ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2017; 13(3):286-294. doi:10.7150/ijbs.15171 This issue Cite
Research Paper
BEX2 promotes tumor proliferation in colorectal cancer
Yeting Hu1,2*, Qian Xiao1,2*, Haiyan Chen1,2*, Jinjie He1,2, Yinuo Tan1,2, Yue Liu1,2, Zhanhuai Wang1,2, Qi Yang1,2, Xiangfeng Shen1,2, Yu Huang3, Ying Yuan2, Kefeng Ding1,2 ![]()
1. Department of Surgical Oncology, Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou, Zhejiang, China;
2. Key Laboratory of Cancer Prevention and Intervention of the China National Ministry of Education, Key Laboratory of Molecular Biology in Medical Sciences of Zhejiang Province, Cancer Institute, Zhejiang University School of Medicine, Hangzhou, Zhejiang, China;
3. Laboratory Animal Research Center, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, China.
*Y. H., Q. X. and H. C. contributed equally to the manuscript and should all be considered first authors.
Abstract

BEX2 has been suggested to promote the tumor growth in breast cancer and glioblastoma, while inhibit the proliferation of glioma cells. Thus, the role of BEX2 in tumor was still in debate. Additionally, the biological functions of BEX2 in colorectal cancer (CRC) have not yet been clarified. Here, we reported that BEX2 was overexpressed in advanced CRC from both the GSE14333 database and fresh CRC tissue specimens, and positively correlated with clinical staging. Knockdown of BEX2 significantly decreased the in vitro proliferation of SW620 colorectal cancer cells, suppressed subcutaneous xenograft growth and enhanced the survival of mice with cecal tumors. These effects were mainly mediated by the JNK/c-Jun pathway. Knockdown of BEX2 inhibited JNK/c-Jun phosphorylation, while BEX2 overexpression activated JNK/c-Jun phosphorylation. Moreover, the administration of the JNK-specific inhibitor SP600125 to SW620 with BEX2 overexpression abolished the effect of BEX2 on SW620 cell proliferation. This study reveals that BEX2 promotes colorectal cancer cell proliferation via the JNK/c-Jun pathway, suggesting BEX2 as a potential candidate target for the treatment of CRC.
Keywords: Colorectal cancer, BEX2, Proliferation, JNK/c-Jun.
Introduction
Colorectal cancer (CRC) is a devastating malignant disease, ranking as the second most common cause of cancer death in Western countries [1] and the fifth most common cause of cancer death in China [2]. Despite the great achievements made in conventional cytotoxic chemotherapy for patients with CRC over last several decades, the overall patient survival rate has not significantly improved. Although the introduction of the anti-EGFR antibodies cetuximab [3] and panitumumab [4] has improved the survival of CRC patients with the wild-type K-Ras gene, there is still an urgent need for new effective therapeutics. It is anticipated that only new drugs with novel targets will improve the state of CRC care [5]. However, our understanding of the mechanisms of colorectal cancer development is still limited. Thus, it is crucial to further explore the molecular events of CRC biology, which will facilitate the discovery of novel therapeutic targets.
Brain-expressed X-linked (BEX) genes are a family of genes that reside on the mammalian X chromosome, and BEX proteins are involved in the cell cycle, cancer and tumor growth [6,7]. BEX2 is highly expressed in the embryonic brain and can interact with transcriptional factor LMO2 to regulate transcriptional activity [8]. In addition, BEX2 has been reported to be involved in tumor development in several types of cancer, such as glioblastoma, glioma and breast cancer [9-11]. Naderi et al. found that BEX2 was up-regulated in a subset of primary breast cancers and that down-regulation of BEX2 induced G1-phase arrest in breast cancer cell lines [10]. BEX2 is also highly expressed in glioblastomas and promotes cell proliferation and survival by mediating nuclear factor-kappa B activity [11]. However, other studies have obtained opposite findings, showing that BEX2 inhibits cell proliferation. For example, Foltz et al. showed that BEX2 was epigenetically silenced in primary glioma cells and exhibited extensive promoter hypermethylation, and BEX2 re-expression resulted in significant suppression of tumor growth [12]. Furthermore, overexpression of BEX2 in mouse pro-B and myeloid cells resulted in decreased FLT3 (FMS-like tyrosine kinase-3)-ITD (Internal tandem duplication)-dependent cell proliferation [13]. Additionally, decreased BEX2 expression in Hs683 oligodendroglioma cells did not lead to changes in cell proliferation [14]. Thus, BEX2 appears to exhibit different expression patterns and functions in different types of tumors. However, the expression and function of BEX2 in colorectal cancer still remain unknown.
Here, to better characterize the role of BEX2 in CRC, we sought to explore the expression of BEX2 in CRC specimens at different tumor stages. BEX2 was shown to be associated with more aggressive characteristics in colorectal cancer. Subsequently, knockdown of BEX2 was proved to inhibit cell growth in a colorectal cancer cell line both in vitro and in vivo. Furthermore, the effects of BEX2 on cell proliferation appeared to be mediated by the JNK/c-Jun pathway. This study reveals that BEX2 plays an important role in cell proliferation in colorectal cancer and suggests that BEX2 might be a novel candidate target for the treatment of CRC.
