ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

Int J Biol Sci 2017; 13(6):735-747. doi:10.7150/ijbs.18153 This issue Cite
Research Paper
Genome-wide Identification and Expression Analysis of Amino Acid Transporters in the Whitefly, Bemisia tabaci (Gennadius)
Jixing Xia1, 2, 3*, Zezhong Yang3, 4*, Cheng Gong3*, Wen Xie3, Huipeng Pan5, Zhaojiang Guo3, Huixin Zheng3, 4, Xin Yang3, Xiaodong Sun3, Shi Kang3, Fengshan Yang6, Qingjun Wu3, Shaoli Wang3, Bin Cong1, Xianfeng Teng2, Youjun Zhang3 ![]()
1. College of Plant Protection, Shenyang Agricultural University, Shenyang, 110866 China;
2. Department of Biocontrol, Institute of Plant Protection, Heilongjiang Academy of Agricultural Sciences, Harbin, 150080 China;
3. Department of Plant Protection, Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences, Beijing, 100081 China;
4. College of Plant Protection, Hunan Agricultural University, Changsha, 41000 China;
5. Department of Entomology, South China Agricultural University, Key Laboratory of Bio-Pesticide Innovation and Application of Guangdong Province, Guangzhou, 510642 China;
6. Key Laboratory of Molecular Biology of Heilongjiang Province, College of Life Sciences, Heilongjiang University, Harbin 150080, China.
* Both authors contributed equally to this work.
Abstract

The whitefly (Bemisia tabaci) is a cosmopolitan and devastating pest of agricultural crops and ornamentals. B. tabaci causes extensive damage by feeding on phloem and by transmitting plant viruses. Like many other organisms, insects depend on amino acid transporters (AATs) to transport amino acids into and out of its cells. We present a genome-wide and transcriptome-wide investigation of the following two families of AATs in B. tabaci biotype B: amino acid/auxin permease (AAAP) and amino acid/polyamine/organocation (APC). A total of 14 putative APCs and 25 putative AAAPs were identified, and a 10-paralog B. tabaci-specific expansion of AAAPs was found by maximum likelihood phylogeny. Detailed gene structure information revealed that 9 members of the B. tabaci-specific AAAP family expansion closely situated on a same scaffold. Expression profiling of the B. tabaci B APC and AAAP genes as affected by stage and plant host showed diverse expression patterns. The analysis of evolutionary rates indicated that purifying selection can explain the B. tabaci-specific AAAP expansion. RNA interference (RNAi)-mediated suppression of two AAAP genes (BtAAAP15 and BtAAAP21) significantly increased the mortality of B. tabaci B adults. The results provide a foundation for future functional analysis of APC and AAAP genes in B. tabaci.
Keywords: Bemisia tabaci, Amino acid/polyamine/organocation, Amino acid/auxin permease, Gene duplication, Genome.
Introduction
Amino acids are essential nutrients and are closely related to the life activities of organisms [1-4]. They are essential for protein synthesis, regulation of cell growth, nerve transmission, hormone metabolism, nitrogen metabolism, and production of metabolic energy [5]. Nevertheless, many animals are incapable of synthesizing 10 amino acids (arginine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine) that are required for normal growth and development; these amino acids are typically acquired through diet [6, 7]. Transport of amino acids into and out of cells depends on specific amino acid transporters (AATs). The following two AAT families, which belong to the APC superfamily, are the main nutritional AATs in eukaryotes and have important nutritional roles in insects [8-18]: the amino acid/polyamine/organocation (APC) family and the amino acid/auxin permease (AAAP) family. The transport proteins of the APC family function as symporters and antiporters, and they also catalyze solute uptake or exchange transport [19]. APC family proteins range from 350 to 800 amino residues in length, and most contain 12 transmembrane domains (TMSs), but 10 or 14 TMSs are sometimes observed by predicted topologies [19]. The transport proteins of the AAAP family catalyze the transmembrane transport of single amino acid, multiple amino acids, auxin (indole-3-acetic acid) or γ-aminobutyric acid [19]. The AAAP family proteins range from 376 to 713 amino residues in length, but most have 400 to 500 residues and contain 11 putative TMSs [19]. In the lepidopteran larval midgut, an apical H+ V-ATPase and an apical K+/2H+ antiporter are responsible for active secretion of K+ and thereby provide the driving force for transport of amino acids [5, 20]. In other insects, the mechanisms of amino acid transport are predominantly driven by Na+ motive force [7, 21]. The roles of AATs have been extensively researched in insects. For example, AATs are potent mediators of growth in Drosophila melanogaster [8-9]. AATs are also involved in the regulation of reproduction in Aedes aegypti [10-11, 22], pupation in Leptinotarsa decemlineata [16], and glutamine supply to intracellular bacterial symbionts in aphids [15]. AAT has been confirmed as a functional receptor of Bombyx mori densovirus type 2 (BmDNV-2) in B. mori [23]. Furthermore, the sex-linked translucent larval skin of Bombyx mori is linked to mutations in an AAT in B. mori [24].
The whitefly Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) is a typical phloem sap-feeding insect and is regarded as one of the most devastating pests of agricultural crops and ornamentals worldwide [25]. The whitefly has the broadest hosts, which is estimated to have 74 families and 500 species [26]. B. tabaci can greatly reduce crop yields by feeding on phloem, excreting honeydew, and transmitting 311 plant viruses [27]. As a diet, phloem sap contains low levels of nitrogen and essential amino acids [6]. The bacterial symbionts in phloem-feeding insects provide the essential amino acids that compensate for the lack of essential amino acids in phloem sap [28-34]. As noted earlier, AATs involved in the transport of amino acids between bacterial symbionts and insect host [15], and the transport of amino acids in insects depends on AATs [5, 21]. Although the interactions between endosymbionts and B. tabaci have been studied [31-35], a genome-wide identification and functional report about AATs in B. tabaci is lacking.
Genome sequencing technology made it possible to study functional genes at the genome level. Genome-wide investigation has been performed for AAT genes in rice and Acyrthosiphon pisum [36, 12]. Our laboratory has recently conducted genome and transcriptome sequencing of B. tabaci biotype B (also termed Middle East-Asia Minor 1; hereafter B. tabaci B). The genomic and transcriptomic information for B. tabaci B enabled the global analysis of APC and AAAP genes in this insect. In the current study, we identified the APC family and AAAP family genes and profiled their expression as affected by life stage and plant host based on the B. tabaci genome, transcriptomic, and RT-qPCR. The results provided an overview of the gene structures, evolutionary relationships, extensive gene duplications, and the expression specificity of these AAT genes. In addition, we found that knockdown of the AAAP15 and AAAP21 genes significantly increased the mortality of B. tabaci B adults.
Materials and Methods
Insects and host plants
A B. tabaci B was maintained on cotton plants at 27 ± 1 °C with an L16:D8 photoperiod and a relative humidity (RH) of 70 ± 10%. Every 3 to 5 generations, the purity of the strain was monitored using the PCR technique and the sequence of mitochondrial cytochrome oxidase I (mtCO I) gene [37]. Samples of eggs, four nymph stages, females, and males were collected from the B. tabaci B population and were rapidly frozen in liquid nitrogen and transferred at -80 °C. All B. tabaci B samples were represented by three biological replicates that were processed independently.
De novo identification of AATs
The AATs were identified using HMMER (v.3.01). In brief, HMMER (v.3.01) was used to search the B. tabaci B genome 1.0 predicted protein data set (unpublished, FTP: http://111.203.21.119/download/B.gene.v3.cds.fa) for proteins containing conserved functional domain defined by Pfam HMM profile AA_permease, PF00324 for APC transporters [19, 38-40] and Aa_trans, PF01490 for AAAP transporters [19, 40-41]. The D. melanogaster and A. pisum AATs were used in tblastn searches against B. tabaci B 1.0 genomic scaffolds to make sure that all the AATs had been identified in the B. tabaci B genome. Putative AAT genes were manually verified by comparison with assembly ESTs [42]. A total of 14 APC family AATs and 25 AAAP family AATs genes were identified in the B. tabaci B genome (shown in Table S1). In addition, the predicted protein sequences from the whole genomes of seven other insect species were searched for APCs and AAAPs based on the methods described above for B. tabaci B. The seven other insects were Aedes aegypti, Apis mellifera, Acyrthosiphon pisum, Drosophila melanogaster, Bombyx mori, Manduca sexta, and Nasonia vitripennis (Table 1).
