ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2017; 13(1):76-84. doi:10.7150/ijbs.17278 This issue Cite
Research Paper
Inhibition of miRNA-210 reverses nicotine-induced brain hypoxic-ischemic injury in neonatal rats
Lei Wang1,2, Jun Ke1,3, Yong Li1, Qinyi Ma1, Chiranjib Dasgupta1, Xiaohui Huang1, Lubo Zhang1, DaLiao Xiao1 ![]()
1. Center for Perinatal Biology, Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, California, USA.
2. Department of Traditional Chinese Medicine, Shanghai Putuo District People's Hospital, Shanghai, China.
3. Guangdong Cardiovascular Institute, Guangdong General Hospital, Guangdong Academy of Medical Sciences, Guangzhou, Guangdong, China.
Abstract

Maternal tobacco use in pregnancy increases the risk of neurodevelopmental disorders and neurobehavioral deficits in postnatal life. The present study tested the hypothesis that perinatal nicotine exposure exacerbated brain vulnerability to hypoxic-ischemic (HI) injury in neonatal rats through up-regulation of miR-210 expression in the developing brain. Nicotine was administered to pregnant rats via subcutaneous osmotic minipumps. Experiments of HI brain injury were performed in 10-day-old pups. Perinatal nicotine treatment significantly decreased neonatal body and brain weights, but increased the brain to body weight ratio. Perinatal nicotine exposure caused a significant increase in HI brain infarct size in the neonates. In addition, nicotine enhanced miR-210 expression and significantly attenuated brain-derived neurotrophic factor (BDNF) and tropomyosin-related kinase isoform B (TrkB) protein abundance in the brain. Of importance, intracerebroventricular administration of a miR-210 inhibitor (miR-210-LNA) significantly decreased HI-induced brain infarct size and reversed the nicotine-increased vulnerability to brain HI injury in the neonate. Furthermore, miR-210-LNA treatment also reversed nicotine-mediated down-regulation of BDNF and TrkB protein expression in the neonatal brains. These findings provide novel evidence that the increased miR-210 plays a causal role in perinatal nicotine-induced developmental programming of ischemic sensitive phenotype in the brain. It represents a potential novel therapeutic approach for treatment of brain hypoxic-ischemic encephalopathy in the neonate-induced by fetal stress.
Keywords: perinatal nicotine, microRNA-210, hypoxic-ischemic injury, neonatal brain
Introduction
Neonatal hypoxic-ischemic (HI) brain injury is a very common malignant complication caused by clinical perinatal asphyxia [1]. Despite continued advances in perinatal medicine, hypoxic-ischemic encephalopathy (HIE) is still one of the leading causes of brain damage, associated with high mortality and morbidity, along with cerebral palsy, epilepsy and cognitive disabilities [2]. However, the etiology and molecular mechanisms of development of HIE remain largely unclear. In addition, although therapeutic hypothermia can reduce neonatal mortality and improve neonatal morbidity in the early stages of HIE [3], some newborns with therapeutic hypothermia die prematurely or have permanent neurological deficits [4]. Therefore, it is very important to investigate the potential molecular therapeutic targets for prevention and effective treatment of HIE.
Numerous fetal insults contribute to the developmental susceptibility of hypoxic-ischemic brain injury. Maternal tobacco smoking is one of the major perinatal insults that can cause aberrant brain development, resulting in increased vulnerability to hypoxic-ischemic brain injury in neonates. Human studies have shown that smokers have a higher incidence of cardiovascular diseases and ischemic stroke [5, 6]. As one of the major active components in cigarettes, nicotine may contribute to the effect of smoking. Indeed, previous studies in animal models have demonstrated that chronic nicotine exposure makes rats more susceptible to ischemic brain injury [7, 8]. Furthermore, our recent studies have demonstrated that perinatal nicotine exposure causes aberrant fetal brain development, leading to an increase in vulnerability to HIE injury, in neonatal rats [9, 10].
The molecular mechanisms underlying perinatal nicotine-mediated exacerbated brain ischemic injury in neonates remain elusive. Previous studies have shown that microRNA (miR)-mediated signaling pathways are significantly involved in various neurological disorders including brain ischemic injury [11, 12]. In addition, growing evidence has indicated that cigarette smoking and nicotine exposure significantly alter the miRNAs profile in human and animal models [13-16]. Furthermore, previous studies have demonstrated that maternal stress induces epigenetic signatures of neurological diseases and cardiovascular disorders in offspring via miRNA pathway [17]. Given the fact that miR-210 has been shown to play a key role in the process of hypoxic-ischemic brain injury [18, 19], in the present study we examined the potential effect of perinatal nicotine exposure on the level of miR-210 in the developing brains and tested the hypothesis that the miR-210 is one of the key therapeutic molecular targets for nicotine-mediated exacerbated brain hypoxic-ischemic injury in the neonatal offspring.
Materials and Methods
Experimental Animals
All procedures and protocols in present study were approved by the Institutional Animal Care and Use Committee of Loma Linda University and followed the guidelines by the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Time-dated (day 2 of gestation) pregnant Sprague-Dawley rats were obtained from Charles River Laboratories (Portage, MI) and housed individually in Plexiglas acrylic plastic cages in air-conditioned rooms (room temperature 22ºC, relative humidity 60%; lights on from 8:00 a.m. to 8:00 p.m.). Pellet food and tap water were available ad libitum. On day 4 of gestation, the rats were randomly divided into two groups: 1) saline control and 2) nicotine group. Nicotine was administered to pregnant rats through an osmotic minipump at 4 μg/kg/min for 28 days, as described in detail previously [9, 10, 20]. The dose of nicotine resulted in blood levels closely resembling those occurring in moderate human smokers [21]. Control rats received saline from the osmotic minpump as the vehicle control. Our previous studies [9, 10] in the same animal models have demonstrated that perinatal nicotine exposure causes heightened brain vulnerability to hypoxic-ischemic (HI) injury in neonatal male but not female pups, therefore the male pups were chosen for present experiments. To see whether nicotine exposure affects postnatal growth, the body weights and brain weights in the 7-days-old pups were measured in both saline control and nicotine-treated groups.