Methods
Validation of the relationship between the expression of BEX2 and clinical manifestations of illness in an independent dataset
The relationship between the expression of BEX2 and clinical manifestations of illness was validated using publicly available independent microarray datasets (GSE14333) by Jorissen et al. [15]. The GSE14333 dataset consisted of 290 patients (Supplementary Table 1) with CRC and was downloaded from the Gene Expression Omnibus dataset (http://www.ncbi.nlm.nih.gov/geo).
Patients and specimens
Thirty-four patients (Supplementary Table 2) who were diagnosed with CRC and underwent colectomy between 2013 and 2014 were enrolled in this study. None of the enrolled patients had received any prior chemotherapy or radiotherapy. The disease stage was determined according to the pathological tumor-node-metastasis (pTNM) classification system [16]. Surgically resected colorectal cancer specimens and paired normal mucosal tissues banked at -80°C were obtained. Tumors were de-identified in accordance with the protocols approved by the Institutional Review Board (IRB) of the Second Affiliated Hospital of Zhejiang University School of Medicine.
Cell culture and SP600125 treatment
All of the cells were maintained in RPMI-1640 (Jinuo Biotech, HangZhou, China) supplemented with 10% fetal bovine serum (FBS) (Life Technologies, Carlsbad, CA, USA), penicillin and streptomycin at 37°C with 5% CO2. SW620 cells were obtained from the American Type Culture Collection (ATCC) (Rockville, MD, USA). The SW620/shBEX2 cell line and the SW620/Ctrl cell line were established by stably expressing shRNA targeting BEX2 or a scramble control shRNA in SW620 cells as described below. SW620/BEX2 cells and SW620/vector cells were established by transfecting the mammalian expression vectors pCMV-Myc-BEX2 and pCMV-Myc as described below. SP600125, a selective inhibitor of JNK [17], was purchased from Selleck Chemicals Company (Houston, TX, USA). SW620/Ctrl cells were treated with 10 µM SP600125 for 72 hours before protein extraction or cell proliferation analysis.
Establishment of the SW620/shBEX2 and SW620/Ctrl cell lines
Lentiviral particles containing a validated short hairpin RNA directed against BEX2 (sc-60271-V) and the corresponding scramble control (sc-108080) were purchased from Santa Cruz Biotechnology, (Santa Cruz, CA). Lentiviral infection was performed according to the manufacturer's instructions. Briefly, SW620 cells were plated at 50% confluence. On the day of infection, the culture medium was replaced with complete medium containing lentiviral particles (MOI=20) and polybrene (5 μg/ml). Following 24 hours of infection at 37°C, the viral supernatant was replaced with fresh medium. After an additional 48 hours, the infected cells were treated with 2.0 µg/ml puromycin dihydrochloride (Santa Cruz Biotechnology) for 2 weeks for stable clone selection. The knockdown efficiency was determined through quantitative real-time PCR and Western blot analyses of BEX2 using a rabbit anti-BEX2 polyclonal antibody (1:1000, Proteintech, Chicago, IL).
BEX2 transfection in SW620 cells
Sense cDNA for BEX2 was introduced into the multi-cloning site of the mammalian expression vector pCMV-Myc to construct sense plasmids. The plasmids were then transfected into SW620 cells using the GeneJet™ Plus reagent (SignaGen Laboratory, Rockville MD, USA) following the manufacturer's protocol. Cells were incubated with the transfection medium for 12 hours, after which the transfection complex-containing medium was gently removed, and fresh culture medium was added. The cells were grown for two days, and the transfection efficiency was analyzed before further use.
Quantitative real-time RT-PCR
Total RNA from cells, fresh CRC tissues or xenograft tissues was isolated using the TRIzol reagent (PuFei Biotech, ShangHai, China) according to the manufacturer's instructions. The quality and quantity of the RNA was evaluated using a NanoDrop 1000 spectrophotometer (Thermo Scientific, Pittsburgh, PA, USA). cDNA was synthesized with the PrimeScript™ II 1st Strand cDNA Synthesis Kit (Takara Biotechnology, Dalian, China). Quantitative real-time RT-PCR (qPCR) was performed using the StepOnePlus Real-Time PCR System (Life Technologies, Foster, CA, USA), and the results were analyzed with StepOne software V2.1. Gene expression was quantified using the TaqMan probe system and the following primers and probes: BEX2 forward: 5'-TGCCTAGAGGAAACCGTAGG-3', reverse: 5'- TCCATCAGCTGTCTCACCTC-3', and probe: 5'-FAM-TTCCGCGTTAGGCAGCCCAT-TAMRA-3'; GAPDH forward: 5'-CCACTCCTCCACCACCTTTGAC-3', reverse: 5'-ACCCTGTTGCTGTAGCCA-3', and probe: 5'- FAM-TTGCCCTCAACGACCACTTTGTC-TAMRA -3'. All PCR assays were performed in triplicate. BEX2 gene expression was normalized to the expression of GAPDH. The primers and probes were synthesized by Sangon Biotech (Shanghai, China).
