ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Result
Discussion
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2017; 13(2):219-231. doi:10.7150/ijbs.16725 This issue Cite
Research Paper
Hyperhomocysteinemia Exacerbates Cisplatin-induced Acute Kidney Injury
Yanjun Long1,2, Xin Zhen1, Fengxin Zhu1, Zheng Hu1, Wenjing Lei1, Shuang Li1, Yan Zha 2, Jing Nie1 ![]()
1. State Key Laboratory of Organ Failure Research, National Clinical Research Center of Kidney Disease, Division of Nephrology, Nanfang Hospital, Southern Medical University, Guangzhou, P.R. China;
2. Division of Nephrology, Guizhou Provincial People's Hospital, Guizhou Provincial Institute of Nephritic & Urinary Disease, Guiyang, P.R. China.
Abstract

Hyperhomocysteinemia (HHcy) has been linked to several clinical manifestations including chronic kidney disease. However, it is not known whether HHcy has a role in the development of acute kidney injury (AKI). In the present study, we reported that HHcy mice developed more severe renal injury after cisplatin injection and ischemia-reperfusion injury shown as more severe renal tubular damage and higher serum creatinine. In response to cisplatin, HHcy mice showed more prevalent tubular cell apoptosis and decreased tubular cell proliferation. Mechanistically, a heightened ER stress and a reduced Akt activity were observed in kidney tissues of HHcy mice after cisplatin injection. Stimulating cultured NRK-52E cells with Hcy significantly increased the fraction of cells in G2/M phase and cell apoptosis together with decreased Akt kinase activity. Akt agonist IGF-1 rescued HHcy-induced cell cycle arrest and cell apoptosis. In conclusion, the present study provides evidence that HHcy increases the sensitivity and severity of AKI.
Keywords: Hyperhomocysteinemia, AKI, apoptosis, cell proliferation, ER stress.
Introduction
Acute kidney injury (AKI), often resulting from ischemic, nephrotoxic, and septic insults, is a devastating clinical condition that is associated with high rates of morbidity and mortality. It has also become increasingly clear that AKI contribute to the development of chronic kidney disease (CKD) [1,2]. Despite the growing incidence of AKI, there is no effective therapy for this condition. CKD, Diabetes mellitus, aging, and heart failure are all strong risk factors for AKI [3-7]. However, aggressive control of these risk factors has not significantly improved the outcomes in patients with AKI, suggesting that other factors need to be evaluated.
Homocysteine (Hcy) is a sulfur-containing amino acid that is formed as the transmethylation product during the metabolic conversion of methionine to cysteine, with S-adenosylmethionine (SAM) and S-adenosylhomocysteine (SAH) as intermediates. Once synthesized, Hcy can be remethylated to methionine or be metabolized to cysteine [8]. In human, plasma level of total Hcy (tHcy) is about 10μM. Elevated plasma Hcy concentrations (tHcy>15μM), a condition known as hyperhomocysteinemia (HHcy), is developed by genetic enzymatic deficiencies and/or nutritional defects that interfere with the proper metabolism of methionine [9,10]. Moderate HHcy is defined as a plasma tHcy level in the range of 16-30μM, which is common in the general population. Genetic variation of the enzymes, or deficiency of nutritional factors, including folic acid, vitamin B6 and B12, impairs Hcy metabolism, can cause moderate HHcy.
Elevated plasma Hcy levels are prevalent in CKD patients and that a lower glomerular filtration rate (GFR) is associated with a higher plasma tHcy concentration [11,12]. There is considerable evidence showing that HHcy is implicated in the progression of CKD [13], however the impact of Hcy on AKI has received little attention. It is not known whether HHcy could increase the susceptibility or severity of AKI. Experimental studies suggest that local oxidative stress, endoplasmic reticulum (ER) stress, and DNA hypomethylation are cellular mechanisms mediating the pathogenic action of Hcy in endothelial and glomerular cells [14-20]. Given the critical role of oxidative stress and ER stress in tubular cell injury, we hypothesize that elevated plasma level of Hcy might increase the susceptibility to AKI. To address this issue, we generated HHcy mice by feeding mice with high methionine diet. We showed that cisplatin induces more severe tubular injury, tubular cell apoptosis and lower proliferation in HHcy mice than in normal mice. Mechanistically, the exacerbation of renal injury in HHcy mice appeared to be mediated by a heightened ER stress and a reduced Akt kinase activity.
Materials and Methods
Animal Experiments
Experiments were performed using 6 to 8 week-old male C57BL/6 mice. Mice were kept on a 12-h light/dark cycle with free access to diet and water. All animal were conducted under a protocol approved by Ethics Committee for Animal Experiments of the Southern Medical University.
To generate HHcy mice, mice were fed with high methionine diet containing 19.56g/kg (2%) methionine for two weeks. Cisplatin (24 mg/kg body weight) was given by a single intraperitoneal injection. Mice were killed at day 1 or day 3 after cisplatin administration, blood and kidney samples were harvested for further analysis.
Renal ischemia-reperfusion was induced in mice as described previously [21,22]. Briefly, mice were kept on a homeothermic station to maintain body temperature at 37°C. Kidneys were exposed by bilateral flank incisions, and the renal pedicles were clamped for 30 minutes, to induce ischemia, and then the clamps were released to allow reperfusion to the kidney. One milliliter of warm saline (37℃) was injected intraperitoneally after surgery for volume supplement. Sham operations were underwent the identical procedure except that clamping of the renal pedicles was omitted. Mice were killed 24h after reperfusion, blood and kidney samples were collected for further analysis.
Cell culture and treatment
Normal Rat Kidney epithelial cells (NRK-52E) were cultured in F12 medium supplemented with 10% fetal bovine serum (Gibco/Life Technologies, NY). When reached approximately 60% confluence, cells were changed to serum free medium overnight and then treated with DL-Hcy (Sigma-Aldrich, MO) at various concentrations for various time periods.
Serum Hcy measurement
Serum Hcy was measured using a Homocysteine Assay Kit (Ausa, CHN) and an automatic clinical analyzer (Beckman Coulter, CA) according to the manufacturer's instructions. The Hcy measurement was standardized by the National Institute of Standards and Technology (NIST) Standard Reference Material (SRM) 1955. Serum creatinine was measured by automatic biochemical analyzer (Beckman Coulter, CA).
Renal function
Renal function was assessed by measuring serum creatinine (Bioassay Systems, USA) using an automatic biochemical analyzer (Beckman Coulter, CA).
Histology
2μm paraffin-embedded kidney sections were subjected to hematoxylin and eosin staining using commercial kits (Sigma-Aldrich, MO) according to the manufacturer's protocol. Tubular injury was scored on a scale of 0-4 on the basis of the percentage of tubules with necrosis, dilatation, or cell swelling: 0, less than 5%; 1, 5-25%; 2, 25-50%; 3, 50-75%; and 4, over 75%. At least ten randomly chosen fields in the cortex under the microscope (×400) were evaluated for each mouse in a blinded manner, and an average score was calculated.