Insect species used for phylogenetic analyses and number of AATs for each species.
| Species | Order | AAAP | APC | Total |
|---|---|---|---|---|
| Bemisia tabaci | Hemiptera | 25 | 14 | 39 |
| Acyrthosiphon pisum | Hemiptera | 23 | 21 | 44 |
| Aedes aegypti | Diptera | 19 | 14 | 33 |
| Drosophila melanogaster | Diptera | 15 | 10 | 25 |
| Apis mellifera | Hymenoptera | 16 | 10 | 26 |
| Nasonia vitripennis | Hymenoptera | 14 | 9 | 23 |
| Bombyx mori | Lepidoptera | 21 | 13 | 34 |
| Manduca sexta | Lepidoptera | 26 | 12 | 38 |
Phylogenetic analysis
Because amino acid sequences are not similar between APC and AAAP family members; the phylogenetic analysis of each family was performed separately. For the APC family, the phylogenetic analysis comprised 103 amino acid sequences in total, the amino acid sequences are shown in Table S7. A total of 14 APC family AATs were identified in the B. tabaci B genome, the CDS sequences are shown in Table S8. For the AAAP family, the phylogenetic analysis comprised 159 amino acid sequences in total, the amino acid sequences are shown in Table S9. A total of 25 AAAP family AATs were identified in the B. tabaci B genome, the CDS sequences are shown in Table S10. Full-length amino acid sequences were aligned with MUSCLE using MEGA 6 (http://www.megasoftware.net/). Phylogenies were estimated using the Maximum Likelihood (ML) method. And the LG with Freqs. (+F) model with a gamma distribution (the best-fit model was determined by MEGA 6, the best-fit model of APC family and AAAP family are the same) were used for phylogenetic analysis with 1000 bootstraps and 95% site coverage cutoff. Apart from B. tabaci B, the analysis included AATs from the seven other insects listed in the previous section.
Molecular evolution analyses
The molecular evolution rate were estimated for a subset of B. tabaci B AAAP transporters that form a B. tabaci B-specific AAAP transporter expansion. We conducted the molecular evolution analyses using the 10 B. tabaci B paralogs that form the AAAP family expansion (BtAAAP16, BtAAAP20, BtAAAP17, BtAAAP19, BtAAAP18, BtAAAP24, BtAAAP15, BtAAAP13, BtAAAP14, and BtAAAP21); insect AAAP orthologs from M. sexta (MsAAAP18, MSEX2.10425-RA), B. mori (BmAAAP16, XP_012544833.1), N. vitripennis (NvAAAP3, NV11499), A. mellifera (AmAAAP16, XP_624573.2), A. aegypti (AaAAAP12, AAEL007191-PA and AaAAAP17, AAEL008913-PA), and D. melanogaster (DmAAAP5, FBPP0075931); and an outgroup sequence from the sister clade of the expansion (BtAAAP4). The amino acid sequences of the 18 AAAP transporters were aligned with MUSCLE and used to construct the phylogenetic trees by MEGA 6. The methods for phylogenetic analysis were described above, and the phylogenetic tree was used as input for the molecular evolution analyses.
The full-length CDS were aligned in MAFFT and were used to estimate non-synonymous (dN)/synonymous (dS) (dN/dS, also denoted ω). dN/dS < 1 demonstrates purifying selection; dN/dS = 1 indicates neutral selection and dN/dS > 1 demonstrates positive selection [12-13].
Branch-based model of molecular evolution was used to detect lineages under accelerated rates of evolution, and site-based model was used to detect specific sites under positive selection. The ω were calculated in the B. tabaci B-specific AAAP transporter expansion by ML in the PAML package for both models [43]. And the molecular evolution analyses of both models were conducted as described previously [12]. In brief, for branch-based models of molecular evolution, one or more ω (model ω = 0 or 2, NSsites = 0) were performed. The parameters ω and κ were calculated starting from 0.2 and 2, respectively. The codon frequency was determined utilizing a 3 × 4 codon table. Various models were used to test the ω along specific branches, and then likelihood ratio test (LRT) were used to compared log likelihoods of different models [44, 13].
For site-based models of molecular evolution [45-46], the B. tabaci B-specific AAAP genes, including other insect orthologs but excluding outgroup sequence were used to analyze the selection at specific sites. Three pairs of models were implemented to test whether candidate sites had experienced positive selection [12]. The models were then compared using an LRT. The first model pair compared M0 with model M3 for testing site-based positive selection [12]. The second and third model pairs, M1a/M2a [47-48] and M7/M8, were used to test site-based positive selection. Model M1a (nearly neutral) constrains 0 < ω0 < 1 (purifying selection) and ω1=1 (neutral selection); model M2a (positive selection) allows for ω2 > 1; model M7 (beta) assumes a beta distribution for 0 < ω < 1; and model M8 (positive selection) allows for ω > 1. A significantly higher likelihood resulting from M2a and M8 demonstrates that the sites undergo positive selection.
RNA isolation and cDNA synthesis
Total RNA was extracted from eggs, nymphs (first, second, third, and fourth instars), and newly emerged (2-day-old) adults of B. tabaci B using TRIzol® reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer's recommendations. The RNA was quantified with spectrophotometer (NanoDrop 2000c, Thermo Fisher Scientific Inc., Waltham, MA, USA), and RNA integrity was determined by 1% TBE agarose gel electrophoresis. The first-strand cDNA was synthesized using the PrimeScript™ II 1st strand cDNA synthesis kit (TaKaRa, Dalian, China) for gene fragments cloning, and PrimeScript RT kit (containing gDNA Eraser, Perfect Real Time) (TaKaRa, Dalian, China) for RT-qPCR analysis. The synthesized cDNA was stored at -20 °C until used.
Gene fragments cloning
To clone B. tabaci B AAT gene fragments, we corrected the full-length sequences of B. tabaci B AAT genes based on their putative CDS in the B. tabaci B genome 1.0 and based on the partial cDNA sequences in our B. tabaci B transcriptome databases [42-49]. Their cDNA fragments were then amplified by designing 400- to 600-bp primer pairs which were designed with Primer Premier 5.0 (Table S2). The reactions (total 25 μl) contained 15.25 μl of ddH2O, 4 μl of dNTP Mix, 2.5 μl of 10×LA Taq buffer, 1 μl of the cDNA template, 1 μl of each specific primer, and 0.25 μl LA Taq polymerase (TaKaRa). The PCR reactions were then performed using a S1000 PCR system (BioRad). The parameters as follows: one cycle of 94°C for 5 min (initial denaturation); 35 cycles of 94°C for 30 s (denaturation), 50-59°C for 1 min (annealing), and 72°C for 1 min (extension); and one cycle of 72°C for 10 min (final extension). The PCR products were separated on 2% agarose gels and visualized with SYBR Safe (Life Technologies) (Figure S1-2).
Reverse transcriptase-quantitative PCR (RT-qPCR) analysis
Gene-specific primers for the AATs of B. tabaci B were designed with Primer Premier 5.0 and used in RT-qPCR analysis. The PCR reactions (total 25 μl) contained 12.5 μl of 2×SuperReal PreMix Plus (SYBR Green) (TIANGEN, Beijing, China), 9.5 μl of ddH2O, 1 μl of cDNA template, 0.75 μl of each specific primer, and 0.5 μl of 50×ROX Reference Dye (TIANGEN, Beijing, China). The RT-qPCR were conducted with an ABI 7500 system (Applied Biosystems) with the following parameters: one cycles of 95°C for 3 min; 40 cycles of 95°C for 15 s, 60°C for 30 s, 72°C for 30 s. Five 2-fold serial dilutions of whitefly cDNA template were used to determine the RT-qPCR primers amplification efficiencies by dissociation curve analysis [50]. Only primers with 90-110% amplification efficiencies were used for the subsequent researches.