Western blot analysis
Western blot analysis was performed as previously described [18]. Briefly, cell proteins were extracted in M-PER (Thermo Scientific) supplemented with a 1% proteinase cocktail and 1% phosphatase inhibitor cocktail (Sigma-Aldrich, Hamburg, Germany). The protein concentration was measured using a BCA protein assay kit (Thermo Scientific). Protein samples (50 µg) were subsequently separated via 12% SDS-PAGE, then transferred to a PVDF membrane (Bio-Rad, Berkeley, CA), and proteins were detected using appropriate primary and secondary antibodies. Protein bands were visualized using the enhanced chemiluminescence substrate (Thermo Scientific) and scanned with a Kodak Image Station (Carestream Health, Rochester, NY). The primary antibodies included a rabbit anti-BEX2 polyclonal antibody (1:1000, Proteintech), a rabbit anti-NF-κB monoclonal antibody (1:1000, Cell Signaling Technology, Beverly, MA), a rabbit anti-phosphorylated-Akt (S473) polyclonal antibody (1:1000, Abcam, Cambridge, UK), a rabbit anti-Akt monoclonal antibody (1:1000, Abcam), a rabbit anti-Erk1/2 polyclonal antibody (1:1000, Abcam), a rabbit anti-phosphorylated-p38 (Thr180/Tyr182) monoclonal antibody (1:1000, Cell Signaling Technology), a mouse anti-JNK monoclonal antibody (1:1000, Santa Cruz Biotechnology), a rabbit anti-phosphorylated-JNK (Thr183/Tyr185) monoclonal antibody (1:1000, Cell Signaling Technology), a rabbit monoclonal c-Jun antibody (1:1000, Santa Cruz Biotechnology, CA USA) and a goat polyclonal phosphorylated-c-Jun antibody (1:1000, Santa Cruz Biotechnology). A rabbit anti-GAPDH monoclonal antibody (1:5000, Cell Signal Technology) was used as a loading control.
Cell proliferation assay
Cell proliferation was analyzed using a Cell Counting Kit-8 (CCK-8) (KeyGEN BioTech, Nanjing, China). All cells were seeded into 96-well plates at a density of 5000 cells/well in a 200 µl volume and incubated at 37°C under 5% CO2 for 24, 48, 72, or 96 hours, followed by the addition of 10 μl of CCK-8 solution. The absorbance in each well was measured at 0 and 2 hours using a microculture plate reader at a test wavelength of 450 nm. Four replicate wells were set up in each group, and three independent experiments were performed.
Clone formation assay
Cells in the exponential phase of growth were digested and resuspended in complete medium. The cell suspension was then serially diluted and inoculated into 6-well plates containing 5 ml of medium at a density of 500 cells/well, quantified using a hemocytometer. The culture medium was changed every 3 days. After 2 weeks, clone spheres had formed in the dishes. The cells were then rinsed with 0.01% PBS and fixed with 4% paraformaldehyde for 15 minutes. Next, crystal violet solution was added for 15 minutes, and the samples were rinsed with water and air-dried. The number of clones was counted using Image J software. [19]
Mice
Balb/c athymic nude mice (SLAC Laboratory Animal Co. Ltd., Shanghai, China) were maintained and subjected to experiments in accordance with the protocols approved by the Animal Care and Use Committee of the Second Affiliated Hospital of Zhejiang University School of Medicine. All of the animal experiments were performed on six-to-eight-week-old female Balb/c athymic nude mice.
Subcutaneous xenograft model
Tumor cells (1×106) were subcutaneously injected into mice. Tumor growth was monitored daily until the tumor was palpable. Then, the tumor diameter was measured with calipers twice a week. Tumor-free survival was measured from the day of tumor inoculation until the long axis of the tumor was shorter than 2 mm and analyzed using the Kaplan-Meier curve. Mice were euthanized at week 5 following tumor inoculation. The long (L) and short (S) axes of each tumor were measured for the harvested tumors with calipers. Tumor volume (V) was calculated as V = (L×S2)/2.
Colorectal cancer orthotopic model
The orthotopic mouse model of colorectal cancer was established using a previously described cecal wall injection technique [20, 21]. In brief, nude mice were anesthetized, and the cecum was exteriorized via laparotomy. A total volume of 50 μl of a cell suspension containing 1×106 tumor cells was injected into the cecal wall using a 27G needle. After injection, the injection point was slightly pressed with a cotton stick and inspected to ensure no leakage. The cecum was subsequently returned to the abdominal cavity and closed with running sutures. All of the mice were maintained until death caused by the neoplastic process or until the end of the experiment (140 days). All mice were monitored twice daily. Overall mouse survival was analyzed using the Kaplan-Meier curve.