TUNEL assay
Renal apoptosis was detected by TUNEL assay using the in situ cell death detection kit POD (Roche, GER) according to the manufacturer's protocol. Briefly, paraffin-embedded renal tissue sections were exposed to the TUNEL reaction mixture. TUNEL-positive nuclei were identified by fluorescence microscopy. The number of TUNEL-positive cells was counted in 10 fields per section and five sections per kidney.
Immunofluorescence and Immunohistochemical Staining
Immunohistochemical staining was performed on 4μm kidney sections as previously described [23]. Briefly, sections were deparaffinized and rehydrated in ethanol. After antigen repairing, sections were incubated with the primary antibody against CD3+ (sc-20047, Santa Cruz Biotechnology, CA) and F4/80 (14-4801, eBioscience, CA) for 14 h at 4℃, followed by incubating with secondary antibodies (Dako, CA) for 30 min at 37ºC. Images were taken by an Olympus BX51 microscope (Olympus, JPN).
Immunofluorescence staining of the kidney was performed on 4μm kidney sections as previously described [24]. Briefly, sections were deparaffinized and rehydrated in ethanol, and then were microwaved in 0.01 mol/L sodium citrate (pH 6.0). After antigen repairing, Sections were incubated with overnight at 4°C with the primary antibodies against Ki67 (ab66155, Abcam, UK), followed by incubation with secondary antibody conjugated with Alexa Fluor 488 (Molecular Probes, Inc., USA). Tissues were then counterstained with 49, 6-diamidino-2-phenylindole (DAPI) to visualize the nuclei. Images were taken by a confocal microscopy (Olympus Corporation, JPN).
Western blotting
Frozen kidney tissues or cells were lysed in PLC lysis buffer containing cocktail inhibitor (Merck Millipore, GER) for 30 minutes on ice. Samples were boiled in SDS loading buffer and then separated on SDS-PAGE gels following standard protocol. Transfer membranes were immunoblotted with primary antibodies against cleaved caspase-3 (#9664S, Cell Signaling Technology, UK), caspase-12 (#2202P, Cell Signaling Technology, UK), CHOP (#2895P, Cell Signaling Technology, UK), GRP78 (#3177S, Cell Signaling Technology, UK), Phospho-Akt (Ser473) (#4060S, Cell Signaling Technology, UK), Akt (#4691S, Cell Signaling Technology, UK), Phospho-p70S6 kinase (#9204S, Cell Signaling Technology, UK), p70S6 kinase (#9202S, Cell Signaling Technology, UK) and GAPDH (#2118S, Cell Signaling Technology, UK) overnight at 4°C. After extensive washing in TBST buffer, the membranes were incubated with secondary antibody for 1h at room temperature. The protein bands on the western blots were then visualized using an ECL Plus (Amersham, IL) according to the manufacturer's instructions.
Quantitative real-time PCR
Total RNA was extracted from the kidney tissue using TRIzol Reagent (Invitrogen, CA) according to the standard protocol, 1μg of RNA was used to synthesize cDNA by PrimeScript® RT reagent Kit With gDNA Eraser (Takara, JP). Real time RT-PCR was performed on an ABI PRISM 7500 Fast sequence detection system (Applied Biosystems, CA). The primers used for evaluation were as follows: (i) TNF-α Forward Primer (5'-CAGGCGGTGCCTATGTCTC-3') and Reverse Primer (5'-CGATCACCCCGAAGTTCAGTAG-3'), (ii) MCP-1 Forward Primer (5'-TAAAAACCTGGATCGGAACCAAA-3') and Reverse Primer (5'-GCATTAGCTTCAGATTTACGGGT-3'), (iii) GAPDH Forward Primer (5'-GGTGAAGGTCGGTGTGAACG-3') and Reverse Primer (5'-CTCGCTCCTGGAAGATGGTG-3'). The mRNA levels of various genes were calculated after normalizing with GAPDH by the comparative CT method (2-DDCT).
MTT assay
MTT assay was performed as follows: 20μL of MTT (5mg/mL) was added to each well and the plates were incubated at 37℃ for 4h. The MTT medium mixture was then removed and 150μl of dimethyl sulfoxide was added to each well. The absorbance was measured at 490 nm using a multi-well spectrophotometer.
Flow cytometry analysis
Cells were washed with cold PBS and then stained with the Cycle TESTTM PLUS DNA Reagent Kit (Becton Dickinson, USA). Cell cycle distribution was evaluated using BD FACSCalibur Flow Cytometer. Data were analyzed using ModFit LT3.3 (BD, Topsham, ME, USA) and represented as percent cell in G0/G1, S, and G2/M.
To analyze cell apoptosis, NRK-52E cells were washed with PBS and then co-stained with Annexin V-APC and 7-AAD (MultiSciences Biotech Co, Ltd). The number of apoptotic cells was evaluated using BD FACSCalibur Flow Cytometer.
Statistical Analyses
Data were expressed as means ± SD, and differences between groups were analyzed using t-test, one-way ANOVA and correlation analysis (SPSS software, version 19.0; SPSS, Inc., IL). P<0.05 was considered significantly different. All experiments were carried out in duplicate.
Result
HHcy mice were susceptible to cisplatin-induced AKI
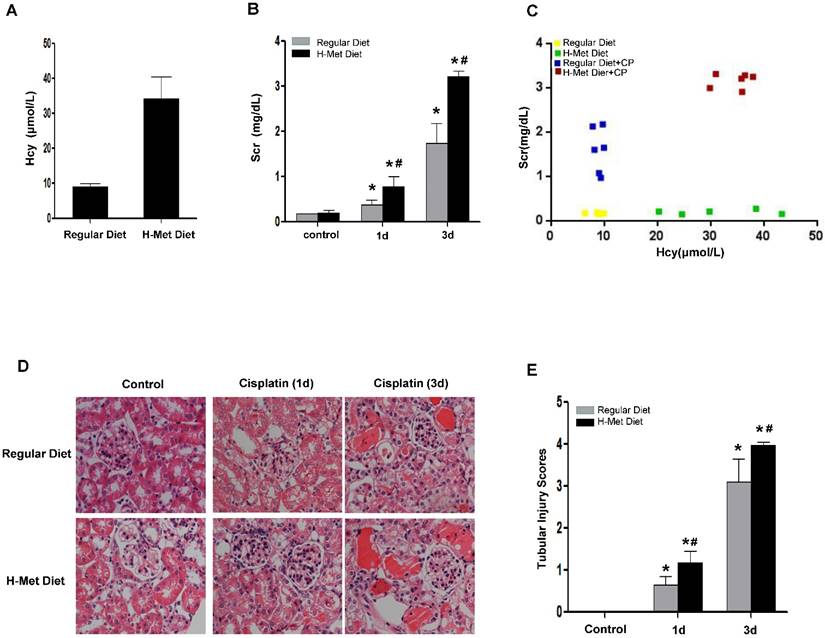
Moderate HHcy is prevalent in general population and CKD patients [25]. To examine the impact of moderate HHcy on the sensitivity of AKI, we generated HHcy mice by feeding male C57BL/6 mice with high methionine (H-Met) diet for 2 weeks when the plasma level of Hcy reached 34.19±6.15μM (Figure 1A). The plasma level of Hcy in control mice fed with regular diet is 8.85±0.96μM. Hematoxylin and eosin (H&E) staining showed no obvious abnormality in kidney morphology in HHcy mice (Figure 1D-E). A single intraperitoneal administration of 24mg/kg cisplatin induced a dramatic increase of serum creatinine (Scr) 24 h and 72 h after injection in both control and HHcy mice, but Scr level was significantly higher in HHcy mice than in control mice (Figure 1B). To address whether HHcy contributes to cisplatin-induced AKI sensitivity, we analyzed the correlation between the plasma levels of Hcy and Scr. As shown in Figure 1C, Scr measured after cisplatin injection was correlated with the plasma level of Hcy with an R value of 0.902, suggesting that HHcy contributes to cisplatin-induced AKI sensitivity. Corroborating with the functional analysis, renal histology by H&E staining revealed that cisplatin-treated HHcy kidney showed more severe renal tubular damage, including loss of brush border, cast formation and tubular cell necrosis (Figure 1D-E).
