ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2017; 13(7):878-887. doi:10.7150/ijbs.18976 This issue Cite
Research Paper
Inhibition of mTOR signaling Confers Protection against Cerebral Ischemic Injury in Acute Hyperglycemic Rats
Changchun Hei1, 2*, Ping Liu3, 2*, Xiao Yang2, 4, Jianguo Niu1, P. Andy Li2 ![]()
1. Key Laboratory of Craniocerebral Diseases of Ningxia Hui Autonomous Region and Department Human Anatomy, Histology and Embryology, Ningxia Medical University, Yinchuan 750004, China;
2. Department of Pharmaceutical Sciences, Biomanufacturing Research Institute Biotechnology Enterprise (BRITE), North Carolina Central University, 1801 Fayetteville Street, Durham, NC 27707, USA;
3. Department of Endocrinology, General Hospital of Ningxia Medical University, Yinchuan 750004, China;
4. Neuroscience Center, General Hospital of Ningxia Medical University, and Key Laboratory of Craniocerebral Diseases of Ningxia Hui Autonomous Region, Yinchuan 750004, China.
* These two authors contributed equally to this work.
Abstract

Hyperglycemia is known to exacerbate neuronal death resulted from cerebral ischemia. The mechanisms are not fully understood. The mammalian target of rapamycin (mTOR) pathway regulates cell growth, division and apoptosis. Recent studies suggest that activation of mTOR may mediate ischemic brain damage. The objective of the present experiment is to explore whether mTOR mediates ischemic brain damage in acute hyperglycemic animals. Rats were subjected to 10 min of forebrain ischemia under euglycemic, hyperglycemic and rapamycin-treated hyperglycemic conditions. The rat brain samples were collected from the cortex and hippocampi after 3h and 16h of reperfusion. The results showed that hyperglycemia significantly increased neuronal death in the cortex and hippocampus and the exacerbation effect of hyperglycemia was associated with further activation of mTOR under control and/or ischemic conditions. Inhibition of mTOR with rapamycin ameliorated the damage and suppressed hyperglycemia-elevated p-MTOR, p-P70S6K and p-S6. In addition, hyperglycemia per se increased the levels of cytosolic cytochrome c and autophagy marker LC3-II, while rapamycin alleviated these alterations. It is concluded that activation of mTOR signaling may play a detrimental role in mediating the aggravating effect of hyperglycemia on cerebral ischemia.
Keywords: Cerebral ischemia, Hyperglycemia, mTOR, Pathology, Rapamycin, Rat.
Introduction
It has been well documented that preischemic hyperglycemia exacerbates brain damage caused by cerebral ischemia [1-5]. The characteristics of the hyperglycemia-exacerbated ischemic brain damage include accelerated cell death maturation time, enhanced tissue damage, recruitment of additional brain structures in to the damage process, increase of hemorrhagic transformation of infarction and development of postischemic seizures [1, 2, 4]. The mechanisms of hyperglycemia-enhanced ischemic brain damage are not fully understood but may include tissue acidosis [6], disturbed calcium homeostasis [7], productions of advanced glycation end products (RAGE), increases of free radicals through activation of the polyol pathway [8-12], activation of apoptotic cell death pathway [13, 14], initiation of neuroinflammation through activation of high-mobility group protein 1 (HMG-1) and toll-like receptors [15-18], early damage to the mitochondria [13, 19, 20], tilting mitochondrial dynamic balance towards fission [19], enhancement of autophagy signaling [19, 21], and increased permeability of the blood-brain barrier due to activation of metalloproteinases and damage to astrocytes and endothelial cells [10, 22, 23]. These adverse effects of hyperglycemia have not only been observed in chronic diabetic models but also in acute hyperglycemia caused by glucose infusion or injection [24, 25]. Recently we observed that mammalian target of rapamycin (mTOR) signaling may mediate hyperglycemia-associated ischemic brain damage in streptozotocin (STZ)-induced chronic (2-month) diabetic rats [26]. The mTOR pathway is an essential cellular signaling pathway involved in cell growth, proliferation, cellular metabolism, protein synthesis, gene transcription, and cell death including apoptosis, autophagy, and necroptosis [27]. Once being activated, mTOR causes phosphorylation of its downstream target proteins P70S6K, S6 and elF4E [28]. The mTOR pathway has received increasingly attention by neuroscientists due to its involvement in neurodegeneration. The role of mTOR in cerebral ischemia is still controversial. For example, Chong and colleagues reported that activation of mTOR protected ischemic brain damage and improved functional recovery cerebral ischemia in animals and oxygen glucose deprivation in cultured neurons [29]. On contrary, we have demonstrated that inhibition of mTOR by rapamycin significantly reduced brain damage in rats subjected to a transient forebrain ischemia and reperfusion [30]. The role of mTOR in hyperglycemia-enhanced ischemic brain damage has barely been examined. The only study was reported by our group, in which the exacerbated ischemic brain damage in STZ-induced chronic diabetic animals was ameliorated by rapamycin treatment [26]. The chronic diabetic model is characterized by hyperglycemia and potential vascular complications. Rapamycin has been shown to pass through the blood-brain barrier [31], making it an attractive therapeutic candidate for neurological diseases. The objective of the present study was to examine the role of mTOR in acute hyperglycemic ischemic rats. To that end, we infused glucose to the animals 40 min prior to the induction of a 10 min transient global ischemia and evaluated the histopathological outcomes in the cerebral cortex and hippocampus after reperfusion of 3h and 16h in euglycemic, hyperglycemic and rapamycin-treated hyperglycemic animals. The results were correlated to key indicators of mTOR signaling proteins. In addition, because cytochrome c release from the mitochondria to cytosol is an initial critical step to activate mitochondria-initiated caspase dependent cell death pathway [32] and rapamycin has been shown to activate autophagy [33], we measured cytosolic cytochrome c and LC3-II. The results showed that acute hyperglycemia increased brain damage, activated mTOR pathway, and increased the release of cytochrome c and levels of LC3-II under control and after ischemic conditions. Treatment with rapamycin reduced brain damage and inhibited the hyperglycemic ischemia induced mTOR alterations.
