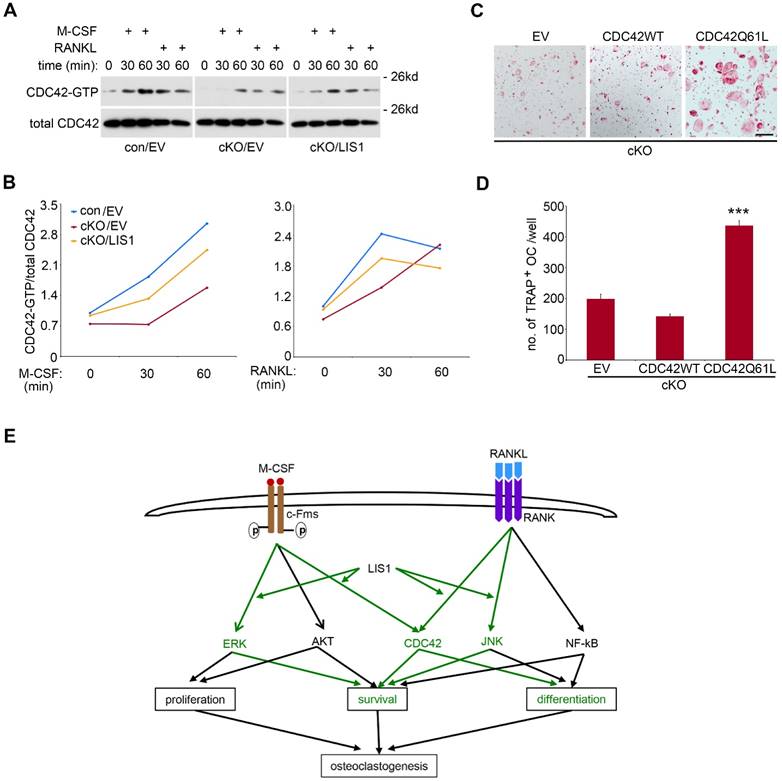
Like other small GTPases, CDC42 switches between GTP-bound active form and GDP-bound inactive form [30]. LIS1 and its binding protein NDEL1 have been shown to regulate CDC42 activation [15, 16]. We have previously reported that M-CSF and RANKL activate CDC42 in pre-osteoclasts and CDC42 is critical for osteoclast survival and formation in vivo and in vitro [17]. We, therefore, asked whether LIS1 modulates CDC42 activation in osteoclast precursor cells and whether CDC42 mediates LIS1's action on osteoclastogenesis. To address these questions, we first performed a CDC42-GTP pull-down assay in M-CSF and RANKL-stimulated control and LIS1-deficient pre-osteoclasts. Shown in Figure 6A and 6B, absence of LIS1 attenuated M-CSF-induced DCc42 activation at 30- and 60-minute time points. LIS1-deficiency also abrogated RANKL-induced CDC42 activation at 30- but not 60-minute stimulation. Retroviral transduction of LIS1, but not empty vector, in LIS1-null pre-osteoclasts partially rescued the decreased CDC42 activation. In consistence with an important role of CDC42 in osteoclastogenesis, retroviral transduction of a constitutively active form of CDC42 (CDC42Q61L), but not the wild type one, in LIS1-deficient bone marrow monocytes markedly increased the number of TRAP positive, multi-nucleated osteoclasts (Figure 6C and 6D). Taken together, these data indicate that LIS1 regulates osteoclast formation, at least in part, through its modulation of M-CSF and RANKL induced CDC42 activation.
LIS1 modulates M-CSF and RANKL induced CDC42 activation and a constitutively active form of CDC42 (CDC42Q61L) partially rescues osteoclastogenic defect in LIS1-deficient osteoclast precursors. (A) Western blots of CDC42-GTP pull-down in empty vector (EV) transduced control (con/EV), LIS1 conditional deletion (cKO/EV), and cKO/LIS1-reconstituted pre-osteoclasts stimulated by 50ng/ml of M-CSF or 100ng/ml of RANKL after a three-hour serum and cytokine starvation. (B) Densitometry measurements of (A) by NIH Image J software. (C) Microscopic images of TRAP-stained osteoclasts cultured from LIS1-deficient bone marrow monocytes transduced with a retro-viral EV or vectors expressing wild type CDC42 (CDC42 WT) and a constitutively active CDC42 (CDC42Q61L). The original magnification: × 20. Scale bar = 200 µm. (D) cell counts of TRAP-positive mature osteoclasts with more than 3 nuclei. Data are presented as mean ± s.d. n = 6. *** p < 0.001 vs EV and CDC42WT by one-way ANOVA. (E) A working model of the role of LIS1 in osteoclastogenesis. Green highlights the molecules and pathways regulated by LIS1 in osteoclast precursor cells.