HHcy mice are more susceptible to cisplatin-induced AKI. (A) The plasma level of Hcy in mice. (B) The level of serum creatinine (Scr) at day 1 or day 3 after cisplatin injection. *P<0.05 versus control mice; #P<0.05 versus mice fed with regular diet. (C) Graphic representation of relative level between serum creatinine and serum Hcy in control and HHcy mice before and after cisplatin injection. (D) Representative micrographs of H&E staining show the morphological injury at day 1 and day 3 after cisplatin injection. (E) Quantification assessment of tubular dilation and atrophy on the basis of H&E staining. Data are expressed as mean±SD, n=5. *P<0.05 versus control, #P<0.05 versus mice fed with regular diet mice.
HHcy mice were susceptible to ischemia-reperfusion-induced AKI
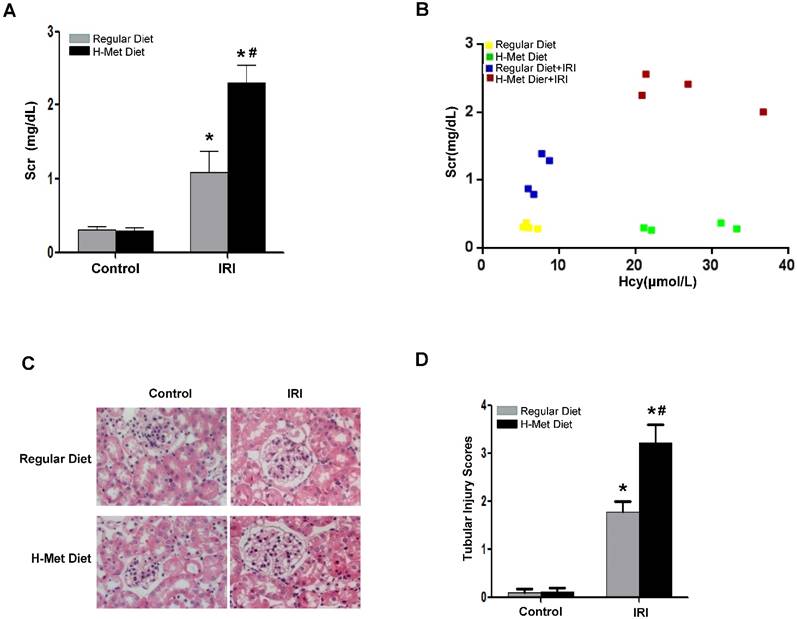
To demonstrate the sensitivity of HHcy to AKI is not specific to cisplatin-induced nephrotoxicity, we next induced acute renal damage by ischemia-reperfusion (I/R). As shown in Figure 2A, a more dramatic increase of Scr 24 h after reperfusion was observed in HHcy mice than that in control mice. Similarly, Scr measured after I/R was correlated well with the plasma Hcy level with an R value of 0.85 (Figure 2B). Consistently, I/R also led to a more severe tubular damage in HHcy mice as shown by H&E staining (Figure 2C-D). Collectively, these observations indicate that HHcy mice are sensitized to both cisplatin-induced and I/R-induced AKI.
HHcy did not aggravate cisplatin-induced inflammation
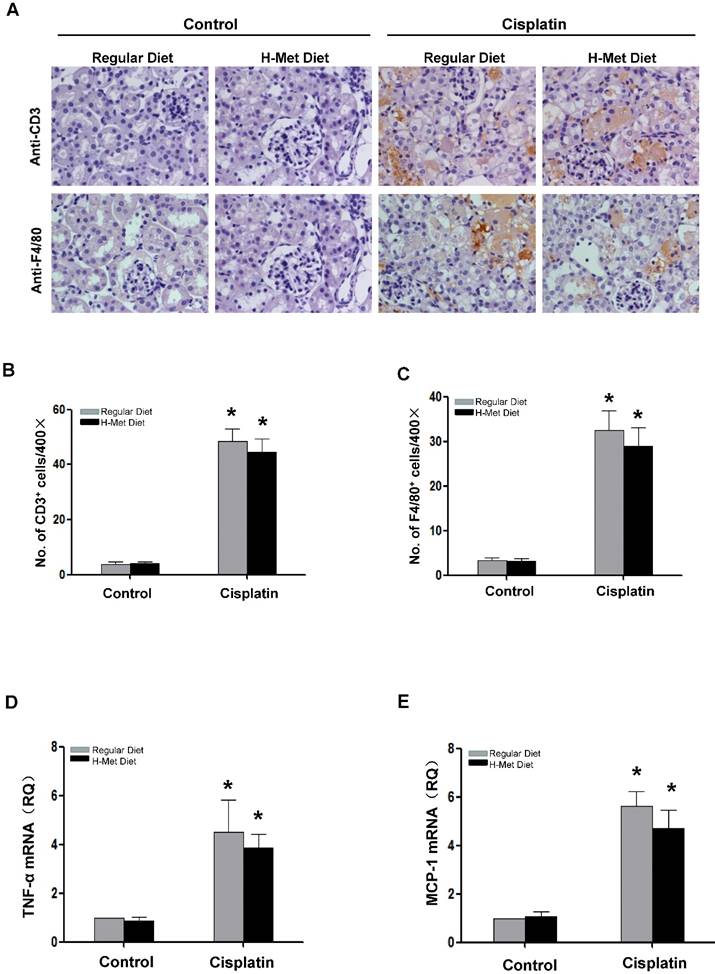
Inflammatory cell infiltration is crucial in AKI induced by various causes [26,27]. To assess the role of HHcy in renal inflammation, we examined pro-inflammatory cytokine expression and the infiltration of inflammatory cells in the kidneys after cisplatin injection. As shown in Figure 3A-C, the infiltration of CD3+ T cells and F4/80+ macrophage in the kidneys was significantly increased after cisplatin injection, whereas, no significant difference was detected between control and HHcy mice. TNF-α is a critical pro-inflammatory cytokine thought to contribute to cisplatin-induced AKI and is produced by T-cells and activated macrophages in renal injury [28,29]. Previous studies have shown that Hcy induces the expression of monocyte chemoattractant protein 1(MCP-1) in cultured human vascular endothelial cells, smooth muscle cells and monocytes [30-32]. We therefore examined whether the upregulation of these cytokines by cisplatin is affected by HHcy. As shown in Figure 3D-E, real time-PCR revealed that cisplatin induced a significant increase of TNF-α and MCP-1 mRNA level in both control and HHcy mice. However, there was no significant difference between control and HHcy mice. Collectively, these data indicate that moderate HHcy does not affect cisplatin-induced inflammatory cell infiltration and inflammatory cytokine production in kidney tissue.