Materials and Methods
Animals
All procedures were approved by the Institutional Animal Care and Usage Committee (IACUC) at North Carolina Central University. Adult male Wistar rats (Charles River Laboratories) weighing between 230 and 260 g were used for all experiments. The animals were housed in the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International approved animal facility on campus. The animal housing room had controlled temperature of 20±2°C, humidity to 40-60%, and a 12-hour light/12-hour dark cycle. Animals had free access to food and water before surgery. The animals fasted overnight with free access to water prior to the surgical day. All efforts were made to minimize animal suffering, to reduce the number of animals, and to use alternatives to in vivo techniques, if available.
Eighty-four animals were randomly divided into 3 groups: euglycemic, hyperglycemic and hyperglycemic + rapamycin (n=28 in each of the 3 groups). Each group contained a sham control (n=8) ischemia plus 3h reperfusion (I/R 3h, n=10) and ischemia plus 16h reperfusion (I/R 16h, n=10). Four animals from sham and 6 animals from both I/R 3h and I/R 16h were perfusion-fixed and used for histology examination. The brain samples from the rest of the animals were snap frozen in liquid nitrogen and used for Western blot analysis. One hundred percentage success rate was achieved in animal model induction.
Rapamycin injection
Rapamycin (LC Laboratories, USA) was dissolved initially in 100% ethanol to a 20 mg/ml stock solution, which was stored at -20°C. Immediately before use, the stock solution was diluted in 5% Tween 80 and 5% polyethyleneglycol 400 to a final concentration of 4% ethanol. Rapamycin (6 mg/kg/d) was intraperitoneally (i.p) injected 7 days prior to the induction of ischemia in rapamycin treated subgroup. Animals in Vehicle group were injected with a solution containing 4% ethanol, 5% Tween 80 and 5% polyethyleneglycol 400.
Operative procedures
The animals were anesthetized by inhalation of 3.0% isoflurane in a mixture of N2O and O2 (70:30) and maintained at 1.0-2.0% during the surgery. A midline incision was made in the ventral cervical area to expose the common carotid arteries. A central catheter was inserted into the right jugular vein for the withdrawal of blood. A tail artery was cannulated for blood pressure monitoring. A rectal thermometer was inserted to monitor body temperature. Head temperature was monitored by placing another needle thermometer subcutaneously on the temporal muscle. Both rectal and head temperatures were maintained close to 37 ± 0.5°C by lamp heating combined with an Automated Thermal Control Unit (DC Temperature Controller, FHS 40-90-8D, Bowdoin, ME) during the surgical procedures. Blood pressure was monitored using a pressure monitor (BP-1, World Precision Instruments).
Induction of hyperglycemia and ischemia
Acute hyperglycemia was induced by i.v. infusion of 25% glucose solution (2.5 to 3.0 ml/h) 40 min prior to the induction of ischemia to yield a plasma glucose concentration of 16-25 mg/L. Blood glucose levels were measured using OneTouch Glucometer (OneTouch®) 5 min after infusion and 5 min prior to the induction of cerebral ischemia. Transient forebrain cerebral ischemia (10 min) was induced using a two-vessel occlusion (2-VO) method plus systemic hypotension, in which both the common carotid arteries were occluded with vascular clamps for 10 min and blood was withdrawn through the central catheter inserted into the jugular vein to maintain the blood pressure at 40 to 50 mmHg during the ischemic period. Ischemia was terminated by reinfusion of the shed blood and removal of the carotid clamps. Animals that experienced the same surgical procedure but without occlusion of the common carotid arteries and withdrawal of blood were used as sham-operated controls. Because previous reports have shown that hyperglycemic ischemia induces postischemic seizure and animal death of status epilepticus after 16-24 h of reperfusion, we have selected 3 and 16 hours of reperfusion as the experimental end points in the present study.
Histology
Upon predetermined end points, the animal brains were perfusion fixed by 4% paraformaldehyde through a transcardiac catheter. The brains were removed from the skull, placed in a pre-cooled rat brain matrix, cut into 3 coronal blocks of 4 mm each and post-fixed with 4% paraformaldehyde for 24 h. The blocks were dehydrated in ascending concentrations of ethanol, embedded in paraffin, and sectioned at 5 μm thickness. For routine histology, the sections were stained with celestine blue and acid fuchsin. Neuronal death was examined under a light microscope (Olympus IX51) at 400× magnification. Bright red-stained acidophilic neurons with shrunken, triangular dense purple nuclei were considered to be dead neurons. The numbers of dead cells were counted in 5 microscopic fields in the parietal cortex and throughout the hippocampal CA1 subregion. The results are presented as percentage of dead neurons.
Western blotting
The cerebral cortex and hippocampi were gently separated, quickly frozen in liquid nitrogen and then stored in a -80ºC freezer until used. The tissues were individually homogenized in a pre-cooled suspension buffer (15 mM Tris base/HCL (pH 7.7), 0.25 M sucrose, 15 mM NaCl, 1.5 mM MgCl2, 2.5 mM EDTA, 0.25 mM Na3VO4, 25 mM NaF, 1 mM EGTA, 2 mM NaPPi) containing a protease inhibitor cocktail (Complete Tablets, Roche), 1 mM DTT, and 0.5 mM PMSF. Cytosolic and nuclear fractions were separated through a series of centrifugations. Protein contents of the samples were measured using a Microplate BCA Protein Assay kit (Thermo Scientific). An equal amount of protein (25 μg) was loaded into each lane, separated in 4%-12% NuPAGE gels (Invitrogen, Carlsbad, CA, USA), transferred to PVDF membranes (Millipore), and probed with the primary antibodies listed in Table 1. Blots were imaged using a LI-COR Biosciences Odyssey Infrared Fluorescent scanner (Lincoln, NE, USA). Both target protein bands and internal loading control protein bands were measured using LI-COR software and are presented as a ratio of the target protein band fluorescent intensity over the loading control protein band intensity.