Brain HI Treatment and Intracerebroventricular Injection (i.c.v)
A modified Rice-Vannucci model was employed as we described previously [9, 10]. Briefly, the offspring at 10-days-old (P10) were anesthetized with inhalation of 2-3% isoflurane. Right common carotid artery (CCA) was exposed, double ligated with a 5.0 silk surgical suture and then cut between two ligation sites. After surgery, pups were recuperated for 1.5 hours, and then placed in a hypoxic incubator containing humidified 8% oxygen balanced with 92% nitrogen for 1.5 hours at 37℃. Inhalation of 8% oxygen-balance nitrogen is the most common concentration used to produce systemic hypoxia in the rat pups [9, 10, 22]. The rationale for using 8% oxygen is that the rat pups are capable of surviving this severity of hypoxia, which are allowed to measure the patho-physiologic variables in the rat pups [22]. Pups were returned to their dams for recovery after the hypoxic treatment. For the sham treatment, CCA was exposed but without ligation and the hypoxic treatment.
In order to determine the functional significance of the enhanced miR-210 expression patterns in nicotine-induced heightened HI brain injury in neonates, miR-210 inhibitor was administered intracerebroventricularly in the pups 48 hours before the HI treatment as described previously [9, 18]. Briefly, miR-210-complementary locked nucleic acid oligonucleotides (miR-210-LNA, a miR-210 inhibitor) and negative control (LNA scramble control) (Exiqon) working solution were prepared according to manufacturer's instructions. Drugs with the total volume of 2 µL were administered into the ipsilateral hemisphere of rat pups via i.c.v. (2 mm posterior, 1.5 mm lateral, 3 mm below the skull surface). The treated animal groups were as following: 1) saline control + miR-210-LNA (50 pmol), 2) saline control + LNA scramble control (50 pmol), 3) nicotine + miR-210-LNA (50 pmol), and 4) nicotine + LNA scramble control (50 pmol). The rationale for using the dose of miR-210-LNA (50 pmol) is that treatment of miR-210-LNA via i.c.v. administration significantly knocks down miR-210 expression in the rat neonatal brain [18]. The HI treatment was performed 48 h after the injection.
Infarct Size Measurement
Pups were anesthetized and killed 48 hours after the HI treatment. Coronal slices of the brain (2 mm thick) were cut and immersed in a 1% solution of 2,3,5-triphenyltetrazolium chloride monohydrate (TTC; Sigma-Aldrich) for 5 minutes at 37℃ and then fixed by 10% formaldehyde overnight. Each slice was weighed, photographed separately and the percentage of infarction area for each slice was analyzed by Image J software (Version 1.40; National Institute of Health, Bethesda, MD), corrected by slice weight, summed for each brain, and expressed as a percentage of whole brain weight.
Western blot analysis
Protein abundance of brain-derived neurotrophic factor (BDNF) and tropomyosin-related kinase isoform B (TrkB) in the neonatal brain was measured with Western blot analysis as described previously [23]. In brief, tissues were homogenized in a lysis buffer containing 150 mM NaCl, 50 mM Tris HCl, 10 mM EDTA, 0.1% Tween 20, 0.1% β-mercapto-ethanol, 0.1 mM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, and 5 μg/ml aprotinin, pH 7.4. Homogenates were then centrifuged at 4°C for 10 min at 10,000g, and supernatants were collected. Samples with equal amount of proteins were loaded on to the 7.5% polyacrylamide gel with 0.1% SDS and were separated by electrophoresis at 100 V for 90 minutes. Proteins were then transferred to the nitrocellulose membrane and incubated with primary antibodies for BDNF and TrkB (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), respectively. After washing, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (GE Healthcare, Chalfont St. Giles, Buckingghamshire, UK). Proteins were visualized with enhanced chemiluminescence reagents, and blots were exposed to Hyperfilm (GE Healthcare). Results were quantified with electrophoresis documentation and analysis. Band intensities were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Real-Time qRT-PCR for miR-210 Quantification
MiR-210 levels in neonatal brain were measured by miScript II RT kit (Qiagen) and miScript SYBR Green PCR kit with miScript Primer Assay kit (Qiagen) according to manufacturer's instructions. The miScript PCR kit is an integral component of the miScript PCR system for miRNA detection and quantification which can reach 100% reverse transcription efficiency. Primers included miScript Universal Primer plus specific primer assays for miR-210 (Rn_miR-210_1; Cat#MS00000644; Qiagen) and SNORD61 (Hs_SNORD61_11; Cat#MS00033705; Qiagen). Briefly, 1 µg of template RNA isolated from either saline control or perinatal nicotine-treated neonatal brain was mixed with reverse-transcription master mix in a final volume of 20 µl and incubated for 60 min at 37 ℃, and the reaction was stopped at 95℃. Two nanograms of template cDNA were then used for miR-210 quantification in a final volume of 25 µl system containing specific primers and QuantiTect SYBR Green PCR master mix following manufacturer's instructions. PCR was done in triplicate and threshold cycle (CT) numbers were averaged for each sample. Comparative CT method was used for the RT-qPCR data analysis. The relative miR-210 levels were calculated as the Ct values of miR-210 to the reference gene (SONORD61) using the formula: 2ΔΔCT*100. SNORD61 was included in this study as housekeeping miRNA (internal control), which was not affected by the treatment.