Cell cycle analysis
Cells were washed twice in ice-cold 10 mM phosphate-buffered saline (pH 7.4) and fixed in 75% ethanol at 4°C for 24 hours. After additional washing, 1×106 cells were digested with RNase (50 mg/ml) and stained with 4 µg/ml Hoechst 33342 (Sigma, Aldrich, Hamburg, Germany) for 30 minutes at 37°C. DNA content was determined using a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA) and Modfit software. All experiments were performed in triplicate and were independently repeated three times.
Cell apoptosis analysis
A total of 1×106 cells were harvested and double-stained with FITC-conjugated annexin V and propidium iodide (PI) using an Annexin V-FITC Apoptosis Detection Kit (Bio-Rad Lab., Hercules, CA, USA), according to the manufacturer's protocol. The cells were analyzed with a BD FACSCalibur Flow Cytometer (Becton Dickinson) within 1 hour of staining. Apoptotic cells were defined as annexinV-FITC-positive and PI-negative cells. All experiments were performed in triplicate and were independently repeated three times.
Statistical analysis
All of the graphing and statistical analyses, except for the trend of BEX2 expression, were performed using GraphPad Prism version 5.0 software (GraphPad Software, La Jolla, CA). The data are presented as the mean ± standard error of the mean (SEM). The qPCR results from paired clinical samples were analyzed using the two-tailed paired Student's t-test. The trend of BEX2 expression across tumors with different pTNM stages was analyzed via the Cuzick nonparametric test, performed using Stata 13 (StataCorp LP, College Station, TX). Comparison of tumor-free survival between groups was performed using the log-rank (Mantel-Cox) test. The other results were analyzed with the two-tailed unpaired Student's t-test. P values <0.05 were considered statistically significant.
Results
BEX2 is highly expressed in more advanced colorectal cancers
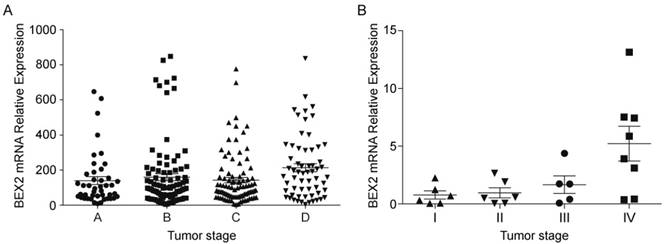
To understand the function of BEX2 in colorectal cancer, we first sought to assess BEX2 expression in colorectal cancer tissues. We analyzed the expression of BEX2 in 290 CRC patients in the GSE14333 sample cohort and observed that the expression of BEX2 was significantly correlated with the aggressive characteristics of CRC (Cuzick nonparametric test for trend, p=0.0147, Figure 1A). The clinical characteristics of this cohort are listed in Supplementary Table 1. To confirm our findings, we verified the expression of BEX2 in 34 colorectal cancer specimens via quantitative real-time PCR. Higher levels of BEX2 were found to be expressed in more advanced tumors (Cuzick nonparametric test for trends, p<0.05; Figure 1B) when the patients were stratified based on the diagnosis of pTNM staging, where stage I was the least advanced, and TNM stage IV was the most advanced.
Down-regulation of BEX2 inhibits colorectal cancer cell growth in vitro.
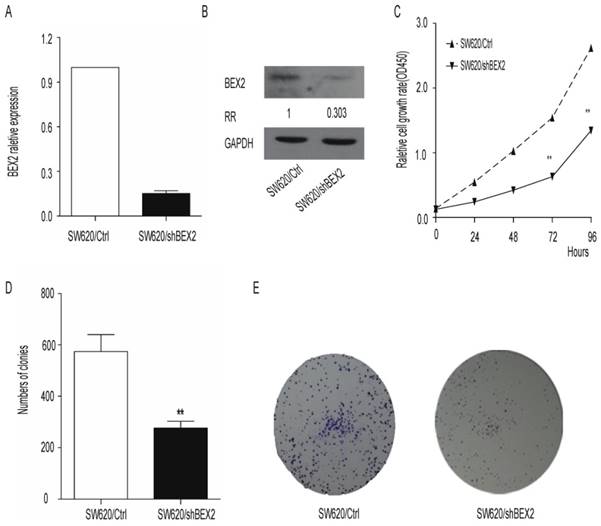
Because BEX2 overexpression was found to be associated with more advanced stages of colorectal cancer, we next examined whether knockdown of BEX2 would inhibit the progression of colorectal cancer in vitro. We first generated the SW620/shBEX2 cell line utilizing a lentivirus shRNA system to inhibit BEX2 expression in the SW620 cell line. BEX2 expression was reduced by 70% in SW620/shBEX2 cells compared with control SW620/Ctrl cells, as assessed via qPCR (Figure 2A) and Western blot (Figure 2B). We next examined the effect of the down-regulation of BEX2 on cell growth using the CCK8 assay. We observed significant growth inhibition in the SW620/shBEX2 cell line (Student's t-test, p<0.01) (Figure 2C). Similarly, the number of cell colonies in monolayer cultures of SW620/shBEX2 cells was smaller than in the SW620/Ctrl group (Student's t-test, P<0.01) (Figure 2D, 2E). These results suggest that BEX2 knockdown may prevent colorectal cancer progression by suppressing cell growth.