Relative quantification was calculated by the 2-ΔΔCt method [51], and data were normalized to EF1-α (GenBank: EE600682) reference gene [52]. Three independent biological replicates and four technical replicates were used for each sample. Significant differences between samples were determined by one-way ANOVA with Holm-Sidak's tests (overall significance level = 0.05).
RNA-seq analysis
RNA-seq samples of different developmental stages of B. tabaci B were prepared as previously described [42-49] and were sequenced on a HiSeq 2500 system according to the manufacturer's instructions with sequenced at 125 bp (PE125, library size is 280-320 bp). RNA-seq libraries from B. tabaci B adults feeding on different host plants were obtained from our previous report [49]. The transcriptome datasets were filtered using Trimmomatic with default parameters [53], and then the Tophat software was used to map the clean data to B. tabaci B genome 1.0 [54]. Cufflinks was used for estimating the expression level (the FPKM value) of each predicted transcript [55]. The results were shown in Table S3-S6.
dsRNA synthesis and RNAi assays
The effects on the mortality of B. tabaci B adults were determined for two AAAP genes (BtAAAP15 and BtAAAP21), which were selected because they have whitefly-specific expansion and are highly expressed in nymphs and adults of B. tabaci B. Specific dsRNA primers of AAAP15, AAAP21, or EGFP (GenBank: KC896843) were designed using Primer Premier 5.0 with a T7 promoter at the 5' end, and the sequences of these primers were listed in Table S2. The T7 Ribomax™ Express RNAiSystem (Promega, Madison, WI, USA) was used for synthesizing dsRNA of AAAP15, AAAP21, and EGFP.
AAT gene expression was silenced by directly feeding dsRNA to B. tabaci B adults in feeding chambers. A 0.2-ml drop of diet solution [30% sucrose and 5% yeast extract (wt/vol)] with dsRNA was placed in the chamber [56]. After about 60 newly emerged B. tabaci B adults (mixed sexes) were introduced, the feeding chambers were placed in an environmental at 25 °C with an L14:D10 photoperiod and 80% RH. Each sample was represented by three biological replicates. Relative expression was assessed after 2 days of feeding. Mortality was assessed after 2 or 7 days of feeding.
Results
Identification of APC and AAAP transporter genes in the genome of B. tabaci B
Predicted APC and AAAP transporters in B. tabaci B were identified by searching for Pfam HMM AA_permease (PF00324) and Aa_trans (PF01490), respectively. We compared the genomic scaffold sequences, intron boundary predictions, and transcriptome sequences manually to confirm that the predicted protein sequences were accurate. A total of 39 putative APC superfamily AATs were identified, and these included 14 APC transporters and 25 AAAP transporters (Table S1). The encoded proteins that contained the PF00324 conserved sequence motif and that showed high levels of similarity to amino acid/polyamine/organocation in other insects were identified as APC transporters, while encoded proteins that contained the PF01490 conserved sequence motif and that showed high levels of similarity to amino acid/auxin permease in other insects were identified as AAAP transporters. To confirm that the AAAT genes are truly present in B. tabaci B, 400- to 600-bp cDNA fragments were cloned from different developmental stages of B. tabaci B, and the results are shown in Figure S1-S2.
Characterization of the B. tabaci B AATs
Most B. tabaci B AAT genes have a multi-exon structure with 7-13 exons, except that BtAAAP23 and BtAAAP24 have 2 and 6 exons, respectively. The lengths of the encoded proteins of APC transporters range from 486 to 792 amino acids, while those of most AAAP transporters range from 442 to 561 amino acids, except BtAAAP5, which encodes a 1081-amino acid protein. The APC family proteins contain 12-14 TMSs, and the AAAP family proteins contain 10-11 TMSs (Table S1).
Phylogenetic analysis of insect AATs
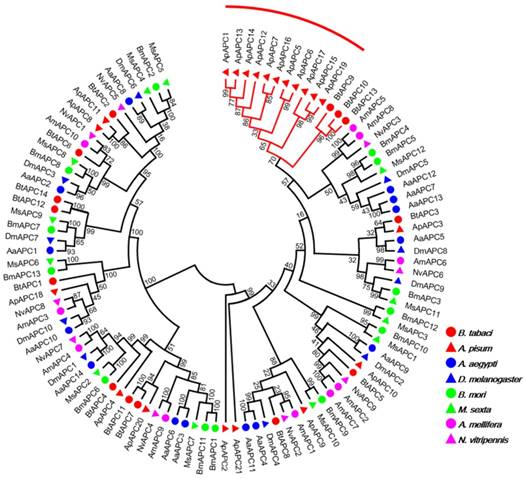
Our investigation of AATs genes from a selection of eight insects with completely sequenced genomes indicated that B. tabaci B contains more AAT genes than six of the species but fewer than A. pisum (Table 1). The results also indicated that B. tabaci B contains more AAAP family genes than six of the species but fewer than M. sexta. To survey phylogenetic relationships between predicted APC transporters in insects, the sequences containing the PF00324 motif were subjected to a MUSLE sequence comparison by the Maximum Likelihood inference method in order to produce a Maximum Likelihood tree (Figure 1). To survey phylogenetic relationships between predicted AAAP transporters in insects, sequences containing the PF01490 motif were selected, and a Maximum Likelihood tree was generated (Figure 2).
Phylogenetic tree of APC transporters in B. tabaci B. The analysis involved 103 amino acid sequences. The phylogenetic analysis was conducted in MEGA6. Bootstrap values were shown at branch points. Hemiptera (Ap, Acyrthosiphon pisum; Bt, Bemisia tabaci); Hymenoptera (Am, Apis mellifera; Nv, Nasonia vitripennis); Diptera (Aa, Aedes aegypti; Dm, Drosophila melanogaster); Lepidoptera (Bm, Bombyx mori; Ms, Manduca sexta). The red branch represents the A. pisum species-specific expansion of APC transporters.
Phylogenetic tree of AAAP transporters in B. tabaci B. The analysis involved 159 amino acid sequences. The phylogenetic analysis was conducted in MEGA6. Bootstrap values were shown at branch points. Hemiptera (Ap, Acyrthosiphon pisum; Bt, Bemisia tabaci); Hymenoptera (Am, Apis mellifera; Nv, Nasonia vitripennis); Diptera (Aa, Aedes aegypti; Dm, Drosophila melanogaster); Lepidoptera (Bm, Bombyx mori; Ms, Manduca sexta). The red branch represents B. tabaci B species-specific expansion of AAAP transporters. The blue branch represents A. aegypti species-specific expansion of AAAP transporters. The green branch represents Lepidoptera species-specific expansion of AAAP transporters.
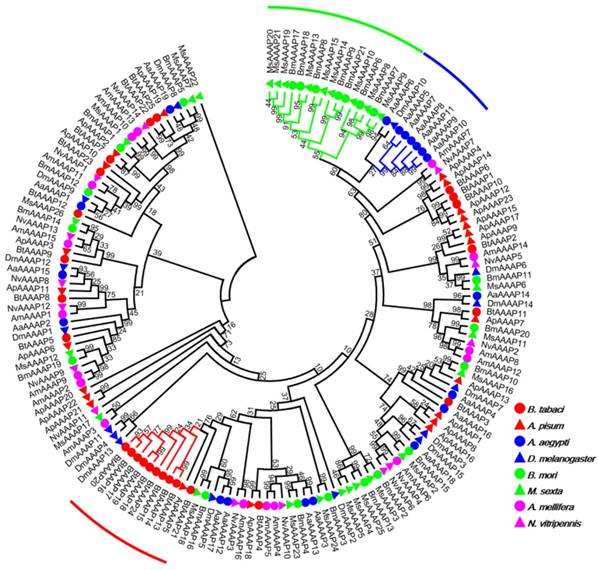
The AAT genes of B. tabaci B grouped together with those of A. pisum. There was an 11 paralog aphid-specific expansion of APC transporters. The analysis of the AAAP transporter family indicated a 10 paralog whitefly-specific expansion, a 7 paralog A. aegypti -specific expansion, a 16 paralog Lepidoptera -specific expansion. The whitefly-specific expansion of AAT genes including BtAAAP13, BtAAAP14, BtAAAP15, BtAAAP16, BtAAAP17, BtAAAP18, BtAAAP19, BtAAAP20, BtAAAP21, and BtAAAP24 (Figure 2).