Discussion
Although LIS1 was first identified as a regulatory subunit of PAFAH 1b complex that metabolites PAF, subsequent genetic and cellular studies have uncovered that most well-known functions of LIS1 in mammalian cells, such as cell cycle progress, migration, and intracellular organelle transportation, are related to its regulatory role in the cytoplasmic dynein, a microtubule-based molecular motor complex [31]. In doing so, LIS1 forms an evolutionary conserved complex with NDE1 and NDEL1 [32, 33]. It has recently been shown that LIS1 regulates dynamic MAPK signaling threshold through a direct interaction with BRAP (BRCA1 associated protein), a MAPK modulator [34]. In our previous studies on PLEKHM1, a lysosomal adaptor protein functioning in osteoclast bone resorption, we have found that LIS1 interacts with PLEKHM1 and regulates microtubule dynamics, dynein function and lysosomal secretion in osteoclasts [18]. Unexpectedly, depletion of LIS1 expression in macrophages by short-hairpin RNAs (shRNAs) markedly decreased osteoclast formation in vitro. To gain better understanding of how LIS1 regulates osteoclastogenesis in vivo and in vitro, we generated conditional knockout mice with specific deletion of LIS1 gene in osteoclast precursor cells by crossing LIS1-floxed mice with LysM-Cre mice. We report here that loss of LIS1 in osteoclast progenitors in mice results in an increase in trabecular bone mass with decreasing number of osteoclasts on bone surface. In vitro mechanistic studies provide evidence that LIS1 regulates the survival of osteoclast precursor cells by modulating the kinetics of M-CSF-induced ERK and RANKL-activated JNK pathways. LIS1 promotes M-CSF and RANKL induced Cdc42 activation which, in turn, is critical for optimized osteoclastogenesis (Figure 6E).
It should be pointed out that the skeletal phenotypes in LIS1 conditional knockout mice are relatively minor given that loss of LIS1 in osteoclast progenitors dramatically inhibits osteoclast formation and function in vitro (present study and reference [18]). The exact explanations and the underlying mechanisms remain unclear and need to be further investigated. One possibility is that Ndel1 and/or Nde1 partially compensate loss of LIS1 functions in osteoclast lineage cells because these three protein have been reported to interact each other and coordinate their functions. Indeed, we found that the expression of NDEL1 and NDE1 was up-regulated in LIS1-deficient pre-osteoclasts and mature cells (supplemental Figure 2).
In the present study, we focused on elucidating the cellular and molecular mechanisms by which LIS1 regulates osteoclastogenesis. Using bone marrow monocytes isolated from control and LIS1 conditional knockout mice and in vitro osteoclast cultures, we have found that loss of LIS1 in osteoclast progenitors attenuates osteoclast formation. Unlike in hematopoietic stem cells, the cell cycle progress in monocytes is not regulated by LIS1. Instead, loss of LIS1 promotes cell death in pre-osteoclasts. All these defects can be partially rescued by LIS1 reconstitution, suggesting that these effects are cell autonomous. Although activation of JNK pathway is required for RANKL-induced osteoclast differentiation [35, 36], prolonged JNK activation results in osteoclast apoptosis [27]. We have found previously [18] and in this study that LIS1 modulates the duration, but not their maximum activation, of M-CSF-induce ERK and RANKL-stimulated JNK activation in osteoclast lineage cells. Whether the function of LIS1 in this regard is mediated by BRAP as LIS1 does in neuronal cells [34] needs further investigation in the future. Together, these changes in the kinetics of M-CSF and RANKL signaling in LIS1-null osteoclast precursor cells lead to increased cell death during osteoclastogenesis.
It has been reported that high-dose of M-CSF and/or RANKL can rescue osteoclastogenesis defects in certain experimental models [28, 29]. It turned out this is not the case in terms of LIS1 deficiency. High-dose of M-CSF and RANKL failed to rescue decreased osteoclast formation in LIS1-null cultures. Similarly, PAF, a potent stimulator of osteoclast formation, could not rescue the defects in LIS1-deficient osteoclast progenitors. Instead, LIS1 participates in the activation of CDC42 induced by M-CSF and RANKL which modulates osteoclastogenesis in vivo and in vitro.
In closing, we have provided evidence indicating that LIS1 promotes M-CSF induced prolonged ERK activation and helps to restrict prolonged JNK activation which induces apoptosis in osteoclast precursor cells. In addition, LIS1 is required for the optimized CDC42 activation stimulated by both M-CSF and RANKL. Thus, LIS1 regulates the cell survival and/or differentiation of osteoclast precursors during osteoclastogenesis in vivo and in vitro (Figure 6E).