BEX2 expression in colorectal cancer tumor tissues. A. The expression of BEX2 is significantly correlated with the aggressive characteristics of CRC (p=0.0147). Dataset GSE14333 was used in this analysis. Colorectal cancer tumor tissues were ordered according to Duckes staging: stage A (n=44), stage B (n=94), stage C (n=91) and stage D (n=61). The Cuzick nonparametric test for trends was employed to evaluate trends. B. Quantitative real-time RT-PCR was used to measure BEX2 expression levels in 34 colorectal cancer specimens. BEX2 is expressed at higher levels in more advanced tumors (p=0.002). Colorectal cancer tumor tissues were ordered according to pTNM staging: stage I (n=0), stage II (n=7), stage III (n=7) and stage IV (n= 10).
Down-regulation of BEX2 inhibits SW620 colorectal cancer cell proliferation in vitro. A. BEX2 mRNA expression was quantified via qPCR. BEX2 expression levels in SW620 cells transduced with BEX2 shRNA (SW620/shBEX2) or with control shRNA (SW620/Ctrl) are shown. GAPDH expression was used for normalization. B. BEX2 expression was examined through Western blot analysis and quantified using ImageJ software. The relative ratio (RR) of BEX2 protein expression in SW620/shBEX2 cells compared with SW620/Ctrl cells is shown. C. The growth curves of SW620/shBEX2 cells and SW620/Ctrl cells were measured using CCK-8 cell proliferation assays. Compared with the control cells, SW620/shBEX2 cells showed significant growth inhibition from the third day onward. D. Quantitative analyses of colony formation were conducted using Image J software. Knockdown of BEX2 in SW620 cells resulted in significant inhibition of colony formation. Three independent experiments were performed in triplicate. E. Representative images of colonies of SW620/shBEX2 cells and SW620/Ctrl cells (**p< 0.01).
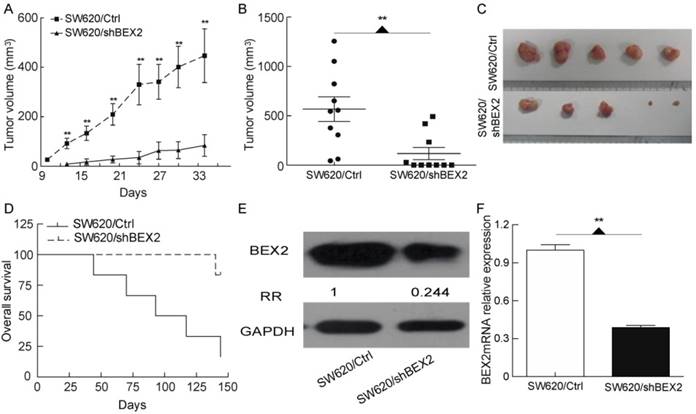
BEX2 knockdown suppresses colorectal cancer proliferation in vivo. A. Subcutaneous xenograft growth curves of SW620/shBEX2 (n=10) and SW620/Ctrl (n=10) cells. Xenograft growth was significantly slowed in the BEX2-knockdown group. B. Measurements of the volumes of xenografts harvested at week 5 after the inoculation of tumor cells (shown in A). Tumor volume was significantly smaller after BEX2 knockdown. Five mice exhibited no palpable tumors in the SW620/shBEX2 group when euthanized. C. Tumors harvested from the experiment quantified in B are shown. The five largest tumors are shown as representatives for each group. D. Overall survival of mice with cecum orthotopic colorectal cancer. The majority of the mice in the SW620/Ctrl cohort (4/5) died within 140 days, whereas none of the mice in the SW620/shBEX2 cohort died within this time (0/5). E. Western blot (E) and q-PCR (F) analyses showing BEX2 expression in the tumor tissues from the subcutaneous xenograft model. Data are expressed as the mean ± SD (**p < 0.01).
Down-regulation of BEX2 suppresses colorectal cancer growth in vivo.
To further assess whether the down-regulation of BEX2 inhibits colorectal cancer growth in vivo, we employed a subcutaneous xenograft model. SW620/shBEX2 cells or SW620/Ctrl cells were subcutaneously inoculated into Balb/c athymic nude mice. The xenografts grew significantly more slowly in the BEX2-knockdown group (p=0.0043) (Figure 3A). Five weeks after inoculation, the xenografts formed by SW620/shBEX2 cells were significantly smaller than those formed by SW620/Ctrl cells (p=0.0047) (Figure 3B, 3C). Within 4 weeks, all of the xenografts in the SW620/Ctrl group had increased in size. However, half of the mice (n=5) showed no palpable tumors in the SW620/shBEX2 group, even after 5 weeks. The tumor-free survival of Balb/c athymic nude mice inoculated with SW620/shBEX2 cells was significantly longer than that of the SW620/Ctrl group (Log-rank test, p=0.0021) (Supplementary Figure 1). We further explored the role of BEX2 in colorectal cancer using a cecum orthotopic model of colorectal cancer. Similar to our previous results, mice injected with SW620/shBEX2 cells survived significantly longer than mice injected with SW620/Ctrl cells (log-rank test, p=0.0169) (Figure 3D). A post-mortem examination was performed for the majority of the animals, and tumor growth was identified as the cause of death. In addition, we validated the down-regulation of BEX2 in tumors from the SW620/shBEX2 group subjected to the subcutaneous xenograft model via both qPCR and Western blotting (Figure 3E, 3F). Taken together, these results indicated that BEX2 knockdown suppressed colorectal cancer proliferation in vivo.