Primary antibodies used in this study.
| Antibodies | Source | Dilutions | Manufacturer | Application |
|---|---|---|---|---|
| p-mTOR (Ser 2448) | Rabbit | 1:200 | Cell Signaling | WB |
| p-P70S6K (T421/S424) | Rabbit | 1:800 | Cell Signaling | WB |
| p-S6 (Ser235+236) | Rabbit | 1:200 | Thermo | WB |
| Cyt c | Mouse | 1:1000 | Cell Signaling | WB |
| LC3-II | Rabbit | 1:800 | Abcam | WB |
| β-actin | Mouse | 1:1000 | Cell Signaling | WB |
WB, Western blot.
Statistical analysis
Data are represented as mean±s.d. Statistical analyses were perform using SPSS statistical software package version 13.0. ANOVA followed by Bonferroni post hoc test to detect differences among the three experimental groups. A p<0.05 was considered statistically significant.
Results
Blood glucose concentrations
Blood glucose concentrations measured at 5 min prior to the induction of cerebral ischemia were presented in Table 2. The average blood glucose levels in the euglycemic group (EG) were 4.53 to 5.00 mM, while those in the hyperglycemic group (HG) were 18.83 to 20.40 mM (p<0.01 vs. EG) and in hyperglycemia plus rapamycin (HG+RAPA) treatment were 18.82 to 20.14 mM (p<0.01 vs. EG).
Blood glucose levels in the rats
| Group | n | Blood glucose (mmol/L) | ||
|---|---|---|---|---|
| Sham (n=8 in each) | I/R 3h (n=10 in each) | I/R 16h (n=10 in each) | ||
| EG | 28 | 4.53±0.54 | 4.79±0.48 | 5.00±0.56 |
| HG | 28 | 18.83±1.59** | 20.40±2.19** | 19.75±1.99** |
| HG+RAPA | 28 | 18.82±1.89** | 20.14±1.20** | 19.76±2.61** |
**p<0.01 vs. EG.
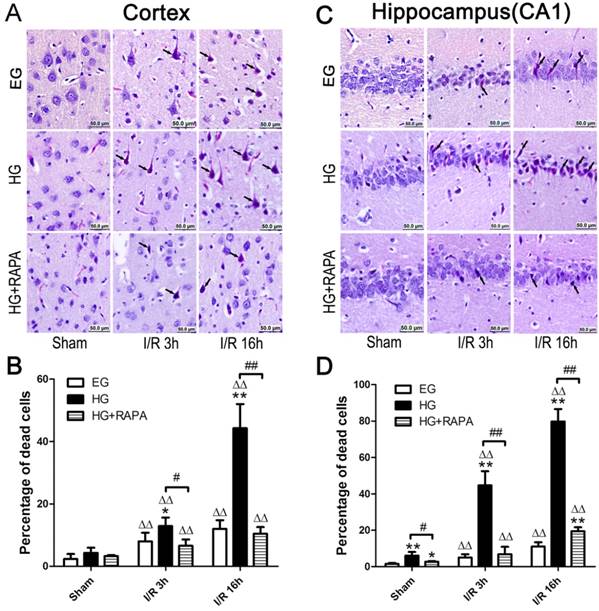
Rapamycin ameliorated hyperglycemia aggravated ischemia-induced neuronal death
The pathological outcomes in the cortex are given in Figure 1A and 1B. As shown in the Figure 1, a few scattered dead neurons were observed in the sham-operated euglycemic animals. Transient cerebral ischemia resulted in a mildly increased number of dead neurons in the cortex after 3h of reperfusion (p<0.01) and the damage remained at high level after 16h of reperfusion in euglycemic animals compared with the sham control (Figure 1A and 1B). Acute hyperglycemia, however, significantly increased the percentage of dead neurons in the cortex after reperfusion. Therefore, the percentage of dead neurons was moderately increased from 4.29±1.68% in sham control to 12.88±2.76% in hyperglycemic ischemia with 3h of recovery (p<0.01), and a more drastic increase (44.32±7.69%) was occurred after 16h of recovery. Treatment with rapamycin in hyperglycemic animals significantly decreased the percentage of damage from 12.88±2.76% to 6.57±2.04% after 3h (p<0.05) and from 44.32±7.69% to 10.46±2.13% after 16h of reperfusion (p<0.01).
The histopathological outcomes in the hippocampal CA1 region (Figure 1C and 1D) followed the same trend as in the cortex. Ten minutes transient forebrain ischemia in euglycemic rats caused a mild increase in the percentage of dead neurons after 3 and 16 h of reperfusion (p<0.01). In contrast, hyperglycemia significantly increased the damage from 6.07±1.98% in control to 44.67±7.77% at 3h and to 79.82±6.69% at 16h of reperfusion (p<0.01). Treatment with rapamycin in hyperglycemic animals reduced the damage to 6.89±4.04% at 3h and 19.50±2.12% at 16h of recovery (p<0.01).