Statistical Analysis
Data were expressed as means ± SEM obtained from the number (n) of experimental animals given. Statistical significance (P < 0.05) was determined by analysis of variance (ANOVA) or Student's t-test, where appropriate.
Results
Perinatal nicotine exposure induced asymmetrical growth restriction and enhanced miR-210 levels in neonatal pups
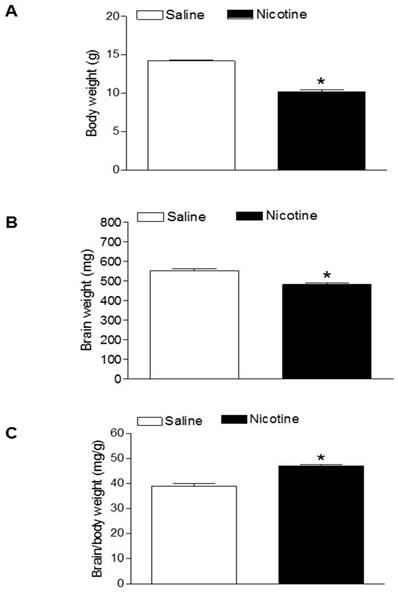
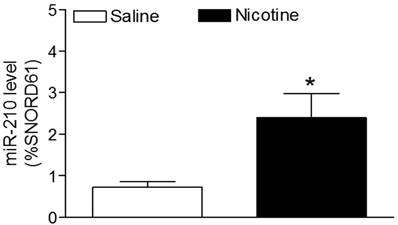
As shown in figure 1, maternal nicotine treatment significantly decreased both body weight and brain weight in the 7-days-old pups. However, nicotine significantly increased the brain to body weight ratio in the pups, which suggests that perinatal nicotine exposure causes an asymmetrical growth restriction in the neonates. By measurement of miR-210 levels in the brain using qRT-PCR analysis, we found that miR-210 levels were significantly higher in nicotine-treated group than in the saline control group (Figure 2).
miR-210 inhibitor reverses nicotine-mediated HI injury
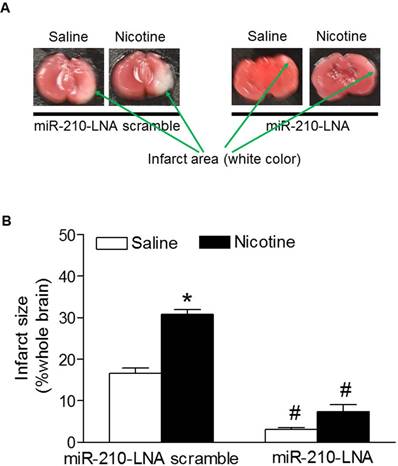
To determine the functional role of the enhanced miR-210 expression patterns in nicotine-induced heightened HI brain injury in neonates, the HI-induced brain infarct sizes were measured after treatment with miR-210 inhibitor. As shown in figure 3, treatment with miR-210-LNA scramble control, the HI-induced brain infarct sizes were significantly higher in nicotine-treated groups than in the saline control group. However, treatment with miR-210-LNA significantly decreased the HI-induced brain infarct sizes in both saline control and nicotine-treated groups. Furthermore, inhibition of miR-210 eliminated the differences of brain infarct size between the saline control and nicotine-treated groups.
Effects of nicotine on body weight, brain weight, and brain to body weight ratio in neonates. Nicotine was administrated to pregnant rats through osmotic pumps, as described under Material and Methods. Control pregnant rats received saline treatment. Body weight (A), brain weight (B), and brain to body weight ratio (C) were measured in offspring at age of 7-day-old. Data are mean ± SEM of animals (n=6-8). *P < 0.05 versus saline control group.
Effects of perinatal nicotine on miR-210 levels in neonatal brains. MicroR-210 levels in the 7-day-old neonatal brains from both nicotine-treated and saline control groups were measured by qRT-PCR analysis, as described under Material and Methods. Data are means ± SEM of animal numbers (n= 4-5) from each group. The pups were randomly chosen from different dams. The expression levels of miR-210 are expressed as percentage of SNOTD61. *P < 0.05 versus saline control group.
Effects of miR-210-LNA on HI-induced brain infarct size in P10 pups. HI-induced brain infarct size was measured in the postnatal pups (10-day-old) from both saline control and nicotine-treated groups after 48 hours of treatment with miR-210-LNA or negative control (Scramble). Panel (A) images represent the HI-induced brain infarct size determined by 1% TTC staining. The summed infarct sizes, expressed as a percentage of the total brain weight, are shown in panel (B). Data are means ± SEM of animal numbers (n=4-6) from each group. The pups were randomly chosen from different dams. Data were analyzed by 2-way ANOVA. *P < 0.05 versus saline control, #P < 0.05 versus miR-210-LNA scramble control. P10 pups indicate 10-day-old pups.