HHcy mice exhibited an increased tubular cell apoptosis after cisplatin injection
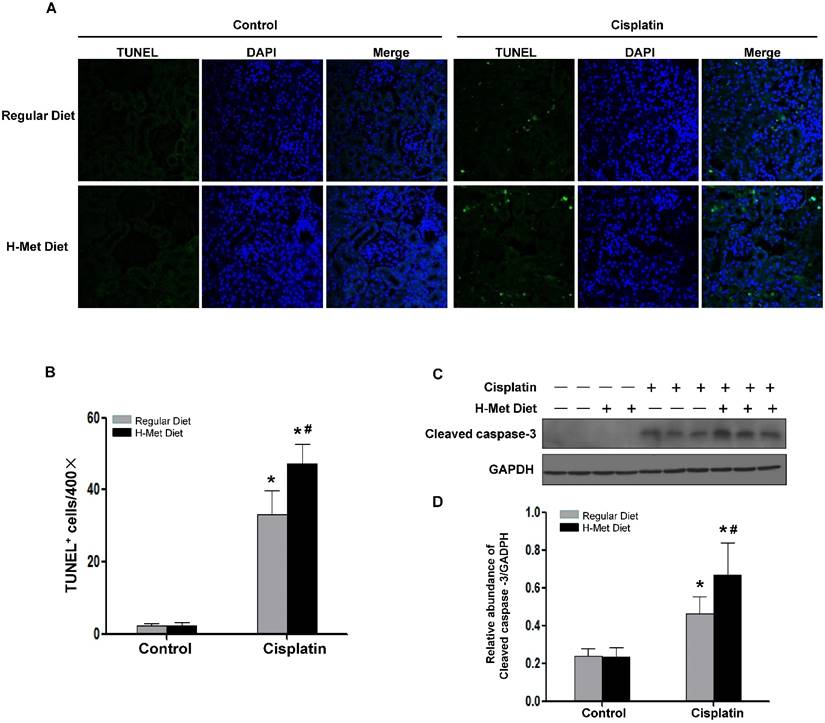
Tubular cell apoptosis plays a crucial pathogenic role in cisplatin-induced AKI[33]. We therefore evaluated tubular cell apoptosis in the renal cortex by TUNEL assay. As shown in Figure 4A-B, very few TUNEL-positive tubular cells in HHcy mice, suggesting that moderate HHcy does not affect cell survival under physiological condition. 72 h after cisplatin injection, the number of TUNEL-positive tubular cells was significantly increased in control mice, whereas, tubular cell apoptosis was exacerbated in HHcy mice. Determination of active cleaved caspase-3 by western blotting further confirmed the more prevalent apoptosis in cisplatin-treated HHcy mice (Figure 4C-D).
HHcy mice are more susceptible to ischemia reperfusion-induced AKI. (A) The level of serum creatinine in normal and HHcy mice 24h after ischemia reperfusion. (B) Graphic representation of relative level between serum creatinine and serum homocysteine in control and HHcy mice before and after IRI. (C) Representative micrographs of H&E show the morphological injury in the kidneys after ischemia reperfusion injury. (D) Quantification assessment of tubular dilation and atrophy on the basis of H&E staining. Data are expressed as mean±SD, n=4. *P<0.05 versus control mice. #P<0.05 versus mice fed with regular diet.
HHcy does not exacerbate cisplatin-induced inflammation. (A) Representative images of the immunohistochemical staining show the infiltration of CD3- and F4/80-positvie cells in the renal interstitium at day 3 after cisplatin injection. (B-C) Quantitative determination of CD3-positive cells (B) and F4/80-positive cells (C) among different groups as indicated. (D-E) Real time-PCR shows the level of TNF-α (D) and MCP-1(E) mRNA in kidney tissues at day 3 after cisplatin injection. Relative levels of TNF-α and MCP-1 mRNA were normalized with GAPDH. *P<0.05 versus control.
HHcy aggravates cisplatin-induced renal tubular epithelial cell apoptosis in mice. (A) Representative micrographs show apoptotic cell death detected by TUNEL staining 3 days after cisplatin injection. (B) Quantitative determination of apoptotic tubular cells at day 3 after cisplatin injection. Data are presented as numbers of apoptotic cells per 400× field. (C) Representative Western blots show the protein level of cleaved caspase-3 in the kidneys at day 3 after cisplatin injection. GAPDH was used to verify equivalent loading. (D) Graphic representation of relative level of cleaved caspase-3 normalized to GADPH. Data are expressed as mean±SD, n=5. *P<0.05 versus control mice. #P<0.05 versus mice fed with regular diet.
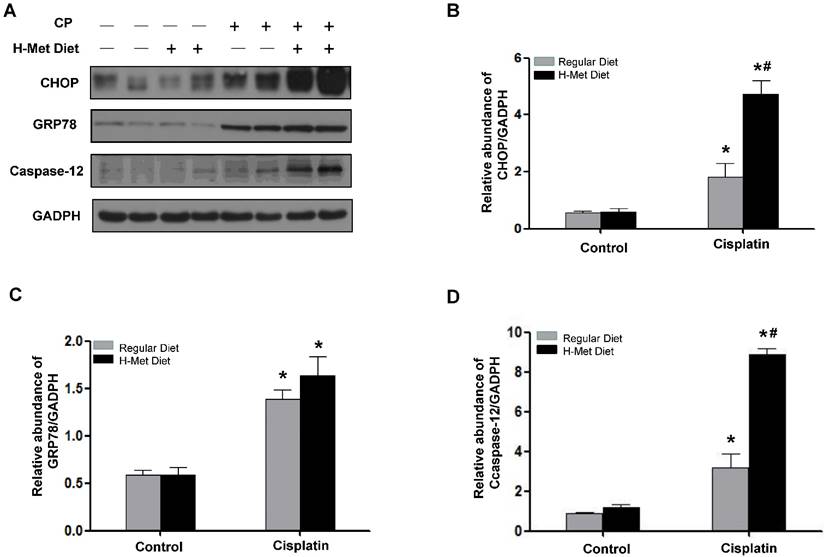
HHcy exacerbated cisplatin-induced ER stress
ER stress is thought to be responsible for cisplatin-induced apoptosis [34-36]. To clarify the molecular mechanism underlying Hcy-augmented apoptosis, the expression of C/EBP homologous protein (CHOP) and protein 78/immunoglobulin chain binding protein (GRP78/BiP), the representative ER stress-responsive genes, was examined. Western blotting showed that, 24 h after cisplatin injection, the expression of CHOP was significantly higher in HHcy mice than those in control mice. The protein level of GRP78/BiP in HHcy mice was slightly higher than that in control mice but the difference was not statistic significant (Figure 5A-C), indicating that HHcy enhanced cisplatin-induced ER stress. However, 72 h after cisplatin, the level of CHOP and GRP78/BiP was similar in control and HHcy mice (Supplementary Figure 1). We assume this might because more injured tubular cells are dying of apoptosis rather than undergoing ER stress at day 3 after cisplatin injection. To address this issue, we measured the number of TUNEL-positive tubular cells. As shown in Supplementary Figure 2, the number of apoptotic cells was significantly increased at day 3 after cisplatin injection compared with that at day 1.