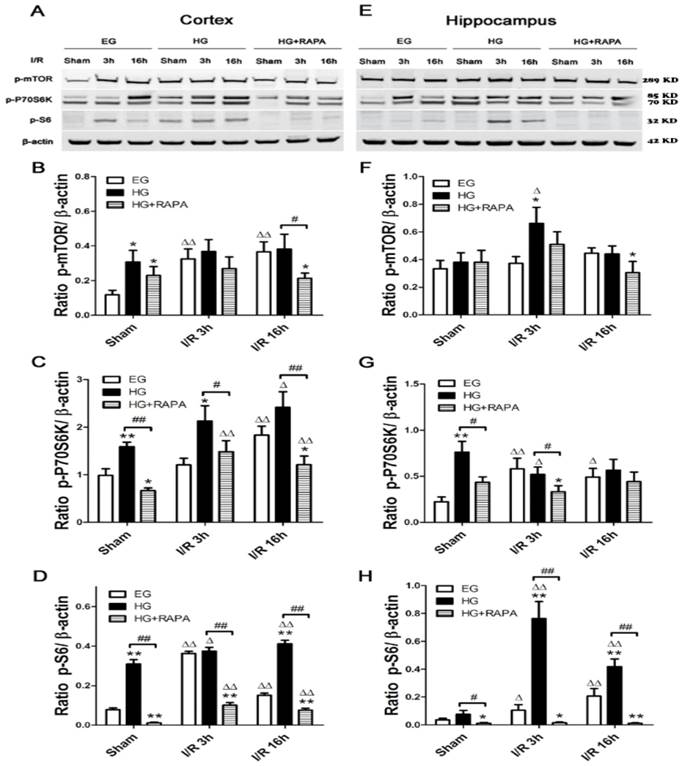
Rapamycin suppressed hyperglycemia-activated mTOR signaling after Ischemia and reperfusion
Protein levels of phospho-mTOR (p-mTOR) and its downstream effectors p-P70S6K and p-S6 were measured in both the cortex and hippocampus. In the cortex (Figure 2 A&B), cerebral ischemia significantly increased the levels of p-mTOR from 0.12±0.03 and p-S6 from 0.08±0.01 in control to 0.32±0.06 and 0.36±0.01 (p<0.01) after 3h of reperfusion in euglycemic animals. However, the levels of p-P70S6K were not significantly changed at 3h. At 16h of reperfusion, p-mTOR remained at high level (0.37±0.06, p<0.01), p-P70S6K significantly elevated (1.83±0.18, p<0.01), while p-S6 levels were declined but remained higher than the control levels (p<0.01). Hyperglycemia per se, without combination of cerebral ischemia, increased the levels of p-mTOR (0.31±0.07 versus 0.12±0.03, P<0.01), p-P70S6K (1.59±0.09 versus 0.99±0.14, p<0.01) and p-S6 (0.31±0.02 versus 0.08±0.01, p<0.01). Ischemia in hyperglycemic rats failed to further increase the already elevated p-mTOR protein after 3 and 16 h of reperfusion, but it further increased the levels of p-P70S6K and p-S6 (p<0.05). Rapamycin treatment suppressed or completely reversed the hyperglycemia-associated elevations of p-P70S6K (0.67±0.06 versus 0.99±0.14, p<0.01) and p-S6 (0.01±0.002 versus 0.08±0.01, p<0.01) under sham control conditions. The p-mTOR level in sham control was lowered by rapamycin but it did not reach statistical significance. Rapamycin also inhibited the hyperglycemic ischemia induced elevations of p-P70S6K (1.48±0.23 versus 2.13±0.32, p<0.05) and p-S6 (0.10±0.01 versus 0.37±0.02, p<0.01) after 3h of reperfusion. The suppression to all three proteins by rapamycin was observed after 16 h of reperfusion.
In the hippocampus (Figure 2 C&D), cerebral ischemia in euglycemic animals increased the levels of p-P70S6K from 0.22±0.05 to 0.58±0.12 after 3h (p<0.01) and to 0.49±0.09 after 16h of reperfusion (p<0.05) and the levels of p-S6 from 0.04±0.01 in control and 0.21±0.05 (p<0.01) after 16h of reperfusion. However, the p-mTOR was not affected by euglycemic ischemia. Hyperglycemia per se increased the level of p-P70S6K from 0.22±0.05 to 0.76±0.12 (p<0.01), but not p-mTOR orp-S6. Ischemia in hyperglycemic animals significantly increased the levels of p-mTOR (0.37±0.05 versus 0.66±0.12, p<0.05) and p-S6 (0.10±0.04 versus 0.76±0.12, p<0.01) at 3h of reperfusion, while p-P70S6K level remained high at this time point compared with the euglycemic control. The levels of p-mTOR at 3h (0.66±0.12) and p-S6 at 3h (0.76±0.12) and 16h (0.42±0.05) were much higher in hyperglycemic animals than in euglycemic samples. Rapamycin treatment suppressed the hyperglycemia-induced increases of p-P70S6K (0.76±0.12 versus 0.43±0.06, p<0.05) and p-S6 (0.08±0.03 versus 0.01±0.005, p<0.05) in sham controls. Similarly, rapamycin deceased the levels of p-mTOR, p-P70S6K and p-S6 at 3h and 16h of reperfusion in general, but more pronounced reductions were observed for p-S6 at 3h and 16h of recovery compared with the hyperglycemic ischemic groups (p<0.01).
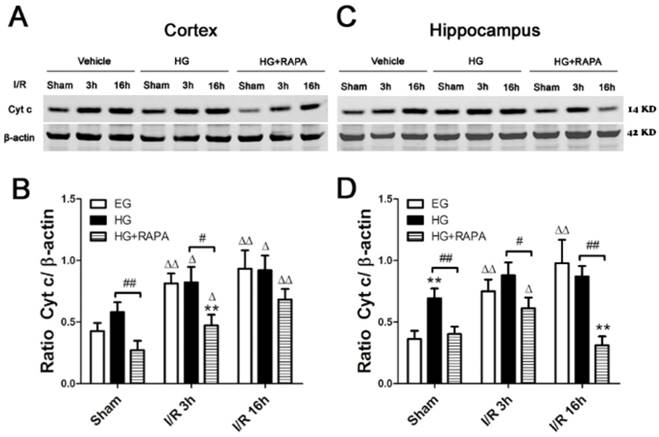
Rapamycin reduced cytosolic cytochrome c after ischemia and reperfusion in hyperglycemic animals
Cytochrome c release is a critical step in activating the caspase-dependent cell death pathway. To examine whether hyperglycemia and rapamycin have any effect on cytochrome c release, we measured cytochrome c levels in the cytosolic fraction in euglycemic, hyperglycemic and hyperglycemic plus rapamycin treated ischemic animals. In the cerebral cortex (Figure 3 A, B), ischemia in euglycemic animals significantly increased the cytosolic cytochrome c from 0.43±0.07 to 0.81±0.08 at 3h (p<0.01) and 0.93±0.15 at 16h (p<0.01). Hyperglycemia per se increased cytosolic cytochrome c, but the increase did not reach statistical significance. Hyperglycemic ischemia increased the cytochrome c levels after 3h and 16h. However, these increases were not significantly higher than those in euglycemic animals that experienced same length of reperfusion periods. Treatment with rapamycin significantly reduced the cytochrome c levels in sham control, ischemia with 3h reperfusion and 16h reperfusion compared with the hyperglycemic animal under the same conditions. In the hippocampus (Figure 3 C, D), transient cerebral ischemia in euglycemic animals significantly increased the levels of cytochrome c from 0.36±0.07 in control to 0.74±0.10 after 3h of reperfusion (p<0.01) and the levels were further increased to 0.98±0.19 at 16h of reperfusion (p<0.01). Hyperglycemia per se significantly increased the levels of cytochrome c compared with the euglycemic control (0.36±0.07 versus 0.69±0.08, p<0.01). After ischemia and reperfusion, the high cytosolic cytochrome c levels were maintained in hyperglycemic groups. Rapamycin significantly reduced the levels of cytochrome c in control and after ischemia and reperfusion of 3h and 16h.