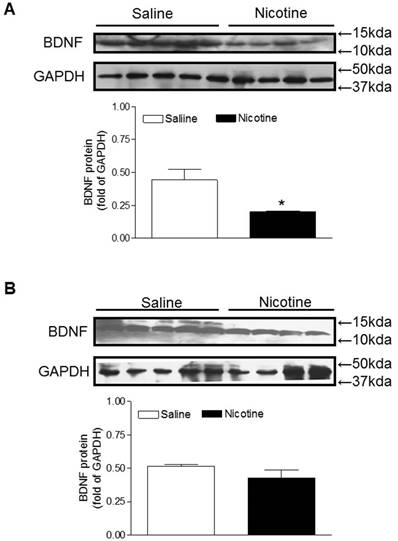
miR-210 inhibitor reverses nicotine-mediated alteration of BDNF and TrkB expressions
BDNF and TrkB are widely distributed throughout the central nervous systems and the BDNF-TrkB signaling plays a key role in neuronal survival and neurodegenerative disorders [24, 25]. Therefore, we examined the protein expression of BDNF to see whether perinatal nicotine exposure alters its expression and to see whether miR-210-LNA reverses the effect of nicotine. As shown in figure 4A, perinatal nicotine exposure significantly decreased brain BDNF protein levels as compared with the saline control. However, treatment with miR-210-LNA abolished the differences of BDNF protein expression between saline control and nicotine-treated animals (Figure 4B).
Effects of miR-210-LNA on protein abundance of BDNF in neonatal brain. Rat pups received via i.c.v. the negative control (A) or miR-210-LNA treatment (B). After 48 hours of treatment, BDNF protein abundance in the brain of 10 day-old pups from both saline control and nicotine-treated groups was determined by Western blot analysis. The protein density was normalized to GAPDH. All of the data expressed as means ± SEM of animal numbers (n=4-5) per group. The pups were randomly chosen from different dams. Data were analyzed by Student's t-test. *P < 0.05 versus saline control.
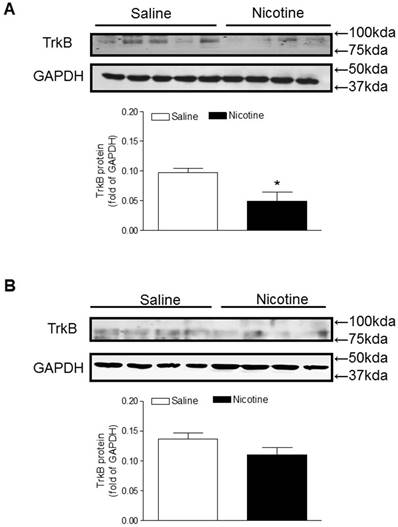
Similar to the BDNF expression, TrkB protein levels in the brain were also significantly lower in the perinatal nicotine-treated offspring than in the saline control offspring (Figure 5A). Furthermore, after treatment with miR-210-LNA there was no significant differences of TrkB protein expression between the saline control and nicotine-treated groups (Figure 5B).
Effects of miR-210-LNA on protein abundance of TrkB in neonatal brain. Rat pups received via i.c.v. the negative control (A) or miR-210-LNA treatment (B). After 48 hours of treatment, TrkB protein abundance in the brain of 10 day-old pups from both saline control and nicotine-treated groups was determined by Western blot analysis. The protein density was normalized to GAPDH. All of the data expressed as means ± SEM of animal numbers (n=4-5) per group. The pups were randomly chosen from different dams. Data were analyzed by Student's t-test. *P < 0.05 versus saline control.
Discussion
The present study provides direct evidence that perinatal nicotine-mediated increased miR-210 plays a causal role in the development of ischemic sensitive brain phenotype in neonates. The major findings in the present study are: (1) perinatal nicotine exposure significantly increased miR-210 levels in the neonatal brain; (2) Inhibition of miR-210 with miR-210-LNA significantly attenuated the HI-induced brain injury and eliminated the differences of HI-induced brain injury between the saline control and nicotine-exposed animals; (3) perinatal nicotine exposure significantly decreased BDNF protein expression in the neonatal brains, which was reversed by miR-210-LNA treatment; (4) perinatal nicotine exposure also suppressed TrkB expression in the neonatal brains as compared with the saline treated group, and miR-210-LNA treatment eliminated the differences of TrkB protein expression between the nicotine-treated and saline control groups.
In the present study, we found that perinatal nicotine exposure significantly decreased the body weight and brain weight of neonates as compared with the saline control groups, which is consistent with previous studies in similar animal models [9, 26]. The current results that brain to body weight ratio in P7 neonates was significantly higher in nicotine-treated group than the saline control group, suggest asymmetrical growth restriction in the neonate in response to perinatal nicotine exposure. Given the fact that the brain is one of the most important organs in the human body, the results that nicotine-mediated increased ratio of brain to body weight further suggest a protective/compensative mechanism for central organ development in response to intrauterine adverse environmental exposure. The growth restriction found in nicotine-exposed offspring provides a potential link between perinatal nicotine exposure and aberrant programming of cardiovascular disease and neurodevelopmental disorders later in life [27-29]. In present studies, we are not sure whether miR-210 inhibition reverses the nicotine-induced asymmetrical growth restriction, but we speculate that relative short-term treatment of miR-210 inhibitor may not restore the nicotine-mediated growth restriction. In our future studies, we will investigate the effect of miR-210 on nicotine-mediated growth restriction.