BEX2 knockdown inhibits colorectal cancer cell proliferation by inactivating the JNK/c-Jun signaling pathway
Next, we focused on identification of the molecular mechanisms through which BEX2 suppresses colorectal cancer proliferation. It is known that the cell cycle duration, growth fraction and cell apoptosis can affect tumor growth. BEX2 was previously shown to interrupt cell apoptosis and reduce the growth fraction in breast cancer cells [8], leukemic cells [21] and glioma cells [22]. However, flow cytometry assays demonstrated that there was no difference in the percentage of cells in each phase of the cell cycle between SW620/shBEX2 cells and SW620/Ctrl cells (Figure 4A) and that the apoptotic ratio was not increased in SW620 cells after BEX2 knockdown (Figure 4B). Therefore, down-regulation of BEX2 may inhibit colorectal cancer cell growth by extending the cell cycle, rather than by inducing cell cycle arrest or activation of cell apoptosis.
Knockdown of BEX2 inhibits colorectal cancer cell proliferation by suppressing JNK/c-Jun phosphorylation. A. Cell cycle analysis via flow cytometry showed no significant differences between SW620/shBEX2 cells and SW620/Ctrl cells. B. Knockdown of BEX2 in SW620 cells resulted in no obvious changes in cell apoptosis. C. Analysis of the NF-κB, Akt and MAPK signaling pathway in BEX2-knockdown SW620 cells. NF-κB (P65) and total and phosphorylated Akt and MAPK kinases, including extracellular signal-regulated kinase 1/2 (Erk1/2), p38 and JNK, were detected through Western blot analysis. The results revealed that down-regulation of BEX2 inactivated JNK, which in turn inhibited the activation of the transcription factor c-Jun. D. Analysis of MAPK/JNK signaling pathway in BEX2-overexpressed SW620 cells. The results showed that SP600125 treatment suppressed BEX2 overexpression-activated JNK/c-Jun phosphorylation. E. Administration of the JNK-specific inhibitor SP600125 to SW620/BEX2 cells suppressed the relatively high phosphorylation of JNK/c-Jun activated by BEX2, thus eliminating the BEX2-induced proliferation advantage. Data are expressed as the mean ± SD (**p < 0.01).
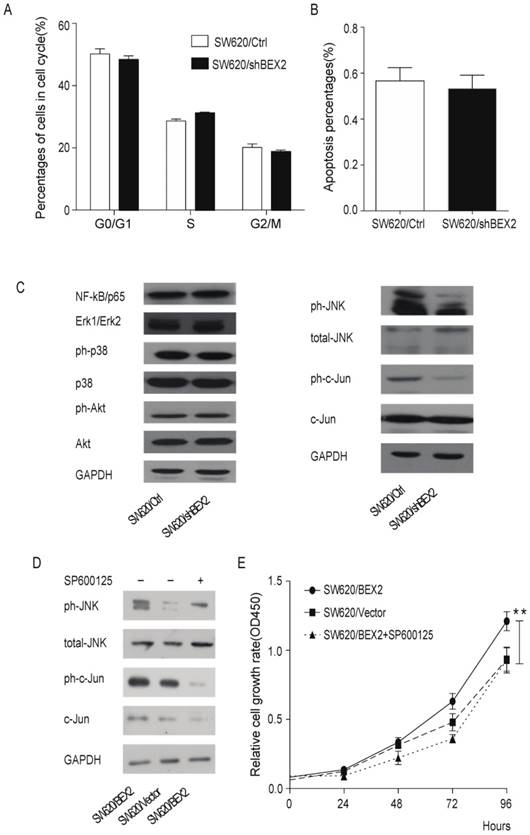
Because the NF-κB, Akt and MAPK signaling pathway is the most relevant pathway in the regulation of cell growth, we evaluated the activity of NF-κB, Akt and MAPK in colorectal cancer cells before and after down-regulation of BEX2. Western blot analysis demonstrated that JNK, a mediator of the MAPK pathway, and its downstream transcription factor c-Jun were inactivated, whereas p65, Akt, Erk1/2 and p38 showed no significant changes in SW620 cells in which BEX2 has been stably knocked down (Figure 4C). To determine whether JNK/c-Jun phosphorylation is required for BEX2-induced proliferation, we overexpressed BEX2 in SW620 cells (SW620/BEX2). BEX2 overexpression was confirmed via qPCR (Supplementary Figure 2A) and Western blotting (Supplementary Figure 2B), which demonstrated a 20-fold increase compared with the control cells (SW620/Vector). BEX2 overexpression promoted cell growth compared with the SW620/Vector (Student's t-test, p<0.01) (Figure 4D). We subsequently treated SW620/BEX2 cells with the JNK-specific inhibitor SP600125 (10 µM). SP600125 treatment suppressed BEX2 overexpression-activated JNK/c-Jun phosphorylation (Figure 4E) and eliminated the BEX2-induced proliferative advantage (Figure 4D). In summary, down-regulation of BEX2 inhibited colorectal cancer cell growth by extending the cell cycle, which was mediated by JNK/c-Jun pathway inactivation.