Genomic distribution of the B. tabaci B AATs
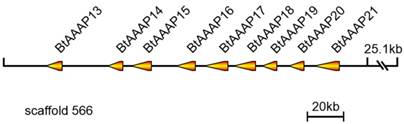
A total of 39 B. tabaci B AAT genes were located on 27 scaffolds. Among them, scaffolds 5, 202, 1270, and 1576 each contained two AATs. A total of nine members of the B. tabaci B AAAP family expansion (BtAAAP13, BtAAAP14, BtAAAP15, BtAAAP16, BtAAAP17, BtAAAP18, BtAAAP19, BtAAAP20 and BtAAAP21) shared a similar genomic location and mapped to a 167-kbp region of genomic scaffold 566 (Figure 3).
Location of B. tabaci B-specific expansion AAAP transporter genes on scaffold 556. The orange arrows indicate transcription orientations of B. tabaci B-specific expansion AAAP transporter genes on the scaffold. Nine B. tabaci B-specific expansion AAAP transporter genes were located on scaffold 556 and had the same orientation.
Expression profiling of B. tabaci B AAT genes
To determine when AATs are expressed during B. tabaci B development, RNA was extracted and sequenced from eggs, nymphs (1st, 2nd, 3th, and 4th instar), and adults). The sequenced fragments obtained from the individual samples were then mapped to the B. tabaci B genome. Figure 4 and Figure 5 show the log2-transformed expression values (FPKM) for the 14 APC transporters and 25 AAAP transporters that were identified in the B. tabaci B genome. Hierarchical clustering was performed by the centroid linkage method with Gene Cluster 3.0. The developmental stage clusters differed substantially between APC and AAAP families. For the APC family, the nymph AATs clustered together. The nymph AATs were more similar to those of males than females. The AATs of eggs clustered separately from those of all other samples. For the AAAP family, the nymph AATs clustered with those of males, 3th- and 4th- instar nymphs were clustered together, while AATs of 1st- and 2nd-instar nymphs and males clustered together; the AATs of these stages then clustered with those of females. Finally, the AAAP of eggs clustered separately from those of all other stages. Some AATs had similar transcription profiles across developmental stages, while other AATs showed stage-specific expression patterns.
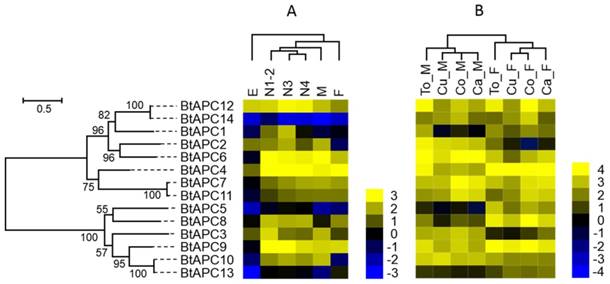
Gene expression of APC transporters as affected by B. tabaci B stage (A) and host plant (B). The phylogenetic tree on the left reflects the phylogenetic relationships of APC transporters in B. tabaci B. The genes correspond to those in Figure 1. A total of 14 amino acid sequences were used for phylogenetic analyses. The phylogenetic analysis was conducted in MEGA6. Bootstrap values were shown at branch points. Values are based on log2-transformed FPKM values relative to the median intensity of all contigs (yellow represents higher expression values; blue represents lower expression values). E, Egg; N1-2, 1st- and 2nd-instar nymphs; N3, 3rd-instar nymph; N4, 4th-instar nymph; M, Male; F, Female; Ca_F, Female B. tabaci B on cabbage; Ca_M, Male B. tabaci B on cabbage; Cu_F, Female B. tabaci B on cucumber; Cu_M, Male B. tabaci B on cucumber; Co_F, Female B. tabaci B on cotton; Co_M, Male B. tabaci B on cotton. To_F, Female B. tabaci B on tomato; To_M, Male B. tabaci B on tomato.
Gene expression of AAAP transporters as affected by B. tabaci B stage (A) and host plant (B). The phylogenetic tree on the left reflects the phylogenetic relationships of AAAP transporters in B. tabaci B. The genes correspond to those in Figure 2. A total of 25 amino acid sequences were used for phylogenetic analyses. The phylogenetic analysis was conducted in MEGA6. Bootstrap values were shown at branch points. Values are based on log2-transformed FPKM values relative to the median intensity of all contigs (yellow represents higher expression values; blue represents lower expression values). E, Egg; N1-2, 1st- and 2nd-instar nymphs; N3, 3rd-instar nymph; N4, 4th-instar nymph; M, Male; F, Female; Ca_F, Female B. tabaci B on cabbage; Ca_M, Male B. tabaci B on cabbage; Cu_F, Female B. tabaci B on cucumber; Cu_M, Male B. tabaci B on cucumber; Co_F, Female B. tabaci B on cotton; Co_M, Male B. tabaci B on cotton. To_F, Female B. tabaci B on tomato; To_M, Male B. tabaci B on tomato.
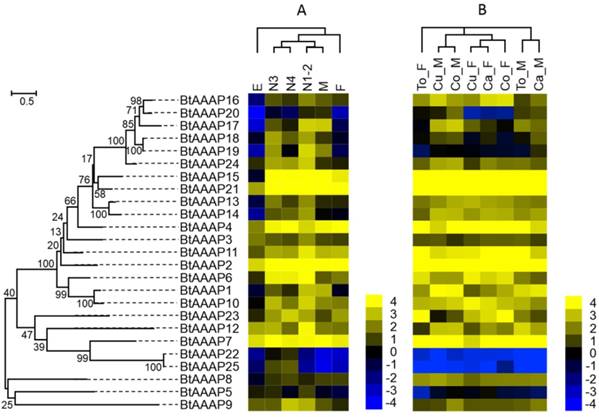
The studies of APC family showed that the BtAPC4 and BtAPC12 were most highly expressed throughout all of the developmental stages, whereas the BtAPC5, BtAPC13, and BtAPC14 were lowly expressed. BtAPC6 and BtAPC9 were down-regulated in eggs but highly expressed in other stages. BtAPC3 expression was highest in 1st- and 2nd-instar nymphs followed by male adults. The expression of BtAPC8 was significantly higher in nymphs and female adults than in other stages. Most APC transporters were highly expressed in 3rd-instar nymphs. In the AAAP family, expression was highest for BtAAAP2 and BtAAAP7 and was lowest for BtAAAP20 and BtAAAP22 across all stages. BtAAAP4, BtAAAP15, and BtAAAP21 were down-regulated in eggs but were highly expressed in other stages. Like BtAPC3, BtAAAP17 was highly expressed in 1st- and 2nd-instar nymphs and male adults. The expression of BtAAAP6 was significantly higher in nymphs than in other stages. Most AAAP transporters were highly expressed in 1st- and 2nd-instar nymphs.
Using previously published RNAseq data for sex and host plant effects on transcriptomes, we compared the expression of AATs in the B. tabaci B [49]. For the APC family, the AATs of males and females clustered together, respectively. (Figure 4 and Figure 5). For the AAAP family, the AATs clustered together for male adults on cucumber and cotton, for cabbage and tomato; and for female adults on cucumber, cabbage, and cotton.