Caspase-12 has been reported as a crucial mediator in ER stress-induced apoptosis [37,38]. Therefore, we examined whether caspase-12 was upregulated by HHcy. Western blotting (Figure 5A) followed by densitometry analysis (Figure 5D) demonstrated that the level of caspase-12 protein was significantly increased after cisplatin injection in HHcy mice compared with control mice, suggesting that Hcy enhanced tubular cell apoptosis, at least in part by exacerbating cisplatin-induced ER stress.
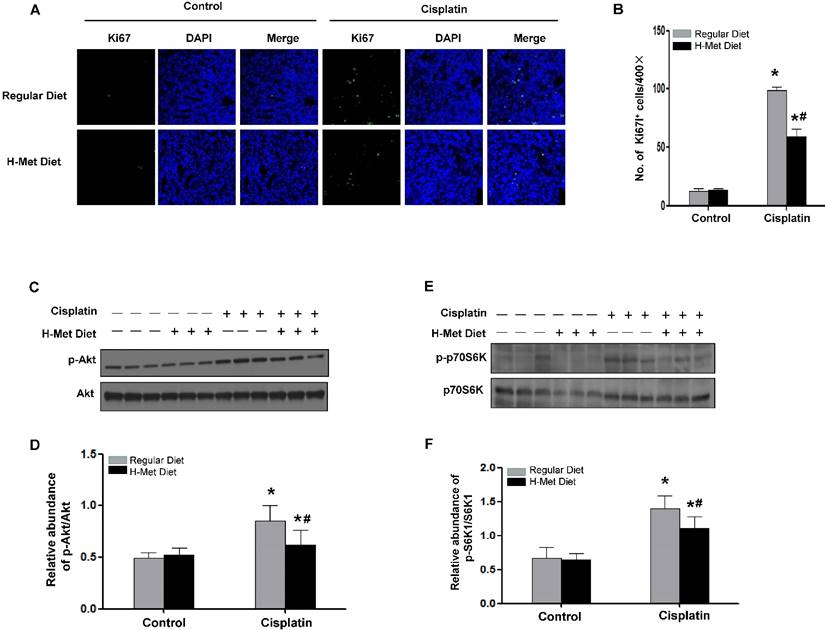
HHcy blunted tubular cell proliferation after CP injection
Except for cell apoptosis, we also examined cell proliferation in mice after cisplatin injection by staining and quantification of Ki67-positive cells. As shown in Figure 6A-B, the expected burst of proliferation 24 h after cisplatin was detected in control mice. However, the number of Ki67-positive tubular cells was significantly fewer in kidney tissue of HHcy mice than those in control mice, suggesting tubules in HHcy mice failed to proliferate normally after cisplatin injection.
HHcy attenuated cisplatin-induced Akt activation
Since Akt activation plays a critical role in promoting cell survival in response to stresses, we thus investigated Akt kinase activity in the kidney tissue after cisplatin injection. As shown in Figure 6C-D, Akt kinase activation exhibited as Akt phosphorylation (p-Akt) was largely induced 24 h after cisplatin injection in control mice, whereas, the abundance of p-Akt in HHcy mice was ~60% of that in control mice, suggesting HHcy suppressed cisplatin-induced Akt kinase activation. A downstream target for activated Akt is p70-S6 kinase (p70S6K). Phosphorylation by p-Akt activates p70S6K, providing increased protein needed for cell division [39]. Similar to the timing of Akt kinase activation, p70S6K was phosphorylated 24 h after cisplatin injection in control mice. In contrast, HHcy mice exhibited a marked reduction of phosphorylated p70S6K (Figure 6E-F), suggesting that attenuated Akt kinase activation might contribute to the delayed cell proliferation in HHcy mice after cisplatin injection.
HHcy promotes ER stress in kidneys during cisplatin-induced AKI. (A) Representative Western blots show the protein level of CHOP, GRP78 and caspase-12 in the kidneys at day 1 after cisplatin injection. GAPDH was used to verify equivalent loading. (B-D) Graphic representation of relative protein level of CHOP (B), GRP78 (C) and caspase-12 (D) normalized to GADPH. Data are expressed as mean±SD, n=5. *P<0.05 versus control mice. #P<0.05 versus mice fed with regular diet.
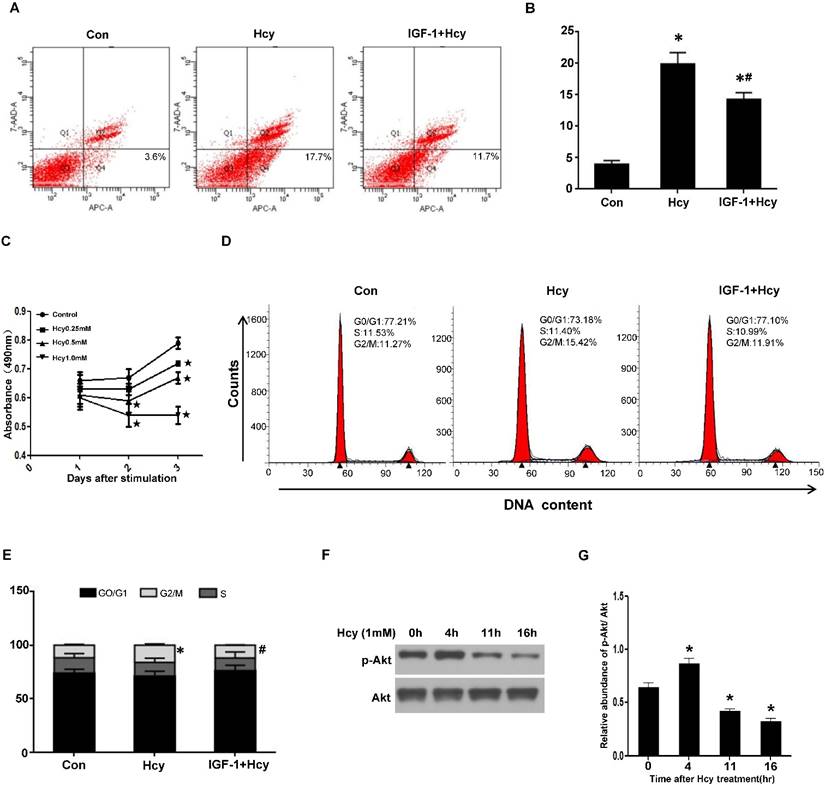
HHcy caused tubular cell apoptosis and cycle arrest via inhibiting Akt activation
The delayed tubular cell proliferation after cisplatin in HHcy mice implies that HHcy might either induce tubular cell apoptosis or delay cell cycle. To confirm the role of HHcy on tubular cell apoptosis, we treated rat proximal tubular (NRK-52E) cells with 1mM of Hcy for 24 h. Flow cytometry analysis demonstrated that 1mM of Hcy led to ~ four-fold increase in cell apoptosis (Figure 7A-B).