A growing body of evidence has shown that cigarette smoking and nicotine exposure significantly alter the miRNA profiles in humans and animal models [15, 16, 30, 31]. In present studies, we found that maternal nicotine exposure enhanced miR-210 expression in the neonatal brain compared with the saline control group, which is consistent with previous studies showing that miRNAs are sensitive to various prenatal insults [17, 32, 33]. However, the epigenetic molecular mechanisms underlying perinatal nicotine-induced up-regulation of miR-210 expression remain unclear. DNA methylation is one of the most common mechanisms of epigenetic modulation that involved various fetal programming of diseases in adulthood. Our previous studies have demonstrated that perinatal exposure nicotine causes a fetal programming of specific gene expression through DNA methylation mechanism, resulting in increased cardiovascular dysfunction and neurodevelopmental disorders later in life [9, 10, 26, 34]. In addition, recent studies suggest that miRNAs could enter the nucleus and be regulated by DNA methylation [35]. Given that the expression of miR-210 has been regulated by DNA methylation in cardiovascular and other diseases [36, 37], we speculate from the present results that nicotine-mediated over-expression of miR-210 may be regulated by DNA demethylation at the miR-210 promoter region. Therefore, our current findings open a broad door for us to further investigate the potential role of DNA methylation in perinatal nicotine-mediated up-regulation of miR-210 expression in neonatal brain.
MicroRNA-210, a master and pleiotropic hypoxia-miRNA, plays multiple roles in brain ischemia [38]. The current finding that pretreatment with miR-210 inhibitor significantly attenuated HI-induced brain infarct size, suggests that miR-210 has a functional significance in the pathophysiology of HI-induced brain injury in the developing brain. This finding is consistent with the previous study, which found that silencing miR-210 with miR-210-LNA via intracerebroventricular or intranasal delivery induced a neuroprotective effect on neonatal brain HI insult [18]. However, the biologic functional role of miR-210 in ischemic disease is inconsistent. It has been reported that miR-210 is involved in the regulation of angiogenesis in response to ischemic injury to the brain [39] and up-regulation of miR-210 protects cells from hypoxia-induced apoptosis [38]. Conversely, miR-210 blockade in the presence of hypoxia induces cell apoptosis [40], and miR-210 activation improves angiogenesis, inhibits apoptosis and improves cardiac function in a murine model of myocardial infarction [41]. Recently, in neonatal rats, hypoxia-ischemia (HI) treatment significantly reduced miR-210 expression and induced brain edema [42]. Furthermore, pretreatment with miR-210 mimic significantly improved HI-induced edema in the neonatal brain [42]. These results suggest that miR-210 may be involved in the pathological process of ischemic disease. However, whether miR-210 plays a protective or detrimental role in the hypoxic-ischemic injury may be dependent on the tissue type and the different insults.
The current finding that inhibition of miR-210 with miR-210-LNA eliminated the differences of HI-induced brain infarct size between the saline control and nicotine-treated neonatal offspring, suggests that nicotine-mediated over-expression of miR-210 may play a causal role in perinatal nicotine-induced HI injury to neonatal brain. Previous studies have provided direct evidence that miR-210 over-expression is a novel biomarker in a variety of ischemia diseases [41, 43, 44]. Of importance, previous studies have demonstrated that treatment with miR-210 mimic or inhibitor could significantly improve or exaggerate the ischemic disease [18, 19, 41]. Taken together, these findings suggest that miR-210 may serve as a potential novel therapeutic approach for treatment of ischemic brain injury-induced by perinatal nicotine exposure.
The molecular mechanisms underlying miR-210-mediated brain injury may be due to an alteration of its target genes. From the gene bank of rodent BDNF mRNA sequence and the database on target miRNAs (www.microrna.org), we have identified that miR-210 has a potential binding site at 3' UTR of gene encoding BDNF mRNA region (see the diagram in figure 6). Furthermore, previous studies have experimentally validated the binding sites of miR-210 at BDNF 3'UTR sequence and demonstrated that miR-210 directly regulates BDNF expression [45]. These observations suggest that the BDNF gene may be one of the important target genes of miR-210. BDNF, a member of the neurotrophin family, plays crucial roles in the developing central nervous system [24]. BDNF, binding with its receptor TrkB, controls the neuronal development, differentiation, proliferation and survival [24]. Altered BDNF/TrkB expression impacts not only the morphological development of neurons as well as their functional activities during brain development [25]. For example, BDNF involves in the formation of appropriate synaptic connections in the developing brain. Deletion of BDNF contributes to disorders of cognitive function [25]. In the present study, in addition to decreased brain weight (Figure 1), maternal nicotine treatment also significantly decreased BDNF and TrkB protein expressions in the developing brains of neonates as compared with the saline control groups, suggesting that the reduced expression of brain BDNF/TrkB may significantly contribute to the nicotine-mediated aberrant brain development and neurological dysfunction. In addition, we observed that miR-210-LNA treatment countered nicotine-induced reduction of BDNF/TrkB expression and eliminated the differences of BDNF/TrkB expression between the nicotine-treated and saline control groups. These findings suggests that miR-210 could directly regulate BDNF/TrkB expression, and the molecular mechanisms underlying miR-210-mediated brain injury in response to perinatal nicotine exposure may be due to decreased BDNF/TrkB expression in neonatal brains. Indeed, previous studies have reported that intravenous treatment with BDNF significantly reduced infarct size and neurological outcome after focal cerebral ischemia, further supporting the notion that BDNF plays a significant role neuroprotection against ischemic injury [46, 47]. Therefore, it's deserved to further investigation of the direct role of BDNF/TrkB signaling in nicotine-mediated HI injury through the techniques of gene manipulation (such as BDNF/TrkB gene knockout and knock-in) in animal model.
Diagram of miR-210 targeting BDNF. The diagram shows that miR-210 has putative binding target sequences at the three prime untranslated region (3'-UTR) in rat BDNF mRNA.