Some APC transporters were expressed differently depending on sex and host. For example, BtAPC2, BtAPC3, and BtAPC6 were up-regulated in male adults, whereas BtAPC1, BtAPC4, BtAPC5, BtAPC8, and BtAPC9 were up-regulated in female adults. BtAPC7 and BtAPC11 were highly expressed in adults on cotton and cabbage. BtAPC12 was highly expressed in adults on cotton and tomato. Expression was high for BtAAAP2, BtAAAP7, BtAAAP15, and BtAAAP21 in all samples but was low for BtAAAP5, BtAAAP19, BtAAAP22, and BtAAAP25. BtAAAP1, BtAAAP12, and BtAAAP17 were up-regulated in male adults, whereas BtAAAP6 and BtAAAP23 were up-regulated in female adults.
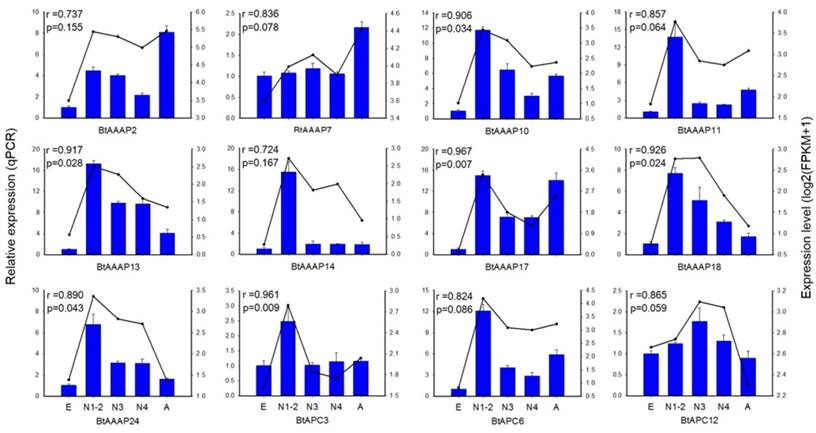
To confirm the gene expression data obtained by transcriptome sequencing, we analyzed the expressions of 12 AAT genes among stages using RT-qPCR. The 12 AAT genes included 3 APC family genes, 4 AAAP genes not belong to whitefly- specific expansion and 5 AAAP genes of whitefly-specific expansion. The expression of these genes were able to reflect the expression of AAT genes in whitefly. The results demonstrated that the FPKM expression patterns of 6 of the 12 AAT genes were highly consistent (p < 0.05) with the qPCR expression profile (Figure 6).
qPCR-based expression profiling of three APC transporter genes and nine AAAP transporter genes in stages of B. tabaci B. Total RNA was extracted from individual eggs, nymphs, and adults of B. tabaci B, and was used for the expression analysis of AAT genes by qPCR (blue bars) and RNA-seq (black lines). The relative expression levels of each AAT gene in each developmental stage were normalized to the references gene EF-1α. The transcript levels of B. tabaci B AAT genes in different development stages were determined by calculating fragments per kilobase of exon model per million fragments mapped (FPKM) values, and results were represented by log2(FPKM+1). E, Egg; N1-2, 1st- and 2nd-instar nymphs; N3, 3rd-instar nymph; N4, 4th-instar nymph; A, Adult.
Molecular evolutionary insights into the evolution of AAAP transporter expansion in B. tabaci B
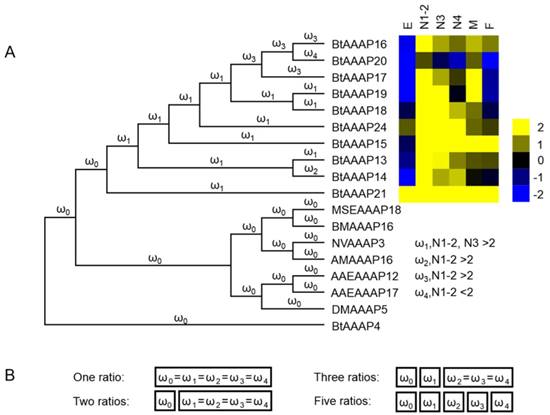
We used AAAP expression profiles of different stages to determine whether branches resting in stage-based paralogs experienced accelerated evolution rates. B. tabaci B-specific AAAP expansion branch models were constructed based on nucleotide coding sequences, and these sequences were used to test the (dN/dS = ω).
The one ratio model set the same ω for all branches of the phylogenetic tree. The two ratios model set different ω for branches between B. tabaci B-specific AAAP expansion and other genes. The three ratios model set an another ω for branches of AAAP expansion based on expression profiles for different stages, and the five ratios model assumed a distinct ω for each branch based on AAAP expression profiles of different stages (log2-transformed FPKM values of 1st- and 2nd-instar nymphs > 2, or log2-transformed FPKM values of 3th-instar nymphs > 2) (Figure 7).
The branch-based models and LRTs results were shown in Table 2. The likelihood of two ratios model did not have a significantly difference compared with one ratio model. The likelihood also did not have significantly improvement between three ratios model and two ratios model, however likelihood was significantly higher for the five ratios model than the two ratios model. Based on the five ratios model, the branches leading to stage-based clades (BtAAAP14) had a lower ω value (0.085721) while BtAAAP16, BtAAAP17 and BtAAAP20 had greater ω values (0.18084 0.103435) than the average branch (ω = 0.087181) within the expansion.
Result of branch-based molecular evolution analysis of B. tabaci B AAAPs. Different ω categories are shown at branch in Fig.7.
| Models | ω Category | ω Parameters for Branches | -InL | CMa |
|---|---|---|---|---|
| One ratio | 1 | ω0=0.10838 | 23387.77905 | - |
| ω1=ω0 | ||||
| ω2=ω0 | ||||
| ω3=ω0 | ||||
| ω4=ω0 | ||||
| Two ratio | 1 | ω0=0.0994538 | 23386.32493 | One ratio |
| 2 | ω1=0.116919 | |||
| ω2=ω1 | ||||
| ω3=ω1 | ||||
| ω4=ω1 | ||||
| Three ratio | 1 | ω0=0.099498 | 23382.97256 | Two ratio |
| 2 | ω1=0.0846088 | |||
| 3 | ω2=0.143378 | |||
| ω3=ω2 | ||||
| ω4=ω2 | ||||
| Five ratio | 1 | ω0=0.0996 | 23379.98694 | Two ratio* |
| 2 | ω1=0.087181 | |||
| 3 | ω2=0.085721 | |||
| 4 | ω3=0.18084 | |||
| 5 | ω4=0.103435 |
aCompared model, i.e., the model that was used for comparison via a likelihood ratio test (e.g., one ratio and two ratios, two ratios and three ratios, and two ratios and five ratios). * indicates P < 0.05.
Molecular evolution models of B. tabaci B -specific AAAP expansion. Phylogenetic analysis for B. tabaci B -specific AAAP family expansion is shown in (A), and each branch of the phylogeny tree is assigned a ω category as indicated. The expression profiles of BtAAAP genes in different stages of B. tabaci B were obtained using RNA-seq; values are based on log2-transformed FPKM values relative to the median intensity of all contigs (yellow represents higher expression values; blue represents lower expression values. E, Egg; N1-2, 1st- and 2nd-instar nymphs; N3, 3rd-instar nymph; N4, 4th-instar nymph; M, Male; F, Female. The models tested are shown in (B). The model names indicate the number of ω categories assumed (e.g., one ratio assumes equal ω across all branches, while two ratios assume two ω across all branches).
We were interested to determine whether the accelerated evolution rates of BtAAAP16, BtAAAP17 and BtAAAP20 resulted from positive selection or relaxed selection. For this reason, site-based analyses were conducted to identify the specific codon sites that might undergo positive selection [45-46]. The results of the first test (shown in Table 3, M0 vs. M3) demonstrated that ω varies across sites. Two other models (M1a vs. M2a, and M7 vs. M8) were used to test the positively selected sites, the results did not show a significantly improved likelihood (Table 3). The results suggested that relaxed selection rather than positive selection explains the elevated ω values observed along some branches (Table 3).