Supplementary Material
Supplementary figures.
Abbreviation
7'AAD, 7-aminoactinomycin-D; BMM, bone marrow macrophage; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; M-CSF, macrophage colony-stimulating factor; NFATc1, nuclear factor of activated T cells, cytoplasmic 1; NF-κB, receptor activator of nuclear factor-κB; PAF, platelet-activating factor; PAFAH, PAF acetylhydrolase; PAFR, PAF receptor; PBS, phosphate buffered saline; RANKL, the receptor activator of NF-κB ligand; TRAP, tartrate-resistant acid phosphatase.
Acknowledgements
The authors would like to thank Dr. O Reiner (The Weizmann Institute of Science, Rehovot, Israel) for providing LIS1 antibody and Andrea Harris for Flow Cytometry. UAMS Flow Cytometry Core Facility is supported in part by the Center for Microbial Pathogenesis and Host Inflammatory Responses grant P20GM103625 through the NIH National Institute of General Medical Sciences Centers of Biomedical Research Excellence. Erin Hogan is grateful for her support in microscopes. The work was supported by grants from National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS, AR062012 and AR068509) and National Institute of Aging (NIA, P01 AG13918).
Competing Interests
All authors claim no competing interest.
References
1. Boyce BF. Advances in the regulation of osteoclasts and osteoclast functions. Journal of dental research. 2013;92:860-7
2. Crockett JC, Rogers MJ, Coxon FP, Hocking LJ, Helfrich MH. Bone remodelling at a glance. Journal of cell science. 2011;124:991-8
3. Novack DV, Teitelbaum SL. The osteoclast: friend or foe?. Annual review of pathology. 2008;3:457-84
4. Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nature reviews Genetics. 2003;4:638-49
5. Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H. et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Developmental cell. 2002;3:889-901
6. Negishi-Koga T, Takayanagi H. Bone cell communication factors and Semaphorins. BoneKEy reports. 2012;1:183
7. Hattori M, Adachi H, Tsujimoto M, Arai H, Inoue K. Miller-Dieker lissencephaly gene encodes a subunit of brain platelet-activating factor acetylhydrolase [corrected]. Nature. 1994;370:216-8
8. Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB. et al. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature. 1993;364:717-21
9. Vallee RB, Tsai JW. The cellular roles of the lissencephaly gene LIS1, and what they tell us about brain development. Genes & development. 2006;20:1384-93
10. Wynshaw-Boris A, Gambello MJ. LIS1 and dynein motor function in neuronal migration and development. Genes & development. 2001;15:639-51
11. Tjoelker LW, Stafforini DM. Platelet-activating factor acetylhydrolases in health and disease. Biochimica et biophysica acta. 2000;1488:102-23
12. Prescott SM, Zimmerman GA, Stafforini DM, McIntyre TM. Platelet-activating factor and related lipid mediators. Annual review of biochemistry. 2000;69:419-45
13. Chao W, Olson MS. Platelet-activating factor: receptors and signal transduction. The Biochemical journal. 1993;292( Pt 3):617-29
14. Hikiji H, Ishii S, Shindou H, Takato T, Shimizu T. Absence of platelet-activating factor receptor protects mice from osteoporosis following ovariectomy. The Journal of clinical investigation. 2004;114:85-93
15. Kholmanskikh SS, Koeller HB, Wynshaw-Boris A, Gomez T, Letourneau PC, Ross ME. Calcium-dependent interaction of Lis1 with IQGAP1 and Cdc42 promotes neuronal motility. Nature neuroscience. 2006;9:50-7
16. Shen Y, Li N, Wu S, Zhou Y, Shan Y, Zhang Q. et al. Nudel binds Cdc42GAP to modulate Cdc42 activity at the leading edge of migrating cells. Developmental cell. 2008;14:342-53
17. Ito Y, Teitelbaum SL, Zou W, Zheng Y, Johnson JF, Chappel J. et al. Cdc42 regulates bone modeling and remodeling in mice by modulating RANKL/M-CSF signaling and osteoclast polarization. The Journal of clinical investigation. 2010;120:1981-93
18. Ye S, Fowler TW, Pavlos NJ, Ng PY, Liang K, Feng Y. et al. LIS1 regulates osteoclast formation and function through its interactions with dynein/dynactin and Plekhm1. PloS one. 2011;6:e27285