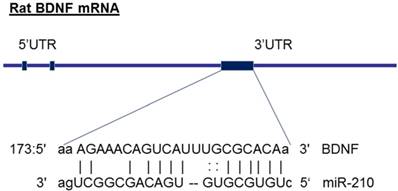
Diagram of potential mechanisms underlying perinatal nicotine exposure-induced exacerbated HI injury in neonatal pups. Perinatal nicotine exposure enhances miR-210 expression and attenuates BDNF-TrkB signaling pathway, leading to exacerbated hypoxic-ischemic (HI) injury in neonatal pups, which is reversed by the knock-down of miR-210 with miR-210-LNA.
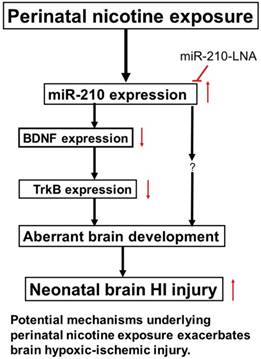
In conclusion, our present study provides new evidence that miR-210 may involve in the nicotine-induced epigenetic mechanism. As shown in Figure 7 (diagram), perinatal nicotine exposure enhances miR-210 expression, but decreases the neurotrophic protein (BDNF/TrkB) expression in neonatal brains, and subsequent development of brain hypoxic-ischemia sensitive phenotype in neonates. Inhibition of miR-210 using the LNA anti-miRNA-mediated miRNA knockdown technique reverses perinatal nicotine-mediated brain ischemic injury in offspring through up-regulation of BDNF/TrkB signaling pathway. Furthermore, in addition to BDNF/TrkB signaling, other molecular signaling pathways may be also regulated by miR-210, which are worthy of further investigation. Given the fact that miRNA-based therapies have already entered clinical trials for treatment of liver disease and cancer [48, 49], our present findings may provide a mechanistic understanding of the pathophysiology of HIE associated with nicotine exposure in pregnancy and have the promising translational implication in the prevention or treatment of HIE in the neonate.
Acknowledgements
This work was supported by National Institutes of Health Grants HL089012 (LZ), HL118861 (LZ), DA032510 (DX), and by the Regents of the University of California Tobacco Related Disease Research Program (TRDRP) grant 22XT-0022 (DX).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Thatipamula S, Ai Rahim M, Zhang J, Hossain MA. Genetic deletion of neuronal pentraxin 1 expression prevents brain injury in a neonatal mouse model of cerebral hypoxia-ischemia. Neurobiol Dis. 2015;75:15-30 Doi: 10.1016/j.nbd.2014.12.016
2. Kelen D, Robertson NJ. Experimental treatments for hypoxic ischemic encephalopathy. Early Hum Dev. 2010;86:369-77 Doi: 10.1016/j.earlhumdev.2010.05.011
3. Jacobs SE, Berg M, Hunt R, Tarnow-Mordi WO, Inder TE, Davis PG. Cooling for newborns with hypoxic ischemic encephalopathy. Cochrane Database Syst Rev. 2013;31:CD003311. Doi: 10.1002/14651858.CD003311.pub3
4. Azzopardi DV, Strohm B, Edwards AD, Dyet L, Halliday HL, Juszczak E, Kapellou O, Levene M, Marlow N, Porter E, Thoresen M, Whitelaw A, Brocklehurst P; TOBY Study Group. Moderate hypothermia to treat perinatal asphyxia encephalopathy. N Engl J Med. 2009;361:1349-58 Doi: 10, 1056/NEJMoa0900854
5. Mainali P, Pant S, Rodriguez AP, Deshmukh A, Mehta JL. Tobacco and cardiovascular health. Cardiovascular Toxicol. 2015;15:107-116 Doi: 10.1007/s12012-014-9280-0
6. Ovbiagele B, Weir CJ, Saver JL, Muir KW, Lees KR. Effect of smoking status on outcome after acute ischemic stroke. Cerebrovasc Dis. 2006;21:260-265 Doi: 10.1159/000091224
7. Li C, Sun H, Arrick DM, Mayhan WG. Chronic nicotine exposure exacerbates transient focal cerebral ischemia-induced brain injury. J Appl Physiol. 2016;120:328-33 Doi: 10.1152/japplphysiol.00663.2015
8. Raval AP, Hirsch N, Dave KR, Yavagal DR, Bramlett H, Saul I. Nicotine and estrogen synergistically exacerbate cerebral ischemic injury. Neuroscience. 2011;181:216-225 Doi: 10.1016/j.neuroscience.2011.02.036
9. Li Y, Xiao D, Dasgupta C, Xiong F, Tong W, Yang S, Zhang L. Perinatal nicotine exposure increases vulnerability of hypoxic-ischemic brain injury in neonatal rats: Role of angiotension II receptors. Stroke. 2012;43:2483-2490 Doi: 10.1161/STROKEAHA.112.664698
10. Li Y, Xiao D, Yang S, Zhang L. Promoter methylation represses AT2R gene and increases brain hypoxic-ischemic injury in neonatal rats. Neurobiol Dis. 2013;60:32-8 Doi: 10.1016/j.nbd.2013.08.011
11. Moon JM, Xu L, Giffard RG. Inhibition of microRNA-181 reduces forebrain ischemia-induced neuronal loss. J Cereb Blood Flow Metab. 2013;33:1976-1982 Doi: 10.1038/jcbfm.2013.157
12. Saugstad JA. MicroRNAs as effectors of brain function with roles in ischemia and injury, neuroprotection, and neurodegeneration. J Cereb Blood Flow Metab. 2010;30:1564-1576 Doi: 10. 1038/jcbfm.2010.101