Discussion
In the present study, we showed that BEX2 expression was associated with the most advanced stages of colorectal cancer, indicating that BEX2 expression is a causal factor in the progression of colorectal cancer. Subsequently, BEX2 knockdown in a colorectal cancer cell line was demonstrated to suppress cell growth and tumor proliferation both in vitro and in vivo. Furthermore, the effects of BEX2 on colorectal cancer cell proliferation appeared to regulate the cell cycle duration through the JNK/c-Jun pathway. To the best of our knowledge, this is the first study to elucidate the role of BEX2 in colorectal cancer, suggesting that BEX2 might be a novel candidate target for the comprehensive treatment of colorectal cancer.
BEX2 is down-regulated in malignant glioma [12] and acute myeloid leukemia [13], and BEX2 re-expression results in significant suppression of tumor growth, supporting the role of BEX2 as a tumor suppressor. However, BEX2 is highly expressed in a subset of estrogen receptor-positive breast cancers [10] and glioblastoma [11] and plays a key role in promoting cell survival and growth in breast cancer cells. The BEX protein family has been reported to contain long regions of intrinsic disorder that may form signaling hubs, and the hubs formed by intrinsically disordered proteins play important roles in cellular differentiation and cancer [6]. Thus, BEX2 appears to play different roles in different tumor types. It is possible that BEX2 expression levels are tightly regulated, and overexpression or down-regulation of BEX2 could lead to unstable cell growth. The present study showed that BEX2 was expressed at higher levels in more advanced tumors (Figure 1A and 1B), indicating that BEX2 is a key modulator in the proliferation of colorectal cancer.
Previous studies have shown that BEX2 plays a critical role in regulating the cell cycle and apoptosis [10, 18, 22, 23]. In breast cancer cells, down-regulation of BEX2 induces mitochondrial apoptosis and results in G1-cell cycle arrest. In glioblastoma cells, BEX2 knockdown also induces apoptosis by activating caspase 9. However, decreasing BEX2 expression was not found to influence apoptosis and the cell cycle in glioma cells [14]. Therefore, BEX2 regulates either the cell cycle or apoptosis in different tumor types. In the present study, neither the cell cycle nor cell apoptosis was affected after BEX2 knockdown in colorectal cancer cells (Figure 4A, 4B), indicating that BEX2 down-regulation can cause growth suppression due to extension of the cell cycle.
Our findings suggested that phospho-JNK and phospho-c-Jun were significantly decreased after BEX2 down-regulation in colorectal cancer cells. This result was consistent with a report by Naderi et al. [24] showing that BEX2 exhibits functional interplay with JNK/c-Jun in breast cancer cells. In particular, BEX2 has been identified as a target gene of c-Jun and is necessary for the phosphorylation of c-Jun and JNK kinase activity.
In summary, our study demonstrates that BEX2, a novel causal factor in the progression of colorectal cancer, promotes colorectal cancer cell proliferation via the JNK/c-Jun signaling pathway. Future studies will be necessary to investigate whether BEX2 expression is associated with the prognosis of colorectal cancer and to assess the potential of BEX2 as an effective therapeutic target for colorectal cancer. Such studies will provide additional insight into colorectal cancer and provide a rationale for the utilization of innovative therapy targeting BEX2 to improve colorectal cancer treatment.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81272455, 81472664), Public Welfare in the Health Industry, 2014, the Health Ministry of China (No. 201402015), Key Projects in the National Science & Technology Pillar Program during the Twelfth Five-year Plan Period (No. 2014BAI09B07) and the Zhejiang Provincial Natural Science Foundation of China (No. LY16H160011). The sponsors of the study had no role in the study design, data collection, data analysis, interpretation of results, writing of the manuscript or the decision to submit the paper for publication.
Competing Interests
The authors declare that they have no competing interests.