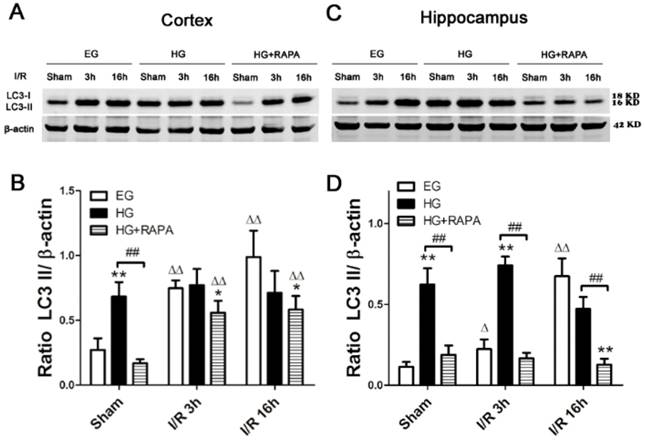
Rapamycin inhibited the hyperglycemia-induced increases of LC3-II in the cortex and hippocampus
To assess the level of autophagy after hyperglycemic ischemia and rapamycin treatment, we measured the most commonly used autophagy markers, LC3-II in the cytosolic fractions. In the cortex (Figure 4 A, B), ischemia in euglycemic animals resulted in significant increases of LC3-II protein level after 3h (0.75±0.06, p<0.01) and 16h (0.99±0.20, p<0.01) compared with control (0.27±0.09). Hyperglycemia per se increased the levels of LC3-II compared with the euglycemic control (0.68±0.11 versus 0.27±0.09, p<0.01). Following hyperglycemic ischemia, the LC3-II levels maintained at high level at 3h and 16h of reperfusion. Rapamycin treatment completely prevented the hyperglycemia-stimulated increase of LC3-II under control condition (0.68±0.11 versus 0.17±0.03, p<0.01). Rapamycin also reduced the levels of LC3-II after 3h and 16h of reperfusion. However, the reduction did not reached statistical significance compared with the levels in hyperglycemic ischemic samples at identical reperfusion endpoint. In the hippocampus (Figure 4 C, D), euglycemic ischemia increased LC3-II after 16h of reperfusion compared with the control (0.67±0.11 versus 0.11±0.03, p<0.01). Similar to the observation in the cortex, hyperglycemia per se significantly increased the level of LC3-II without ischemia and reperfusion compared with the euglycemic control (0.62±0.10 versus 0.11±0.03, p<0.01). The LC3-II level was kept at high levels after 3h (0.74±0.06) and 16h (0.47±0.07) of reperfusion. Rapamycin treatment blocked the hyperglycemia-induced elevation of LC3-II in sham control and ischemia with 3h and 16h of reperfusion.
Discussion
In the present study, we have demonstrated that acute preischemic hyperglycemia enhanced ischemic brain damage. The exacerbation effects of hyperglycemia on ischemic stroke was associated with activation of mTOR signaling, release of cytochrome c, and increase in autophagy marker LC3-II. Treatment with mTOR inhibitor rapamycin significantly reduced the hyperglycemia-exaggerated ischemic damage, suppressed the mTOR signaling, inhibited the release of cytochrome c, and reduced the levels of LC3-II.
Preischemic hyperglycemia induced either by chronic diabetes, acute elevation of glucose by food ingestion, or glucose infusion increases ischemia and reperfusion induced brain damage. When glucose levels increase to above 144mg/dL in stroke patients, their mortality and rate of permanent disability increases 3-fold compared with euglycemic patients [34]. Our results demonstrated that hyperglycemia enhanced ischemic brain damage in early reperfusion stage in both the cerebral cortex and hippocampus. These findings are consistent with the long-standing documentation that hyperglycemia shortens damage maturation period and enhances brain damage caused by transient cerebral ischemia [2, 35]. Although the exacerbation effects of hyperglycemia on ischemic brain damage is well documented, the underlying mechanisms by which hyperglycemia enhances ischemic brain damage remain unclear. In this experiment, we studied the participation of mTOR signaling in mediating the adverse effects of hyperglycemia on ischemia by measuring the key proteins in the mTOR pathway and by applying an mTOR inhibitor in hyperglycemic ischemic animals. The results showed that ischemia in euglycemic animals increased the levels of p-mTOR, p-P70S6K and p-S6 in the cortical and/or hippocampal structures. This is consistent with our previous study in euglycemic animals [30], confirming that cerebral ischemia activates the mTOR pathway. Hyperglycemia further increased the levels of p-P70S6K and p-S6 in the cortex and hippocampus and p-mTOR in the hippocampus. Interestingly, hyperglycemia per se caused significant increases of p-mTOR, p-P70S6K in the cortex and p-S6 in both the cortex and hippocampus. Similar findings were also observed in streptozotocin induced 2-month diabetic rats [26]. Thus, the p-P70S6K was increased in diabetic sham control and reperfusion of 16h following a 10-min global ischemia in diabetic rats compared with the normoglycemic rats. Taken together, these findings suggest that hyperglycemic ischemia exerts a highly pronounced influence on mTOR signaling compared with euglycemic ischemia and that hyperglycemia per se is capable of activating the mTOR pathway, which may predispose the brain tissue to be vulnerable to ischemic injury. The pitfall for this part of the study is that the total protein levels of mTOR, P70S6K and S6 were not measure although the phosphorylation of these proteins indicates the mTOR pathway activation.