19. Zhao H, Ito Y, Chappel J, Andrews NW, Teitelbaum SL, Ross FP. Synaptotagmin VII regulates bone remodeling by modulating osteoclast and osteoblast secretion. Developmental cell. 2008;14:914-25
20. Zhou J, Fujiwara T, Ye S, Li X, Zhao H. Ubiquitin E3 Ligase LNX2 is Critical for Osteoclastogenesis In Vitro by Regulating M-CSF/RANKL Signaling and Notch2. Calcified tissue international. 2015;96:465-75
21. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nature protocols. 2008;3:1101-8
22. Zhou J, Fujiwara T, Ye S, Li X, Zhao H. Downregulation of Notch modulators, tetraspanin 5 and 10, inhibits osteoclastogenesis in vitro. Calcified tissue international. 2014;95:209-17
23. Zhou J, Ye S, Fujiwara T, Manolagas SC, Zhao H. Steap4 plays a critical role in osteoclastogenesis in vitro by regulating cellular iron/reactive oxygen species (ROS) levels and cAMP response element-binding protein (CREB) activation. The Journal of biological chemistry. 2013;288:30064-74
24. Zimdahl B, Ito T, Blevins A, Bajaj J, Konuma T, Weeks J. et al. Lis1 regulates asymmetric division in hematopoietic stem cells and in leukemia. Nature genetics. 2014;46:245-52
25. Nakashima T, Hayashi M, Takayanagi H. New insights into osteoclastogenic signaling mechanisms. Trends in endocrinology and metabolism: TEM. 2012;23:582-90
26. Takeshita S, Faccio R, Chappel J, Zheng L, Feng X, Weber JD. et al. c-Fms tyrosine 559 is a major mediator of M-CSF-induced proliferation of primary macrophages. The Journal of biological chemistry. 2007;282:18980-90
27. Vaira S, Alhawagri M, Anwisye I, Kitaura H, Faccio R, Novack DV. RelA/p65 promotes osteoclast differentiation by blocking a RANKL-induced apoptotic JNK pathway in mice. The Journal of clinical investigation. 2008;118:2088-97
28. Faccio R, Takeshita S, Zallone A, Ross FP, Teitelbaum SL. c-Fms and the alphavbeta3 integrin collaborate during osteoclast differentiation. The Journal of clinical investigation. 2003;111:749-58
29. Faccio R, Zou W, Colaianni G, Teitelbaum SL, Ross FP. High dose M-CSF partially rescues the Dap12-/- osteoclast phenotype. Journal of cellular biochemistry. 2003;90:871-83
30. Melendez J, Grogg M, Zheng Y. Signaling role of Cdc42 in regulating mammalian physiology. The Journal of biological chemistry. 2011;286:2375-81
31. Reiner O, Sapir T. LIS1 functions in normal development and disease. Current opinion in neurobiology. 2013;23:951-6
32. Pawlisz AS, Mutch C, Wynshaw-Boris A, Chenn A, Walsh CA, Feng Y. Lis1-Nde1-dependent neuronal fate control determines cerebral cortical size and lamination. Human molecular genetics. 2008;17:2441-55
33. Sasaki S, Shionoya A, Ishida M, Gambello MJ, Yingling J, Wynshaw-Boris A. et al. A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron. 2000;28:681-96
34. Lanctot AA, Peng CY, Pawlisz AS, Joksimovic M, Feng Y. Spatially dependent dynamic MAPK modulation by the Nde1-Lis1-Brap complex patterns mammalian CNS. Developmental cell. 2013;25:241-55
35. Ikeda F, Nishimura R, Matsubara T, Tanaka S, Inoue J, Reddy SV. et al. Critical roles of c-Jun signaling in regulation of NFAT family and RANKL-regulated osteoclast differentiation. The Journal of clinical investigation. 2004;114:475-84
36. Kawaida R, Ohtsuka T, Okutsu J, Takahashi T, Kadono Y, Oda H. et al. Jun dimerization protein 2 (JDP2), a member of the AP-1 family of transcription factor, mediates osteoclast differentiation induced by RANKL. The Journal of experimental medicine. 2003;197:1029-35
Author contact
![]() Corresponding author: Haibo Zhao, MD, PhD. Center for Osteoporosis and Metabolic Bone Diseases, Division of Endocrinology and Metabolism, University of Arkansas for Medical Sciences, 4301 West Markham Street, Slot 587, Little Rock, AR 72205. Phone: 501-686-5130 Fax: 501-686-8148 Email: hzhaoedu.
Corresponding author: Haibo Zhao, MD, PhD. Center for Osteoporosis and Metabolic Bone Diseases, Division of Endocrinology and Metabolism, University of Arkansas for Medical Sciences, 4301 West Markham Street, Slot 587, Little Rock, AR 72205. Phone: 501-686-5130 Fax: 501-686-8148 Email: hzhaoedu.
Received 2016-3-18
Accepted 2016-9-11
Published 2016-11-25