Results of site-based molecular evolution analysis of B. tabaci B AAAPs. Models are grouped and numbered. Group 1 is used to detect variable ω among sites; groups 2 and 3 are used to detect positive selection.
| Nested Models | Parameters estimated | -InL | |
|---|---|---|---|
| 1 | M0:Oneωamong sites | ω=0.06568 | 18430.96981 |
| M3: Variable ω among sites | p0 =0.33815, p1=0.45409 (p2=0.20776) | 18018.503214* | |
| ω0=0.01425, ω1=0.06931 , ω2=0.19404 | |||
| 2 | M1a:Nearly neutral | p0=0.9060 (p1=0.09396) | 18332.01618 |
| ω0=0.09299 (ω1=1.00000) | |||
| M2a:Postitive selection | p0 =0.90604, p1=0.09396 (p2=0.00000) | 18332.01618 | |
| ω0=0.09299 (ω1=1.00000), ω2=3.88083 | |||
| 3 | M7:Beta | p=0.99519, q=11.52044 | 18012.81774 |
| M8:Beta, ω>1 | p0=0.99999 (p1=0.00001) | 18012.81954 | |
| p=0.99522, q=11.52108, ω=1.00000 | |||
* indicates P < 0.05.
RNAi of the AAAP family genes in B. tabaci B adults
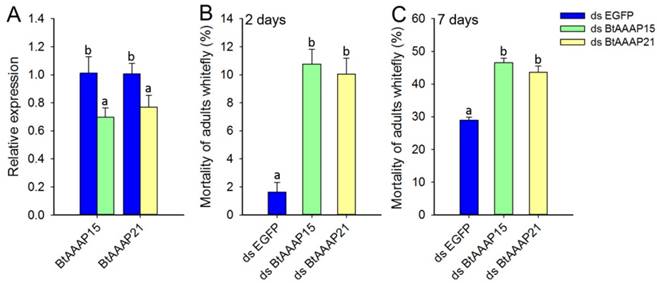
To investigate the effects of whitefly-specific expansion of AAAP genes on B. tabaci B, we used in vivo RNAi knockdown of BtAAAP15 and BtAAAP21 genes by feeding dsRNA of AAAP family genes to adults. Specific suppression of AAAP genes expression were confirmed by qPCR after the adults had fed for 48 h on dsRNA (Figure 8A). The qPCR results revealed that feeding on the dsRNA of AAAP genes significantly reduced transcript levels of BtAAAP15 and BtAAAP21 by about 30% and 23%, respectively, relative to control (P < 0.05; Holm-Sidak's test; n=3). The 2 days mortality and 7 days mortality of B. tabaci B adults fed with dsRNA of AAAP family genes significantly higher than that fed with dsRNA of EGFP (P < 0.05; Holm-Sidak's test; n=3) (Figure 8B and Figure 8C).
Discussion
In this study, our analysis of the B. tabaci B genomic sequence has identified a series of genes encoding putative AATs of the APC superfamily. These AAT genes were validated by comparison with the corresponding transcriptome of B. tabaci B. The proteins in the APC superfamily were predicted by searching Pfam for PF00324 in the case of APC family transporters and for PF01490 in the case of AAAP family transporters. A total of 14 putative APCs and 25 putative AAAPs were identified in the B. tabaci B genome by bioinformatics analysis. Expression profiling of APC and AAAP genes as affected by B. tabaci B stage, sex, and host plant revealed diverse expression patterns. The RNAi-mediated knockdown of two AAAP genes (BtAAAP15 and BtAAAP21) significantly increased the mortality of B. tabaci B adults.
Silencing of AATs genes expression and their effect on B. tabaci B. (A) Effect of feeding of B. tabaci B adults with dsEGFP, dsAAAP15, or dsAAAP21 on the relative expression of AAT genes at 48 h post-RNAi. The mRNA levels are shown as a ratio relative to the levels of the reference gene EF-1α. (B and C) Mortality of B. tabaci B adults after feeding on dsEGFP, dsAAAP15, or dsAAAP21 for (B) 2 days or (C) 7 days. Within each panel, different letters indicate significant differences between treatments (P < 0.05; Holm-Sidak's test; n = 3).
In our phylogenetic analysis of APC family genes in insects, BtAPC3 clustered with AaAPC5 (AAEL009362), which plays a key role in nutritional signaling during mosquito reproduction [10]. BtAPC12 and BtAPC14 clustered with DmAPC7 (FBPP0088598), which helps regulate larval crawling behavior in Drosophila [57]. For AAAP family genes, BtAAAP2 clustered with DmAAAP6 (CG3424), and BtAAAP11 clustered with ApAAAP7 (ACYPI001018). DmAAAP6 (CG3424) play an important role in maintaining growth maintain growth during Drosophila development as local nutrient levels fluctuate [9]. ApAAAP7 (ACYPI001018) can regulate the supply of glutamine to intracellular bacterial symbionts in aphids [15]. Our phylogenetic analysis of insects also demonstrated that the B. tabaci B APC superfamily genes cluster with those in A. pisum. There is a 10 paralog whitefly-specific expansion in the AAAP transporter family, and this expansion is grouped with A. pisum ApAAAP5 (ACYP1000550). An 11 paralog aphid-specific expansion of the APC transporter family was detected. Our phylogenetic analysis also detected the specific expansion of the AAAP family in A. aegypti and in Lepidopterans. The A. aegypti expansion of the AAAP family is grouped with the Lepidoptera-specific expansion. Previous phylogenetic analysis based on 20 genes of 13 species suggested that the Lepidoptera are sister to the Diptera [18], which is consistent with our results on the species-specific expansion of the AAAP family. Our results demonstrate that species-specific expansion of AATs is common in insects. Previously, gene annotation and comparative transcriptomics analyses of AATs in the sap-feeding suborder Sternorrhyncha demonstrated that gene duplication of AATs could mediate host/symbiont interactions [14]. Phylogenetic analysis suggested the presence of AAT expansions in sap-feeding insects, and the authors proposed that these expansions might be associated with the evolution of obligate endosymbiosis in the host insects [12, 18] Recent studies have suggested that endosymbiosis is not the only factor selecting for the retention of AAT paralogs in sap-feeding insects (Hemiptera: Sternorrhyncha) [58]. The different expansions of AATs in different insects may result from differences in feeding characteristics in general and in amino acid requirements in particular.
In insect genomes, genes in same family are commonly arranged as tandem duplicates in the same scaffold or same chromosome [59-61]. The tandem duplication is a type of duplication where the duplicated segment is contiguous with the original duplication [62]. The duplication of individual genes or chromosomal segments has long been recognized as a major source for the origin of evolutionary novelties, including new genes and gene functions [63-65]. Previous study showed that aphid-specific expansion of AAT genes exhibit tandem repeat distribution on the scaffolds [12]. In our study, a total of nine members of the whitefly AAAP family expansion are arranged on a 167-kbp region of scaffold 566, and these genes have the same orientation. Therefore, these genes probably arose by gene tandem duplication, and these genes are likely involved in whitefly-specific amino acid transport. The B. tabaci B genome contains unresolved sequence redundancy between different scaffolds, and whether other AATs are located on neighboring scaffolds remains uncertain.
Our data from different B. tabaci B stages revealed high expression of BtAPC4, BtAPC12, BtAAAP2, and BtAAAP7 and low expression of BtAPC5, BtAPC13, BtAPC14, BtAAAP20, and BtAAAP22. Given that amino acids are important to organisms and that AATs move amino acids into and out of cells, it is possible that the highly expressed AATs may play a role in fundamental life processes, while the less expressed AATs may be involved in specific processes. More AATs exhibited low levels of expression in eggs than in other stages, indicating that less amino acid transport events occur in eggs. Because nymphs and adults must uptake amino acids from hosts to meet the needs of amino acids for growth and development, it is reasonable to expect that the transcript levels of AATs are increased in these stages. The data from different plant hosts and sexes showed that most AATs had similar expression profiles among different hosts but that AAT expression profiles differed between B. tabaci B females and males. Genome sequences and RNA-Seq recently indicated that B. tabaci and bacterial symbionts could cooperate in synthesizing a variety of essential amino acids [32]. The similarity in the expression of AATs among different plant hosts might be due to the interaction between B. tabaci B and bacterial symbionts in that the transport of amino acids mainly occurs between the whitefly and the bacterial symbionts. Differences in the expression of AATs between females and males, on the other hand, may be caused by differences in the requirements for amino acids by females and males.