To explore whether HHcy directly inhibits cell proliferation, we treated NRK-52E cells with various concentrations of Hcy and performed MTT assay. As shown in Figure 7C, incubating NRK-52E cells with 1mM of Hcy for 3 days dramatically inhibited cell growth compared with control cells. Furthermore, flow cytometry analysis revealed that treating NRK-52E cells with 1mM of Hcy for 40h significantly increased the percentage of G2/M population from 11.27% to 15.42% (Figure 7D-E), indicating that HHcy causes cell cycle arrest.
Given the critical role of Akt signaling in cell survival, we examined the activation of Akt kinase. Western blotting revealed that HHcy decreased the level of p-Akt in a time-dependent manner (Figure 7F-G). To evaluate the role of Akt activation in HHcy-induced tubular cell apoptosis and cell cycle arrest, NRK-52E cells were incubated with 1mM of Hcy together with 100ng/ml of Akt agonist IGF-1. Flow cytometry analysis demonstrated that IGF-1 rescued HHcy-induced cell apoptosis and cell cycle arrest (Figure 7A-B and Figure 7D-E). Collectively, these data suggest that elevated concentration of Hcy causes cell cycle arrest and apoptosis by inhibiting Akt kinase activation.
HHcy inhibits tubular cells proliferation and Akt activation after cisplatin injection. (A) Immunofluorescence staining shows the staining of Ki67 to detect proliferating tubular cells at day 3 after cisplatin injection. Nuclei are visualized with DAPI (blue). (B) Quantitative determination of proliferating tubular cells at day 3 after cisplatin injection. Data are presented as numbers of proliferating tubular cells per 400× field. n=5. *P<0.05 versus control mice. #P<0.05 versus mice fed with regular diet. (C) Representative western blot shows the level of p-Akt at day 1 after cisplatin injection. Total Akt was used to verify equivalent loading. (D) Graphic representation of relative abundance of p-Akt to total Akt. (E) Representative western blot shows the level of p-p70S6K at day 1 after cisplatin injection. Total p70S6K was used to verify equivalent loading. (F) Graphic representation of relative abundance of p-p70S6K to total p70S6K. Data are expressed as mean±SD, n=5. *P<0.05 versus control mice. #P<0.05 versus mice fed with regular diet.
HHcy inhibits cell proliferation by inhibiting Akt activation in vitro. (A) NRK-52E cells were treated with 1mM of Hcy with or without IGF-1 for 24h. Apoptosis was analyzed by flow cytometry. (B) Bar graph depicted the percentage of apoptotic cells. *P<0.05 versus control cells. #P<0.05 versus cells treated with Hcy. (C) NRK-52E cells were incubated with indicated amount of Hcy for indicated time period, and then washed for MTT assay. Data are expressed as means±SD of three independent experiments. (D) NRK-52E cells were treated with 1mM of Hcy with or without IGF-1 for 40h. Cell cycle was analyzed by flow cytometry. (E) Bar graph depicted the percentage of cells in the different stages of the cell cycle. Symbols in the cell cycle data panels refer to the comparison of G2/M phases. *P<0.05 versus control cells. #P<0.05 versus cells treated with Hcy. (F) NRK-52E cells were treated with 1mM of Hcy for indicated time period and whole cell lysate were harvested. Representative western blot shows the level of p-Akt in NRK-52E cells after Hcy stimulation. Total Akt was used to verify equivalent loading. (G) Graphic representation of relative abundance of p-Akt to total Akt. Data are means±SD of three experiments. *P<0.05 versus control cells.
Discussion
In the present study, we generated HHcy mice with plasma level of Hcy around 30μM. The HHcy mice did not show any functional and structural abnormality in the kidney. However, cisplatin-induced AKI was dramatically exacerbated in HHcy mice, and the injury sensitivity correlates with the plasma level of Hcy in mice. To demonstrate that the sensitivity of HHcy to AKI is not specific to cisplatin-induced nephrotoxicity, we also generated an ischemia-reperfusion injury model and obtained the same result, suggesting common mechanisms mediating the pathogenic effect of elevated Hcy.
Inflammatory response is known to play a crucial role in AKI induced by various causes [40]. It has also been reported that HHcy induces inflammatory response [41-43]. However, in the present study, both the infiltration of inflammatory cells and the production of inflammatory cytokines did not increase after cisplatin injection in HHcy mice, which is different from previous report [44]. We assume that the concentration of Hcy in our model might not be high enough to affect cisplatin-induced inflammatory response.
Besides inflammation, local oxidative stress and ER stress have been proposed to mediate the pathogenic action of elevated Hcy [19]. In the current study, we provided evidence that HHcy promotes cisplatin-induced ER stress. Thus we believe that HHcy-induced ER stress is one of the common cellular mechanisms which underlie the sensitivity of HHcy mice to both IRI and cisplatin induced AKI. Regarding to cisplatin-induced AKI, HHcy might also increase the toxicity of cisplatin to tubular cells. Cisplatin is a low molecular weight compound, can filter freely in the glomerulus and penetrates the tubular cells. Once enters tubular cells, cisplatin is metabolic activated to form a glutathione-conjugate that is metabolized to a cysteinyl-glycine-conjugate, to a cysteine conjugate, and finally to a reactive thiol. It has been reported that pre-incubation of cisplatin with cysteinyl-glycine, or N-acetyl-cysteine to allow for the formation of cysteine conjugate increased the toxicity of cisplatin [45]. The oxidized Hcys in human and animal plasma exists as a disulfide form, including homocystine (Hcys-S-S-Hcys) and cysteinylhomocysteine (Cys-S-S-Hcys) [19]. Therefore, increased plasma concentration of Hcy in HHcy mice might augment the amount of cysteine which promotes the formation of cysteinyl-glycine-conjugate to enhance the toxicity of cisplatin.
Tubular cell apoptosis is well recognized as the final common pathway in cisplatin induced renal injury, and ER stress contributes to cell apoptosis [46]. It has been shown that Hcy causes ER stress and activates the unfolded protein response (UPR) by disruption of disulfide bond formation and misfolding protein in the endoplasmic reticulum [19]. Moreover, intracellular Hcy has been reported to increase the expression of the ER stress-response genes in cultured hepatic cells, vascular endothelial cells, adipocyte as well as cardiomyoblasts [47-52]. Notably, the induction of the UPR in Hcy-treated cells used very high concentrations (1mM) of the amino acid. The increased ER stress was detected in the liver and myocardium of HHcy rats in which the plasma concentration of Hcy is over 50μM. In the present study, the HHcy mice did not show any sign of ER stress under normal condition. This discrepancy with previous studies might be due to lower concentration of Hcy in our model. Besides, we could not exclude the possibility that the sensitivity to HHcy might be tissue-specific. Noteworthy, the expression of CHOP, a key marker of ER stress, was significantly higher in HHcy mice than that of control mice after cisplatin injection, suggesting HHcy exacerbates cisplatin-induced ER stress. The expression of GRP78 was also increased but not statistic significant. This might because the level of GRP78 is not at its highest level at this time point. Previous study reported that the highest level of GRP78 was detected at 4h after ischemia-reperfusion injury and after that the level of GRP78 gradually decreased although it is still higher than that of normal mice [53].