13. Banerjee A, Luettich K. MicroRNAs as potential biomarkers of smoking-related diseases. Biomark Med. 2012;6:671-84 Doi: 10.2217/bmm.12.50
14. Takahashi K. Yokota S, Tatsumi N, Fukami T, Yokoi T, Nakajima M. Cigarette smoking substantially alters plasma microRNA profiles in healthy subjects. Toxicol Appl Pharmacol. 2013;272:154-60 Doi: 10.1016/j.taap.2013.05.018
15. Taki FA, Pan X, Zhang B. Chronic nicotine exposure systemically alters microRNA expression profiles during post-embryonic stage in Caenorhabditis elegant. J Cell Physiol. 2014;229:79-89 Doi: 10.1002/jcp.24419
16. Zhang Y, Pan T, Zhong X, Cheng C. Nicotine upregulates microRNA-21 and promotes TGF-β-dependent epithelial-mesenchymal transition of esophageal cancer cells. Tumour Biol. 2014;35:7063-72 Doi: 10.1007/s13277-014-1968-z
17. Zucchi FC, Yao Y, Ward ID, Ilnytskyy Y, Olson DM, Benzies K, Kovalchuk I, Kovalchuk O, Metz GA. Maternal stress induces epigenetic signatures of psychiatric and neurological diseases in the offspring. PLoS One. 2013;8:e56967. Doi: 10.1371/journal.pone.0056967
18. Ma Q, Dasgupta C, Li Y, Bajwa NM, Xiong F, Harding B, Hartman R, Zhang L. Inhibition of microRNA-210 provides neuroprotection in hypoxic-ischemic brain injury in neonatal rats. Neurobiol Dis. 2016;89:202-212 Doi: 10.1016/j.nbd.2016.02.011
19. Qiu J, Zhou XY, Zhou XG, Cheng R, Liu HY, Li Y. Neuroprotective effects of microRNA-210 on hypoxic-ischemic encephalopathy. Biomed Res Int. 2013;2013:350419. Doi:10.1155/2013/350419
20. Xiao D, Huang X, Yang S, Zhang L. Antenatal nicotine induces heightened oxidative stress and vascular dysfunction in rat offspring. Br J Pharmacol. 2011;164(5):1400-1409 Doi: 10.1111/j.1476-5381.2011.01437.x
21. Slotkin TA. Fetal nicotine or cocaine exposure: which one is worse?. J Pharmacol Exp Ther. 1998;285:931-945
22. Vannucci RC, Connor JR, Mauger DT, Palmer C, Smith MB, Towfighi J, Vannucci SJ. Rat model of perinatal hypoxic-ischemic brain damage. J Neurosci Res. 1999;55:158-63 Doi: 10.1002/(SICI)1097-4547(19990115)55:2<158::AID-JNR3>3.0.CO;2-1
23. Li Y, Ma Q, Dasgupta C, Halavi S, Hartman RE, Xiao D, Zhang L. Inhibition of DNA methylation in the developing rat brain disrupts sexually dimorphic neurobehavioral phenotypes in adulthood. Mol Neurobiol. 2016 Jun 16. [Epub ahead of print]. Doi: 10.1007/s12035-016-9957-4
24. Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677-736 Doi: 10.1146/annurev.neuro.24.1.677
25. Cohen-Cory S, Kidane AH, Shirkey NJ, Marshak S. Brain-derived neurotrophic factor and the development of structural neuronal connectivity. Dev Neurobiol. 2010;70:271-88 Doi: 10, 1002/dneu.20774
26. Lawrence J, Xaio D, Xue Q, Rejali M, Yang S, Zhang L. Prenatal Nicotine Exposure Increases Heart Susceptibility to Ischemia/Reperfusion Injury in Adult Offspring. J Pharmacol Exp Ther. 2008;324:331-341 Doi: 10.1124/jpet.107.132175
27. Wickstrom R. Effects of nicotine during pregnancy: human and experimental evidence. Curr Neuropharmacol. 2007;5:213-222 Doi: 10.2174/15701590778169555
28. Pauly JR, Slotkin TA. Maternal tobacco smoking, nicotine replacement and neurobehavioural development. Acta Paediatr. 2008;97:1331-1337 Doi: 10.1111/j.1651-2227.2008.00852.x
29. Xiao D, Xu Z, Huang X, Longo LD, Yang S, Zhang L. Prenatal gender-related nicotine exposure increases blood pressure response to angiotensin II in adult offspring. Hypertension. 2008;51:1239-1247 Doi: 10.1161/HYPERTENSIONAHA.107.106203
30. Badrnya S, Baumgartner R, Assinger A. Smoking alters circulating plasma microvesicle pattern and microRNA signatures. Thromb Haemost. 2014;112:128-36 Doi: 10.1160/TH13-11-0977
31. Banerjee A, Luettich K. MicroRNAs as potential biomarkers of smoking-related diseases. Biomark Med. 2012;6:671-84 Doi: 10.2217/bmm.12.50
32. Khorram O, Han G, Bagherpour R, Magee TR, Desai M, Ross MG, Chaudhri AA, Toloubeydokhti T, Pearce WJ. Effect of maternal undernutrition on vascular expression of micro and messenger RNA in newborn and aging offspring. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1366-74 Doi: 10.1152/ajpregu.00704.2009
33. States JC, Singh AV, Knudsen TB, Rouchka EC, Ngalame NO, Arteel GE, Piao Y, Ko MS. Prenatal arsenic exposure alters gene expression in the adult liver to a proinflammatory state contributing to accelerated atherosclerosis. PLoS One. 2012;7:e38713. Doi: 10.1371/journal.pone.0038713