To test whether the accelerated evolution rates are related to the evolution of stage-based expression in the AAAP expansion, four nested branch models of molecular evolution were constructed (Figure 7). The results showed that likelihood of two ratios model was not significantly improved compared with the one ratio model, which demonstrates that the branches within the B. tabaci B AAAP expansion are not evolving at a significantly higher rate than other branches (Table 2). Likelihood was also not improved by the three ratios model but was significant improved by the five ratios model (Table 2). The results of five ratios model showed that two of the four branches of this model have evolved at a significantly higher rate than other expansion branches. The first branch (branch ω 3 in Figure 7) indicated that BtAAAP16 and BtAAAP17 and second branch (branch ω 4 in Figure 7) indicated that BtAAAP20 accelerated rates of evolution with stag-based expression in the AAAP expansion. The site-based analysis demonstrated that the elevation of some branches (Table 3) can be best explained by relaxed selective constraints. Previous studies showed that Drosophila and aphid slimfast undergo purifying selection [8, 12-13]. Our molecular evolution analyses show that B. tabaci B AAAP expansion undergoes purifying selection, as indicated by values < 1 for all ω.
AATs can transport amino acids into and out of cells and can have multiple roles in insects [9-12]. Knockdown of AATs affect the oviposition amount of A. aegypti [10, 22]. Knockdown of LdNAT1 (nutrient amino acid transporter gene) in L. decemlineata retards larval growth and impairs pupation [16]. In the current study, the whitefly-specific expansion genes BtAAAP15 and BtAAAP21 were highly expressed in B. tabaci B nymphs and adults, suggesting that these genes may be important in these stages of B. tabaci B. In addition, expression levels of these genes were relatively similar when B. tabaci B fed on different host plants, i.e., these genes are evidently unaffected by host plant. Our RNAi experiment suggested that knockdown of BtAAAP15 and BtAAAP21 in B. tabaci B significantly increased the mortality of B. tabaci B adults. Therefore, whitefly-specific expansion of AAAP genes may be important to the development and survival of B. tabaci B.
The AATs play important roles in many life processes in insects. Previous studies showed that the AAT (ApGLNT1) regulated the supplement of glutamine to the bacterial symbionts in Aphid [15]. The AaSlif gene has been identified as Na+ dependent amino transporter in A. aegypti and could transport essential amino acids during reproduction [21]. Not all the essential amino acid could be synthesized in the B. tabaci, although the coevolution of symbiosis and whitefly could improve the ability to synthesis of essential amino acids [32]. The essential amino acids were acquired by feeding phloem sap or provided by symbiotic in B. tabaci. The AATs involved in the amino acid transport processes in B. tabaci. Our annotation of AAT genes in B. tabaci provided fundamental material for these researches.
In conclusion, our study is the first genome-wide analysis of APCs and AAAPs in B. tabaci B. We also determined how the expression of these APCs and AAAPs is affected by life stage and host plant. Finally, RNAi knockdown of BtAAAP15 and BtAAAP21 demonstrated that expression of these genes is required by B. tabaci B adults. These results will facilitate future research on the physiological functions of AATs and may also suggest new ways to manage B. tabaci populations.
Supplementary Material
Supplementary figures and tables.
Abbreviations
AAT: amino acid transporter; APC: amino acid/polyamine/organocation; AAAP: amino acid/auxin permease; TMS: transmembrane domains; mtCOI: mitochondria cytochrome oxidase one; LRT: likelihood ratio test; FPKM: fragments per kilobase of exon model per million fragments mapped; RT-qPCR: Reverse transcriptase-quantitative polymerase chain reaction (RT-qPCR) analysis; RNAi: RNA interference.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (31420103919 and 31672032), the China Agriculture Research System (CARS-26 10), the Beijing Training Project for the leading talents in S & T (LJRC201412), and the Beijing Key Laboratory for Pest Control and Sustainable Cultivation of Vegetables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Curis E, Crenn P, Cynober L. Citrulline and the gut. Curr Opin Clin Nutr Metab Care. 2007;10:620-6
2. Manna P, Sinha M, Sil PC. Taurine plays a beneficial role against cadmium-induced oxidative renal dysfunction. Amino Acids. 2009;36:417-28
3. Wu G. Amino acids: metabolism, functions, and nutrition. Amino Acids. 2009;37:1-17
4. Wu G, Bazer FW, Dai Z. et al. Amino acid nutrition in animals: protein synthesis and beyond. Annu Rev Anim Biosci. 2014;2:387-417
5. Castagna M, Shayakul C, Trotti D. et al. Molecular characteristics of mammalian and insect amino acid transporters: implications for amino acid homeostasis. J Exp Biol. 1997;200:269-86
6. Hansen A, Moran NA. Aphid genome expression reveals host-symbiont cooperation in the production of amino acids. Proc Natl Acad Sci USA. 2011;108:2849-54
7. Boudko DY. Molecular basis of essential amino acid transport from studies of insect nutrient amino acid transporters of the SLC6 family (NAT-SLC6). J Insect Physiol. 2012;58:433-49
8. Colombani J, Raisin S, Pantalacci S. et al. A nutrient sensor mechanism controls Drosophila growth. Cell. 2003;114:739-49
9. Goberdhan DCI, Meredith D, Boyd CAR. et al. PAT-related amino acid transporters regulate growth via a novel mechanism that does not require bulk transport of amino acids. Development. 2005;132:2365-75
10. Attardo GM, Hansen IA, Shiao SH. et al. Identification of two cationic amino acid transporters required for nutritional signaling during mosquito reproduction. J Exp Biol. 2006;209:3071-8
11. Evans AM, Aimanova KG, Gill SS. Characterization of a blood-meal-responsive proton-dependent amino acid transporter in the disease vector, Aedes aegypti. J Exp Biol. 2009;212:3263-71
12. Price DRG, Duncan RP, Shigenobu S. et al. Genome expansion and differential expression of amino acid transporters at the aphid/Buchnera symbiotic interface. Mol Biol Evol. 2011;28:3113-26
13. Duncan RP, Nathanson L, Wilson ACC. Novel male-biased expression in paralogs of the aphid slimfast nutrient amino acid transporter expansion. BMC Evol Biol. 2011;11:253
14. Duncan RP, Husnik F, Van Leuven JT. et al. Dynamic recruitment of amino acid transporters to the insect-symbiont interface. Mol Ecol. 2014;32:1608-23
15. Price DRG, Feng HL, Baker JD. et al. Aphid amino acid transporter regulates glutamine supply to intracellular bacterial symbionts. Proc Natl Acad Sci USA. 2014;111:320-5
16. Fu KY, Guo WC, Ahmat T. et al. Knockdown of a nutrient amino acid transporter gene LdNAT1 reduces free neutral amino acid contents and impairs Leptinotarsa decemlineata pupation. Sci Rep. 2015;5:18124
17. Price DRG, Wilson ACC, Luetje CW. Proton-dependent glutamine uptake by aphid bacteriocyte amino acid transporter ApGLNT1. BBA-Biomembranes. 2015;1848:2085-91
18. Dahan RA, Duncan RP, Wilson ACC. et al. Amino acid transporter expansions associated with the evolution of obligate endosymbiosis in sap-feeding insects (Hemiptera: sternorrhyncha). BMC Evol Biol. 2015;15:52
19. Saier MH. Families of transmembrane transporters selective for amino acids and their derivatives. Microbiology. 2000;146:1775-95