It has been shown that, after acute injury, injured tubules are repaired by repopulation of remaining tubular cells through dedifferentiation and proliferation. PI3K-Akt signaling is crucial for renal tubular proliferation both in vivo and in vitro [54,55]. Moreover, it has been shown that inactivation of the PI3K/Akt pathway inhibits tubular cell dedifferentiation in vitro and in vivo[55,56]. In the present study, we found that, after cisplatin injection, the level of p-Akt was lower in HHcy mice than that of control mice. Consistently, 1mM of Hcy decreased the level of p-Akt in NRK-52E cells. Moreover, Akt agonist IGF-1 rescued HHcy-induced cell cycle arrest. Collectively, these data suggest that HHcy blunts tubular cell repair after acute injury by inhibiting Akt activation.
Akt have been reported to inhibit apoptosis by inhibiting caspase-9 activation [57]. Kuwana et al. showed that the PI3K-Akt pathway is activated after cisplatin administration and blockage of the PI3K/Akt pathway accelerates renal tubular cell death and leads to poor prognoses [58]. In support of this concept, Zhuang SG et al. demonstrated that inhibition of EGFR activity results in Akt dephosphorylation and an increase in apoptotic renal tubular cells induced by folic acid [56]. In the present study, we demonstrated that Akt agonist IGF-1 partially rescued HHcy-induced NRK-52E cell apoptosis, indicating that attenuated Akt kinase activity contributes to the extended tubular cell apoptosis in HHcy mice.
In conclusion, the present study provides evidence that those mice with moderate HHcy is sensitized to cisplatin-induced AKI shown as exacerbated tubular cell apoptosis and inhibited tubular cell proliferation. At the cellular and molecular level, the injury sensitivity can be tracked down to enhanced ER stress and attenuated Akt kinase activativity in the kidney of HHcy mice. According to our acknowledgement, this study, for the first time, revealed a novel role of HHcy in the development of AKI. In addition, these findings further highlight the importance of diet as a major contributor to the pathophysiological outcome of HHcy. Since moderate HHcy is prevalent in general population, especially in old people, clinical study is required to investigate whether people with elevated level of Hcy present a higher risk of developing AKI.
Supplementary Material
Supplementary figures.
Acknowledgements
This work is supported by grants from National 973 Program (2012CB517700), Nature and Science Foundation of China (81288001, 81521003), and Natural Science Foundation of Guangdong province (S2013020012748) to Dr. Jing Nie.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ishani A, Xue JL, Himmelfarb J. et al. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol. 2009;20:223-228
2. Hsu CY. Yes, AKI truly leads to CKD. J Am Soc Nephrol. 2012;23:967-969
3. Hsu CY, Ordonez JD, Chertow GM. et al. The risk of acute renal failure in patients with chronic kidney disease. Kidney Int. 2008;74:101-107
4. Grams ME, Astor BC, Bash LD. et al. Albuminuria and estimated glomerular filtration rate independently associate with acute kidney injury. J Am Soc Nephrol. 2010;21:1757-1764
5. Kane-Gill SL, Sileanu FE, Murugan R. et al. Risk factors for acute kidney injury in older adults with critical illness: A retrospective cohort study. Am J Kidney Dis. 2015;65:860-869
6. Siew ED, Parr SK, Abdel-Kader K. et al. Predictors of recurrent AKI. J Am Soc Nephrol. 2016;27:1190-1200
7. Dupont M, Shrestha K, Singh D. et al. Lack of significant renal tubular injury despite acute kidney injury in acute decompensated heart failure. Eur J Heart Fail. 2012;14:597-604
8. Ubbink JB, van der Merwe A, Delport R. et al. The effect of a subnormal vitamin B-6 status on homocysteine metabolism. J Clin Invest. 1996;98:177-184
9. Mikael LG, Wang XL, Wu Q. et al. Hyperhomocysteinemia is associated with hypertriglyceridemia in mice with methylenetetrahydrofolate reductase deficiency. Mol Genet Metab. 2009;98:187-194
10. Suszynska-Zajczyk J, Utyro O, Jakubowski H. Methionine-induced hyperhomocysteinemia and bleomycin hydrolase deficiency alter the expression of mouse kidney proteins involved in renal disease. Mol Genet Metab. 2014;112:339-346
11. Francis ME, Eggers PW, Hostetter TH. et al. Association between serum homocysteine and markers of impaired kidney function in adults in the United States. Kidney Int. 2004;66:303-312
12. Sabanayagam C, Shankar A. Association between plasma homocysteine and microalbuminuria in persons without hypertension, diabetes mellitus, and cardiovascular disease. Clin Exp Nephrol. 2011;15:92-99
13. Zhang C, Xia M, Boini KM. et al. Epithelial-to-mesenchymal transition in podocytes mediated by activation of NADPH oxidase in hyperhomocysteinemia. Pflugers Arch. 2011;462:455-467
14. Lang D, Kredan MB, Moat SJ. et al. Homocysteine-induced inhibition of endothelium-dependent relaxation in rabbit aorta: Role for superoxide anions. Arterioscler Thromb Vasc Biol. 2000;20:422-427
15. Tyagi N, Moshal KS, Sen U. et al. Ciglitazone ameliorates homocysteine-mediated mitochondrial translocation and matrix metalloproteinase-9 activation in endothelial cells by inducing peroxisome proliferator activated receptor-gamma activity. Cell Mol Biol (Noisy-le-grand). 2006;52:21-27
16. Scull CM, Tabas I. Mechanisms of ER stress-induced apoptosis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:2792-2797
17. Jamaluddin MS, Yang X, Wang H. Hyperhomocysteinemia, DNA methylation and vascular disease. Clin Chem Lab Med. 2007;45:1660-1666
18. Lei W, Long Y, Li S. et al. Homocysteine induces collagen i expression by downregulating histone methyltransferase g9a. Plos One. 2015;10:e130421
19. Yi F, Li PL. Mechanisms of homocysteine-induced glomerular injury and sclerosis. Am J Nephrol. 2008;28:254-264
20. Xie D, Yuan Y, Guo J. et al. Hyperhomocysteinemia predicts renal function decline: A prospective study in hypertensive adults. Sci Rep. 2015;5:16268
21. Brooks C, Wei Q, Cho SG. et al. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119:1275-1285
22. Yang L, Besschetnova TY, Brooks CR. et al. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16:535-543 1p-143p