Histopathological outcomes in the neocortex (A,B) and hippocampal CA1 region (C,D) after cerebral ischemia and reperfusion (I/R) in euglycemic (EG), hyperglycemic (HG) and rapamycin treated hyperglycemic (HG+RAPA) animals. A and C, representative microphotographs showing histological outcomes in the cortex (A) and hippocampal CA1 (C) area; B and D, summarized percentage of death in each group in the cortex (B) and CA1 (D). Euglycemic ischemia moderately increased neuronal death, which was significantly enhanced by hyperglycemia. Celestine blue and acid fuchsin staining. Arrows indicate dead neurons. Bar = 50 μm. Data are presented as means ± s.d. ΔΔ p< 0.01 versus sham group within each group. * p<0.05 and **p<0.01 versus EG at the same reperfusion endpoint. # p<0.05 and ## p<0.01 HG versus HG+RAPA. Statistical annotations are the same for Figures 2-4.
Western blot analyses of p-mTOR, p-P70S6K and p-S6 in the cytosolic fractions of the cortical and hippocampal samples. A and C, representative Western blots in the cortex (A) and hippocampus (C). B and D, summarized target band densities of the cortical (B) and hippocampal (D) samples. Euglycemic ischemia activated the mTOR and its downstream proteins in the cortex and/or hippocampus. Hyperglycemia per se increased the levels of detected mTOR pathway proteins and ischemia maintained the high levels of these proteins in hyperglycemic animals. Rapamycin suppressed the hyperglycemia-elevated mTOR pathway proteins in control and after ischemia and reperfusion injury. I/R, ischemia and reperfusion; EG, euglycemia; HG, hyperglycemia; HG+RAPA, rapamycin treated hyperglycemic animals.
The participation of m-TOR in mediating hyperglycemia-enhanced ischemic brain damage is further supported by the results showing that inhibition of mTOR by rapamycin significantly reduced ischemic brain damage in hyperglycemic animals. The percentage of neuronal death in the cortex was reduced from 44.32% in hyperglycemic ischemic animals to 10.46% by rapamycin at 16h of reperfusion. Similarly, the percentages of neuronal death in the hippocampal CA1 were reduced from 44.67% and 79.82% at 3h and 16h in hyperglycemic animals to 6.89% and 19.50% by rapamycin treatment. These agree with previous reports in ischemic animals under both hyperglycemic and normoglycemic conditions [26, 30]. The observed protecting effect of rapamycin against ischemic brain injury in the hyperglycemic animals was associated with suppression of m-TOR signaling. Therefore, rapamycin suppressed not only elevations of p-mTOR, p-P70S6K and p-S6 caused by hyperglycemic ischemia but also normalized these protein levels in hyperglycemic control samples. Because the protective effects of rapamycin have also been observed in ischemic animals under normo- and chronic hyperglycemic conditions[26, 30], it is likely that inhibition of mTOR signaling is one of the major mechanisms that rapamycin reduces ischemic brain damage under both euglycemic and hyperglycemic conditions. This study extended the model to acute hyperglycemia as seen in clinic due feeding, stress and intravenous infusion caused elevation of plasma glucose levels.
Western blot analyses of cytochrome c in the cytosolic fractions of the cortical and hippocampal samples. A and C, representative Western blots in the cortex (A) and hippocampus (C). B and D, summarized cytochrome c band optical densities of the cortical (B) and hippocampal (D) samples. Euglycemic (EG) ischemia increased the cytosolic cytochrome c. Hyperglycemia (HG) increased the baseline cytochrome c levels. After ischemia and reperfusion, the cytochrome c further increased in hyperglycemic animals compared with the control but not significantly higher than euglycemic ischemic samples. Rapamycin treatment in hyperglycemic animals (HG+RAPA) significantly reduced the levels of cytochrome c in the both control and ischemic samples.
Western blot analyses of LC3-II in the cytosolic fractions of the cortical and hippocampal samples. A and C, representative Western blots in the cortex (A) and hippocampus (C). B and D, summarized LC3-II band optical densities of the cortical (B) and hippocampal (D) samples. Euglycemic ischemia increased the cytosolic LC3-II. Hyperglycemia increased the baseline levels of LC3-II and maintained the high levels after ischemia and reperfusion. Rapamycin treatment significantly reduced the levels of LC3-II in the both hyperglycemic control and hyperglycemic ischemic samples.
Cytochrome c release from the mitochondria to the cytosol plays a pivotal role in activating caspase-dependent apoptotic cell death pathway. In present study, we measured the level of cytochrome c. The results showed that cytosolic cytochrome c increased after I/R in the euglycemic group and the level increased as the reperfusion hours progressed. Hyperglycemia did not further increase the cytochrome c in control and after I/R compared with the euglycemic animals in the cortex. However, hyperglycemia resulted in a significant elevation of cytosolic cytochrome c in sham control of the hippocampal samples compared with the euglycemic animals. These data suggest that 1) cytochrome c release may be involved in mediating cell damage in euglycemic ischemia, 2) hyperglycemia-enhanced ischemia may not be associated with further exacerbation of cytochrome c release, and 3) the elevated cytochrome c in sham control of the hyperglycemic hippocampal samples may predispose the brain tissue to be vulnerable to ischemic injury. The present findings are at variance with previous studies showing that hyperglycemia increases cytochrome c release and activates capases-9 and -3 following forebrain ischemia [13, 24] and focal ischemia [14]. Nevertheless, rapamycin significantly inhibited the cytochrome c levels in hyperglycemic control and ischemic samples. This suggests that rapamycin is capable of inhibiting intrinsic cell death pathway through an undefined mechanism. Jing and colleagues [36] reported that rapamycin administration decreased cytochrome c release from the mitochondria to the cytosol in a rat model of subarachnoid hemorrhage and alleviated brain injury, which is consistent with our observation.