34. Xiao D, Dasgupta C, Li Y, Huang X, Zhang L. Perinatal nicotine exposure increases angiotensin II receptor-mediated vascular contractility in adult offspring. PLoS One. 2014;9:e1-8161 doi: 10.1371/journal.pone.0108161
35. Sato F, Tsuchiya S, Meltzer SJ, Shimizu K. MicroRNAs and epigenetics. FEBS J. 2011;278:1598-1609 Doi: 10.1111/j.1742-4658.2011.08089.x
36. Chen KC, Liao YC, Wang JY, Lin YC, Chen CH, Juo SH. Oxidized low-density lipoprotein is a common risk factor for cardiovascular diseases and gastroenterological cancer via epigenenomical regulation of microRNA-210. Oncotarget. 2015;6:24105-18 Doi: 10.18632/oncotarget.4152
37. Kiga K. Mimuro H, Suzuki M, Shinozaki-Ushiku A, Kobayashi T, Sanada T, Kim M, Ogawa M, Iwasaki YW, Kayo H, Fukuda-Yuzawa Y, Yashiro M, Yashiro M, Fukayama M, Fukao T, Sasakawa C. Epigenetic silencing of miR-210 increases the proliferation of gastric epithelium during chronic helicobacter pylori infection. Nat Commun. 2014;5:4497. Doi: 10.1038/ncomms5497
38. Kulshreshtha R, Ferracin M, Wojcik SE, Garzon R, Alder H, Agosto-Perez FJ, Davuluri R, Liu CG, Croce CM, Negrini M, Calin GA, Ivan M. A microRNA signature of hypoxia. Mol Cell Biol. 2007;27:1859-67 Doi: 10.1128/MCB.01395-06
39. Lou YL, Guo F, Liu F, Gao FL, Zhang PQ, Niu X, Guo SC, Yin JH, Wang Y, Deng ZF. miR-210 activates notch signaling pathway in angiogenesis induced by cerebral ischemia. Mol Cell Biochem. 2012;370:45-51 Doi: 10.1007/s11010-012-1396-6
40. Fasanaro P, D'Alessandra Y, Di Stefano V, Melchionna R, Romani S, Pompilio G, Capogrossi MC, Martelli F. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand ephrin-A3. J Biol Chem. 2008;283:15878-83 Doi: 10.1074/jbc.m800731200
41. Hu S, Huang M, Li Z, Jia F, Ghosh Z, Lijkwan MA, Fasanaro P, Sun N, Wang X, Martelli F, Robbins RC. Wu JC. MircoRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation. 2010;122:S124-31 Doi: 10.1161/CIRCULATIONAHA.109.928424
42. Zhao L, Zhou XY, Zhou XG, Cheng R, Li Y, Qiu J. Role of miRNA-210 in hypoxic-ischemic brain edema in neonatal rats. Zhongguo Dang Dai Er Ke Za Zhi. 2016;18:770-4 Doi: 10.7499/j.issn.1008-8830.2016.08.019
43. Liu F, Lou YL, Wu J, Ruan QF, Xie A, Guo F, Cui SP, Deng ZF, Wang Y. Upregulation of microRNA-210 regulates renal angiogenesis mediated by activation of VEGF signaling pathway under ischemia/perfusion injury in vivo and in vitro. Kidney Blood Press Res. 2012;35:182-91 Doi: 10.1159/000331054
44. Zeng L, Liu J, Wang Y, Wang L, Weng S, Tang Y, Zheng C, Cheng Q, Chen S, Yang GY. MicroRNA-210 as a novel blood biomarker in acute cerebral ischemia. Front Biosci (Elite Ed). 2011;3:1265-72
45. Fasanaro P, Greco S, Lorenzi M, Pescatori M, Brioschi M, Kulshreshtha R, Banfi C, Stubbs A, Calin GA, Ivan M, Capogrossi MC, Martelli F. An integrated approach for experimental target identification of hypoxia-induced miR-210. J Biol Chem. 2009;284:35134-43 Doi: 10. 1074/jbc.m109.052779
46. Schabitz WR, Schwab S, Spranger M, Hacke W. Intraventricular brain-derived neurotrophic factor reduces infarct size after focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1997;17:500-506 Doi: 10.1097/00004647-199705000-00003
47. Schabitz WR, Sommer C, Zoder W, Kiessling M, Schwaninger M, Schwab S. Intravenous brain-derived neurotrophic factor reduces infarct size and counter-regulates Bax and Bcl-2 expression after temporary focal cerebral ischemia. Stroke. 2000;31:2212-7
48. Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Qrum H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198-201 Doi: 10.1126/science.1178178
49. Stenvang J, Petri A, Lindow M, Obad S, Kauppinen S. Inhibition of microRNA function by antimiR oligonucleotides. Silence. 2012;3:1. Doi: 10.1186/1758-907x-3-1
Author contact
![]() Corresponding author: Daliao Xiao, Ph.D. Center for Perinatal Biology, Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, CA 92350; Tel: 909-558-4325; Fax: 909-558-4029; E-mail: Dxiaoedu
Corresponding author: Daliao Xiao, Ph.D. Center for Perinatal Biology, Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, CA 92350; Tel: 909-558-4325; Fax: 909-558-4029; E-mail: Dxiaoedu
Received 2016-8-19
Accepted 2016-9-27
Published 2017-1-1