23. Zhou L, Li Y, Hao S. et al. Multiple genes of the renin-angiotensin system are novel targets of Wnt/beta-catenin signaling. J Am Soc Nephrol. 2015;26:107-120
24. Zhu F, Liu W, Li T. et al. Numb contributes to renal fibrosis by promoting tubular epithelial cell cycle arrest at G2/M. Oncotarget. 2016
25. Bostom AG, Culleton BF. Hyperhomocysteinemia in chronic renal disease. J Am Soc Nephrol. 1999;10:891-900
26. Akcay A, Nguyen Q, Edelstein CL. Mediators of inflammation in acute kidney injury. Mediators Inflamm. 2009;2009:137072
27. Friedewald JJ, Rabb H. Inflammatory cells in ischemic acute renal failure. Kidney Int. 2004;66:486-491
28. Zhang B, Ramesh G, Norbury CC. et al. Cisplatin-induced nephrotoxicity is mediated by tumor necrosis factor-alpha produced by renal parenchymal cells. Kidney Int. 2007;72:37-44
29. Ramesh G, Reeves WB. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest. 2002;110:835-842
30. Poddar R, Sivasubramanian N, DiBello PM. et al. Homocysteine induces expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human aortic endothelial cells: Implications for vascular disease. Circulation. 2001;103:2717-2723
31. Wang G, O K. Homocysteine stimulates the expression of monocyte chemoattractant protein-1 receptor (CCR2) in human monocytes: Possible involvement of oxygen free radicals. Biochem J. 2001;357:233-240
32. Wang G, Siow YL, O K. Homocysteine induces monocyte chemoattractant protein-1 expression by activating NF-kappaB in THP-1 macrophages. Am J Physiol Heart Circ Physiol. 2001;280:H2840-H2847
33. Li J, Xu Z, Jiang L. et al. Rictor/mTORC2 protects against cisplatin-induced tubular cell death and acute kidney injury. Kidney Int. 2014;86:86-102
34. Peyrou M, Hanna PE, Cribb AE. Cisplatin, gentamicin, and p-aminophenol induce markers of endoplasmic reticulum stress in the rat kidneys. Toxicol Sci. 2007;99:346-353
35. Mandic A, Hansson J, Linder S. et al. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J Biol Chem. 2003;278:9100-9106
36. Liu H, Baliga R. Endoplasmic reticulum stress-associated caspase 12 mediates cisplatin-induced LLC-PK1 cell apoptosis. J Am Soc Nephrol. 2005;16:1985-1992
37. Morishima N, Nakanishi K, Tsuchiya K. et al. Translocation of Bim to the endoplasmic reticulum (ER) mediates ER stress signaling for activation of caspase-12 during ER stress-induced apoptosis. J Biol Chem. 2004;279:50375-50381
38. Nakagawa T, Zhu H, Morishima N. et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98-103
39. Liu R, Liu D, Trink E. et al. The Akt-specific inhibitor MK2206 selectively inhibits thyroid cancer cells harboring mutations that can activate the PI3K/Akt pathway. J Clin Endocrinol Metab. 2011;96:E577-E585
40. Havasi A, Borkan SC. Apoptosis and acute kidney injury. Kidney Int. 2011;80:29-40
41. Zhang D, Jiang X, Fang P. et al. Hyperhomocysteinemia promotes inflammatory monocyte generation and accelerates atherosclerosis in transgenic cystathionine beta-synthase-deficient mice. Circulation. 2009;120:1893-1902
42. Zhu S, Li J, Bing Y. et al. Diet-Induced hyperhomocysteinaemia increases intestinal inflammation in an animal model of colitis. J Crohns Colitis. 2015;9:708-719
43. Meng S, Ciment S, Jan M. et al. Homocysteine induces inflammatory transcriptional signaling in monocytes. Front Biosci (Landmark Ed). 2013;18:685-695
44. Zhang D, Fang P, Jiang X. et al. Severe hyperhomocysteinemia promotes bone marrow-derived and resident inflammatory monocyte differentiation and atherosclerosis in LDLr/CBS-deficient mice. Circ Res. 2012;111:37-49
45. Townsend DM, Deng M, Zhang L. et al. Metabolism of Cisplatin to a nephrotoxin in proximal tubule cells. J Am Soc Nephrol. 2003;14:1-10
46. Linkermann A, Chen G, Dong G. et al. Regulated cell death in AKI. J Am Soc Nephrol. 2014;25:2689-2701
47. Li Y, Zhang H, Jiang C. et al. Hyperhomocysteinemia promotes insulin resistance by inducing endoplasmic reticulum stress in adipose tissue. J Biol Chem. 2013;288:9583-9592
48. Wei H, Zhang R, Jin H. et al. Hydrogen sulfide attenuates hyperhomocysteinemia-induced cardiomyocytic endoplasmic reticulum stress in rats. Antioxid Redox Signal. 2010;12:1079-1091
49. Werstuck GH, Lentz SR, Dayal S. et al. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J Clin Invest. 2001;107:1263-1273
50. Yu X, Lv J, Zhu Y. et al. Homocysteine inhibits hepatocyte proliferation via endoplasmic reticulum stress. Plos One. 2013;8:e54265
51. Zhang C, Cai Y, Adachi MT. et al. Homocysteine induces programmed cell death in human vascular endothelial cells through activation of the unfolded protein response. J Biol Chem. 2001;276:35867-35874
52. Ron D. Hyperhomocysteinemia and function of the endoplasmic reticulum. J Clin Invest. 2001;107:1221-1222
53. Noh MR, Kim JI, Han SJ. et al. C/EBP homologous protein (CHOP) gene deficiency attenuates renal ischemia/reperfusion injury in mice. Biochim Biophys Acta. 2015;1852:1895-1901
54. Xing J, Zhang Z, Mao H. et al. Src regulates cell cycle protein expression and renal epithelial cell proliferation via PI3K/Akt signaling-dependent and -independent mechanisms. Am J Physiol Renal Physiol. 2008;295:F145-F152
55. Zhuang S, Duan M, Yan Y. Src family kinases regulate renal epithelial dedifferentiation through activation of EGFR/PI3K signaling. J Cell Physiol. 2012;227:2138-2144
56. He S, Liu N, Bayliss G. et al. EGFR activity is required for renal tubular cell dedifferentiation and proliferation in a murine model of folic acid-induced acute kidney injury. Am J Physiol Renal Physiol. 2013;304:F356-F366
57. Zhou H, Li XM, Meinkoth J. et al. Akt regulates cell survival and apoptosis at a postmitochondrial level. J Cell Biol. 2000;151:483-494
58. Kuwana H, Terada Y, Kobayashi T. et al. The phosphoinositide-3 kinase gamma-Akt pathway mediates renal tubular injury in cisplatin nephrotoxicity. Kidney Int. 2008;73:430-445
Author contact
![]() Corresponding author: Dr. Jing Nie, Division of Nephrology, Nanfang Hospital, North Guangzhou Ave 1838, Guangzhou 510515, P.R. China, Phone: 86-20-62787972; Fax: 86-20-87281713; E-mail: niejingedu.cn.
Corresponding author: Dr. Jing Nie, Division of Nephrology, Nanfang Hospital, North Guangzhou Ave 1838, Guangzhou 510515, P.R. China, Phone: 86-20-62787972; Fax: 86-20-87281713; E-mail: niejingedu.cn.
Received 2016-7-4
Accepted 2016-10-21
Published 2017-2-6