Autophagy is a lysosome degradation pathway that plays an important role in maintaining cell homeostasis. The existence of autophagy in ischemic stroke has been found for many years; however, it is not certain whether autophagy plays a protective or detrimental role in ischemic cerebral injury [37].Treatment with an autophagy inhibitor decreased the number of neuronal cells, whereas an autophagy inducer increased the number of cells under hypoxic stress, suggesting that autophagy promotes neuronal cell survival under hypoxic stress [38]. On the other hand, high glucose decreased LC3-II in endothelial cells [39] and inhibition of autophagy by RNA interference-mediated downregulation of Beclin 1 attenuated cerebral ischemic injury in rats [40]. In present study, we observed that ischemia in euglycemic animals significantly increased the levels of LC3-II and the neuroprotective effect of rapamycin was associated with inhibition of LC3-II, suggesting that autophagy may likely be associated with cell death in the present experimental setting. We have also observed that hyperglycemia per se significantly increased the levels of LC3-II in both the cortex and hippocampus and that the LC3-II remained at high level after ischemia and reperfusion in hyperglycemic animals. Compared with the euglycemic ischemic animals, hyperglycemia increased the level of LC3-II in the hippocampus after 3h of reperfusion but not in other time point or in the cortex. This suggest that hyperglycemia itself may affect the autophagy signaling and hyperglycemia-enhanced ischemic brain damage may or may not be associated with autophagy. However, since treatment with rapamycin blocked or ameliorated the LC3-II in hyperglycemic groups, this suggests that hyperglycemia may activate autophagy and inhibition of this process is protective in against hyperglycemic ischemia. It is interesting though that rapamycin is known to activate autophagy under physiological conditions [33], yet in this study and one of our previous experiments [30], we found that rapamycin inhibited the autophagy under cerebral ischemic condition. The complicated cross-talk between the survival kinases, different models, dosages, the administration routes of drugs and other sealed factors could be the reasons resulting these diverse outcomes. Kurdi and colleagues [41] administrated everolimus (rapamycin analogs) to mice for short term and long term respectively, and discovered that short term use of everolimus induced autophagy whereas continuous use of everolimus inhibited autophagy. This may explain controversial results of rapamycin in autophagy.
Taken together, the present study suggests that hyperglycemia enhances ischemic brain damage and activates mTOR signaling. Rapamycin apparently alleviates ischemic brain damage in hyperglycemic rats and this beneficial effect is associated with inhibition of mTOR.
Acknowledgements
The authors appreciate Mr. Brent Caligan for proofread and editing. This study is supported by American Epileptic Society to PAL, National Natural Science Foundation of China (81560226) to XY, National Natural Science Foundation of China (81660206) to PL and National Natural Science Foundation of China 973-Project (2014CB560711) to JN. The Biomanufacturing Research Institute and Technology Enterprise (BRITE) is partially funded by the Golden Leaf Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Contributions
XY, JN, PAL conceived and designed the experiments. CH, PL, XY, performed the experiments. CH, PL, XY, PAL analyzed the data. PAL contributed reagents and materials. CH, XY, PAL wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Li PA, Uchino H, Elmer E, Siesjo BK. Amelioration by cyclosporin A of brain damage following 5 or 10 min of ischemia in rats subjected to preischemic hyperglycemia. Brain Res. 1997;753:133-40
2. Siesjö BK, Katsura KI, Kristian T, Li PA, Siesjö P. Molecular mechanisms of acidosis-mediated damage. Acta Neurochir Suppl. 1996;66:8-14
3. Luitse MJ, Biessels GJ, Rutten GE, Kappelle LJ. Diabetes, hyperglycaemia, and acute ischaemic stroke. Lancet Neurol. 2012;11:261-71
4. Luitse MJ, Velthuis BK, Kappelle LJ, van der Graaf Y, Biessels GJ. Chronic hyperglycemia is related to poor functional outcome after acute ischemic stroke. Int J Stroke. 2016 DOI: 10.1177/1747493016676619. [Epub ahead of print]
5. Rehni AK, Nautiyal N, Perez-Pinzon MA, Dave KR. Hyperglycemia / hypoglycemia-induced mitochondrial dysfunction and cerebral ischemic damage in diabetics. Metab Brain Dis. 2015;30:437-47
6. Li P-A, Shamloo M, Katsura K-i, Smith M-L, Siesjö BK. Critical values for plasma glucose in aggravating ischaemic brain damage: correlation to extracellular pH. Neurobiology of disease. 1995;2:97-108
7. Moreira T, Cebers G, Pickering C, Ostenson CG, Efendic S, Liljequist S. Diabetic Goto-Kakizaki rats display pronounced hyperglycemia and longer-lasting cognitive impairments following ischemia induced by cortical compression. Neuroscience. 2007;144:1169-85
8. Li PA, Liu GJ, He QP, Floyd RA, Siesjo BK. Production of hydroxyl free radical by brain tissues in hyperglycemic rats subjected to transient forebrain ischemia. Free Radic Biol Med. 1999;27:1033-40
9. Muranyi M, Li PA. Hyperglycemia increases superoxide production in the CA1 pyramidal neurons after global cerebral ischemia. Neurosci Lett. 2006;393:119-21
10. Prasad S, Sajja RK, Naik P, Cucullo L. Diabetes Mellitus and Blood-Brain Barrier Dysfunction: An Overview. J Pharmacovigil. 2014;2:125