References
1. Jemal A, Siegel R, Xu J. et al. Cancer statistics, 2010. CA Cancer J Clin. 2010;60(5):277-300
2. Sung JJ, Lau JY, Young GP. et al. Asia Pacific consensus recommendations for colorectal cancer screening. Gut. 2008;57(8):1166-76
3. Jonker DJ, O'Callaghan CJ, Karapetis CS. et al. Cetuximab for the treatment of colorectal cancer. New Engl J Med. 2007;357(20):2040-8
4. Saltz L, Easley C, Kirkpatrick P. Panitumumab. Nat Rev Drug Discov. 2006;5(12):987-8
5. Saltz L. Colorectal Cancer Treatment: What's Next? (or: Is There Life After EGFR and VEGF?). Gastrointestinal cancer research: GCR. 2008;2(4 Suppl):S20-2
6. Fernandez EM, Diaz-Ceso MD, Vilar M. Brain Expressed and X-Linked (Bex) Proteins Are Intrinsically Disordered Proteins (IDPs) and Form New Signaling Hubs. Plos One. 2015;10(1):e0117206
7. Alvarez E, Zhou W, Witta SE, Freed CR. Characterization of the Bex gene family in humans, mice, and rats. Gene. 2005;357(1):18-28
8. Han C, Liu H, Liu J. et al. Human Bex2 interacts with LMO2 and regulates the transcriptional activity of a novel DNA-binding complex. Nucleic Acids Res. 2005;33(20):6555-65
9. Kazi JU, Kabir NN, Ronnstrand L. Brain-Expressed X-linked (BEX) proteins in human cancers. Biochimica et biophysica acta. 2015;1856(2):226-33
10. Naderi A, Liu J, Bennett IC. BEX2 regulates mitochondrial apoptosis and G1 cell cycle in breast cancer. Int J Cancer. 2010;126(7):1596-610
11. Meng QM, Zhi TL, Chao YW. et al. Bex2 Controls Proliferation of Human Glioblastoma Cells Through NF-kappa B Signaling Pathway. J Mol Neurosci. 2014;53(2):262-70
12. Foltz G, Ryu GY, Yoon JG. et al. Genome-wide analysis of epigenetic silencing identifies BEX1 and BEX2 as candidate tumor suppressor genes in malignant glioma. Cancer Res. 2006;66(13):6665-74
13. Lindblad O, Li T, Su X. et al. BEX1 acts as a tumor suppressor in acute myeloid leukemia. Oncotarget. 2015;6(25):21395-405
14. Le Mercier M, Fortin S, Mathieu V. et al. Galectin 1 proangiogenic and promigratory effects in the Hs683 oligodendroglioma model are partly mediated through the control of BEX2 expression. Neoplasia. 2009;11(5):485-96
15. Jorissen RN, Gibbs P, Christie M. et al. Metastasis-Associated Gene Expression Changes Predict Poor Outcomes in Patients with Dukes Stage B and C Colorectal Cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15(24):7642-51
16. Wittekind C, Compton CC, Greene FL. et al. TNM residual tumor classification revisited. Cancer. 2002;94(9):2511-6
17. Bennett BL, Sasaki DT, Murray BW. et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. P Natl Acad Sci USA. 2001;98(24):13681-6
18. Ding K, Su Y, Pang L. et al. Inhibition of apoptosis by downregulation of hBex1, a novel mechanism, contributes to the chemoresistance of Bcr/Abl+ leukemic cells. Carcinogenesis. 2009;30(1):35-42
19. Cai Z, Chattopadhyay N, Liu WJ. et al. Optimized digital counting colonies of clonogenic assays using ImageJ software and customized macros: Comparison with manual counting. Int J Radiat Biol. 2011;87(11):1135-46
20. Tseng W, Leong X, Engleman E. Orthotopic mouse model of colorectal cancer. JOVE-J Vis Exp. 2007(10):484
21. Virtudes Cespedes M, Espina C, Angel Garcia-Cabezas M. et al. Orthotopic microinjection of human colon cancer cells in nude mice induces tumor foci in all clinically relevant metastatic sites. Am J Pathol. 2007;170(3):1077-85
22. Xiao Q, Hu Y, Liu Y. et al. BEX1 promotes imatinib-induced apoptosis by binding to and antagonizing BCL-2. PLoS One. 2014;9(3):e91782
23. Zhou X, Meng Q, Xu X. et al. Bex2 regulates cell proliferation and apoptosis in malignant glioma cells via the c-Jun NH2-terminal kinase pathway. Biochem Biophys Res Commun. 2012;427(3):574-80
24. Naderi A, Liu J, Hughes-Davies L. BEX2 has a functional interplay with c-Jun/JNK and p65/RelA in breast cancer. Mol Cancer. 2010;9:111
Author contact
![]() Corresponding author: Kefeng Ding, M.D., Ph.D. dingkefengedu.cn; 88 Jiefang Road, Building 9 Room 1314, Hangzhou, Zhejiang, China 310009. Tel: 86-571-87784720; Fax: 86-571-87214404.
Corresponding author: Kefeng Ding, M.D., Ph.D. dingkefengedu.cn; 88 Jiefang Road, Building 9 Room 1314, Hangzhou, Zhejiang, China 310009. Tel: 86-571-87784720; Fax: 86-571-87214404.
Received 2016-2-1
Accepted 2016-9-23
Published 2017-2-12