20. Wieczorek H, Putzenlechner M, Zeiske W. et al. A vacuolar-type proton pump energizes K+/H+ antiport in an animal plasma membrane. J Biol Chem. 1991;266:15340-7
21. Boudko DY, Tsujimoto H, Rodriguez SD. et al. Substrate specificity and transport mechanism of amino-acid transceptor Slimfast from Aedes aegypti. Nat Commun. 2015;6:8546
22. Carpenter VK, Drake LL, Aguirre SE. et al. SLC7 amino acid transporters of the yellow fever mosquito Aedes aegypti and their role in fat body TOR signaling and reproduction. J Insect Physiol. 2012;58:513-22
23. Ito K, Kidokoro K, Sezutsu H. et al. Deletion of a gene encoding an amino acid transporter in the midgut membrane causes resistance to a Bombyx parvo-like virus. Proc Natl Acad Sci USA. 2008;105:7523-7
24. Kiuchi T, Banno Y, Katsuma Susumu K. et al. Mutations in an amino acid transporter gene are responsible for sex-linked translucent larval skin of the silkworm, Bombyx mori. Insect Biochem Mol Biol. 2011;41:680-7
25. Ghanim M, Czosnek H. Interactions between the whitefly Bemisia tabaci and begomoviruses: biological and genomic perspectives. Czosnek H, Ghanim M. Management of Insect Pests to Agriculture. Switzerland: Springer International Publishing. 2016:181-200
26. Brown JK, Frohlich DR, Rosell RC. The sweetpotato or silverleaf whiteflies: biotypes of Bemisia tabaci or a species complex?. Annu Rev Entomol. 1995;40:511-34
27. Gilbertson RL, Batuman O, Webster CG. et al. Role of the insect supervectors Bemisia tabaci and Frankliniella occidentalis in the emergence and global spread of plant viruses. Annu Rev Virol. 2015;2:67-93
28. Douglas AE. The nutritional quality of phloem sap utilized by natural aphid populations. Ecol Entomol. 1993;18:31-8
29. Wilkinson TL, Douglas AE. Phloem amino acids and the host plant range of the polyphagous aphid, Aphis fabae. Entomol Exp Appl. 2003;106:103-13
30. Douglas AE. Phloem-sap feeding by animals: problems and solutions. J Exp Bot. 2006;57:747-54
31. Rao Q, Rollat-Farnier PA, Zhu DT. et al. Genome reduction and potential metabolic complementation of the dual endosymbionts in the whitefly Bemisia tabaci. BMC Genomics. 2015;16:226
32. Luan JB, Chen WB, Hasegawa DK. et al. Metabolic coevolution in the bacterial symbiosis of whiteflies and related plant sap-feeding insects. Genome Biol Evol. 2015;7:2635-47
33. Su Q, Xie W, Wang S. et al. The endosymbiont Hamiltonella increases the growth rate of its host Bemisia tabaci during periods of nutritional stress. PLoS ONE. 2014;9:e89002
34. Su Q, Xie W, Wang S. et al. Location of symbionts in the whitefly Bemisia tabaci affects their densities during host development and environmental stress. PLoS ONE. 2014;9:e0091802
35. Su Q, Oliver KM, Xie W. et al. The whitefly-associated facultative symbiont Hamiltonella defensa suppresses induced plant defences in tomato. Funct Ecol. 2015;29:1007-18
36. Zhao H, Ma H, Yu L. et al. Genome-wide survey and expression analysis of amino acid transporter gene family in rice (Oryza sativa L.). PLoS ONE. 2012;7:e49210
37. Chu D, Wan FH, Zhang YJ. et al. Change in the biotype composition of Bemisia tabaci in Shandong Province of China from 2005 to 2008. Environ Entomol. 2010;39:1028-36
38. Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755-63
39. Jack DL, Paulsen IT, Saier MH. The amino acid/polyamine/organocation (APC) superfamily of transporters specific for amino acids, polyamines and organocations. Microbiology. 2000;146:1797-814
40. Saier MH, Yen MR, Noto K. et al. The transporter classification database: recent advances. Nucleic Acids Res. 2009;37:D274-D278
41. Young GB, Jack DL, Smith DW. et al. The amino acid/auxin: proton symport permease family. BBA-Biomembranes. 1999;1415:306-22
42. Xie W, Guo L, Jiao X. et al. Transcriptomic dissection of sexual differences in Bemisia tabaci, an invasive agricultural pest worldwide. Sci Rep. 2014;4:4088
43. Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586-91
44. Yang Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol. 1998;15:568-73
45. Nielsen R, Yang Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148:929-36
46. Yang Z, Nielsen R, Goldman N. et al. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431-49
47. Wong WSW, Yang Z, Goldman N. et al. Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics. 2004;168:1041-51
48. Yang Z, Wong WSW, Nielsen R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005;22:1107-18
49. Xie W, Wu Q, Wang S. et al. Transcriptome analysis of host-associated differentiation in Bemisia tabaci (Hemiptera: Aleyrodidae). Front Physiol. 2014;5:487
50. Guo Z, Kang S, Chen D. et al. MAPK signaling pathway alters expression of midgut ALP and ABCC genes and causes resistance to Bacillus thuringiensis Cry1Ac toxin in diamondback moth. PLoS Genet. 2015;11:e1005124
51. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using realtime quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402-8
52. Li R, Xie W, Wu Q. et al. Reference gene selection for qRT-PCR analysis in the sweetpotato whitefly, Bemisia tabaci (Hemiptera: Aleyrodidae). PLoS ONE. 2013;8:e53006
53. Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114-20
54. Trapnell C, Pachter L, Salzberg SL. Tophat: Discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105-11
55. Trapnell C, Williams BA, Pertea G. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511-5
56. Yang X, He C, Xie W. et al. Glutathione S-transferases are involved in thiamethoxam resistance in the field whitefly Bemisia tabaci Q (Hemiptera: Aleyrodidae). Pestic Biochem Physiol. 2016;134:73-8
57. Ziegler AB, Augustin H, Clark NL. et al. The amino acid transporter JhI-21 coevolves with glutamate receptors, impacts NMJ physiology, and influences locomotor activity in Drosophila larvae. Sci Rep. 2016;6:19692
58. Duncan RP, Feng HL, Nguyen DM. et al. Gene family expansions in aphids maintained by endosymbiotic and nonsymbiotic traits. Genome Biol Evol. 2016;8:753-64
59. Liu S, Zhou S, Tian L. et al. Genome-wide identification and characterization of ATP-binding cassette transporters in the silkworm, Bombyx mori. BMC Genomics. 2011;12:491
60. Dermauw W, Osborne EJ, Clark RM. et al. A burst of ABC genes in the genome of the polyphagous spider mite Tetranychus urticae. BMC Genomics. 2013;14:317
61. Lin H, Xia X, Yu L. et al. Genome-wide identification and expression profiling of serine proteases and homologs in the diamondback moth, Plutella xylostella (L.). BMC Genomics. 2015;16:1054
62. Leister D. Tandem and segmental gene duplication and recombination in the evolution of plant disease resistance genes. Trends Genet. 2004;20:116-22
63. Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151-9
64. Kaessmann H. Origins, evolution, and phenotypic impact of new genes. Genome Res. 2010;20:1313-26
65. Hanada K, Zou C, Lehti-Shiu MD. et al. Importance of lineage-specific expansion of plant tandem duplicates in the adaptive response to environmental stimuli. Plant Physiol. 2008;148:993-1003
Author contact
![]() Corresponding author: Prof. Youjun Zhang, Department of Plant Protection, Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences, No. 12, South Street of Zhong-Guan-Cun, Haidian District, Beijing 100081, China; Tel: +86-10-82109518; Fax: +86-10-82109518; E-mail: zhangyoujuncn
Corresponding author: Prof. Youjun Zhang, Department of Plant Protection, Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences, No. 12, South Street of Zhong-Guan-Cun, Haidian District, Beijing 100081, China; Tel: +86-10-82109518; Fax: +86-10-82109518; E-mail: zhangyoujuncn
Received 2016-10-31
Accepted 2017-3-7
Published 2017-5-16