11. Wei J, Huang NC, Quast MJ. Hydroxyl radical formation in hyperglycemic rats during middle cerebral artery occlusion/reperfusion. Free Radical Biology and Medicine. 1997;23:986-95
12. Yorek MA. The role of oxidative stress in diabetic vascular and neural disease. Free Radic Res. 2003;37:471-80
13. Li PA, Rasquinha I, He QP, Siesjo BK, Csiszar K, Boyd CD. et al. Hyperglycemia enhances DNA fragmentation after transient cerebral ischemia. J Cereb Blood Flow Metab. 2001;21:568-76
14. Muranyi M, Fujioka M, He Q, Han A, Yong G, Csiszar K. et al. Diabetes activates cell death pathway after transient focal cerebral ischemia. Diabetes. 2003;52:481-6
15. Ding C, He Q, Li PA. Diabetes increases expression of ICAM after a brief period of cerebral ischemia. J Neuroimmunol. 2005;161:61-7
16. Huang J, Liu B, Yang C, Chen H, Eunice D, Yuan Z. Acute hyperglycemia worsens ischemic stroke-induced brain damage via high mobility group box-1 in rats. Brain Res. 2013;1535:148-55
17. Jing L, Wang JG, Zhang JZ, Cao CX, Chang Y, Dong JD. et al. Upregulation of ICAM-1 in diabetic rats after transient forebrain ischemia and reperfusion injury. J Inflamm (Lond). 2014;11:35
18. Pahwa R, Jialal I. Hyperglycemia Induces Toll-Like Receptor Activity Through Increased Oxidative Stress. Metab Syndr Relat Disord. 2016;14:239-41
19. Kumari S, Mehta SL, Li PA. Glutamate induces mitochondrial dynamic imbalance and autophagy activation: preventive effects of selenium. PLoS One. 2012;7:e39382
20. Sanderson BA, Sowersby DS, Crosby S, Goss M, Lewis LK, Beall GW. Charge density and particle size effects on oligonucleotide and plasmid DNA binding to nanosized hydrotalcite. Biointerphases. 2013;8:8
21. Xia CY, Zhang S, Chu SF, Wang ZZ, Song XY, Zuo W. et al. Autophagic flux regulates microglial phenotype according to the time of oxygen-glucose deprivation/reperfusion. Int Immunopharmacol. 2016;39:140-8
22. Kamada H, Yu F, Nito C, Chan PH. Influence of hyperglycemia on oxidative stress and matrix metalloproteinase-9 activation after focal cerebral ischemia/reperfusion in rats: relation to blood-brain barrier dysfunction. Stroke. 2007;38:1044-9
23. Jing L, Mai L, Zhang JZ, Wang JG, Chang Y, Dong JD. et al. Diabetes inhibits cerebral ischemia-induced astrocyte activation - an observation in the cingulate cortex. Int J Biol Sci. 2013;9:980-8
24. Li PA, He QP, Csiszar K, Siesjo BK. Does long-term glucose infusion reduce brain damage after transient cerebral ischemia?. Brain Res. 2001;912:203-5
25. Tsai MJ, Lin MW, Huang YB, Kuo YM, Tsai YH. The Influence of Acute Hyperglycemia in an Animal Model of Lacunar Stroke That Is Induced by Artificial Particle Embolization. Int J Med Sci. 2016;13:347-56
26. Liu P, Yang X, Hei C, Meli Y, Niu J, Sun T. et al. Rapamycin Reduced Ischemic Brain Damage in Diabetic Animals Is Associated with Suppressions of mTOR and ERK1/2 Signaling. Int J Biol Sci. 2016;12:1032-40
27. Maiese K. Cutting through the Complexities of mTOR for the Treatment of Stroke. Current Neurovasc Res. 2014;11:177-86
28. King MA, Hands S, Hafiz F, Mizushima N, Tolkovsky AM, Wyttenbach A. Rapamycin inhibits polyglutamine aggregation independently of autophagy by reducing protein synthesis. Mol Pharmacol. 2008;73:1052-63
29. Chong ZZ, Yao Q, Li HH. The rationale of targeting mammalian target of rapamycin for ischemic stroke. Cell Signal. 2013;25:1598-607
30. Yang X, Hei C, Liu P, Song Y, Thomas T, Tshimanga S. et al. Inhibition of mTOR Pathway by Rapamycin Reduces Brain Damage in Rats Subjected to Transient Forebrain Ischemia. Int J Biol Sci. 2015;11:1424-35
31. Banerjee S, Gianino SM, Gao F, Christians U, Gutmann DH. Interpreting mammalian target of rapamycin and cell growth inhibition in a genetically-engineered mouse model of Nf1-deficient astrocytes. Mol Cancer Ther. 2011:10 (2). doi:10.1158/1535-7163.MCT-10-0654
32. Niizuma K, Yoshioka H, Chen H, Kim GS, Jung JE, Katsu M. et al. Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim Biophys Acta. 2010;1802:92-9
33. Buckley KM, Hess DL, Sazonova IY, Periyasamy-Thandavan S, Barrett JR, Kirks R. et al. Rapamycin up-regulation of autophagy reduces infarct size and improves outcomes in both permanent MCAL, and embolic MCAO, murine models of stroke. Exp Transl Stroke Med. 2014;6:8
34. Capes SE, Hunt D, Malmberg K, Pathak P, Gerstein HC. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: a systematic overview. Stroke. 2001;32:2426-32
35. Myers RE, Yamaguchi S-i. Nervous system effects of cardiac arrest in monkeys: preservation of vision. Arch Neurol. 1977;34:65-74
36. Jing CH, Wang L, Liu PP, Wu C, Ruan D, Chen G. Autophagy activation is associated with neuroprotection against apoptosis via a mitochondrial pathway in a rat model of subarachnoid hemorrhage. Neuroscience. 2012;213:144-53
37. Smith CM, Chen Y, Sullivan ML, Kochanek PM, Clark RS. Autophagy in acute brain injury: feast, famine, or folly?. Neurobiol Dis. 2011;43:52-9
38. Tanabe F, Yone K, Kawabata N, Sakakima H, Matsuda F, Ishidou Y. et al. Accumulation of p62 in degenerated spinal cord under chronic mechanical compression: functional analysis of p62 and autophagy in hypoxic neuronal cells. Autophagy. 2011;7:1462-71
39. Liang X, Zhang T, Shi L, Kang C, Wan J, Zhou Y. et al. Ampelopsin protects endothelial cells from hyperglycemia-induced oxidative damage by inducing autophagy via the AMPK signaling pathway. Biofactors. 2015;41:463-75
40. Zheng YQ, Liu JX, Li XZ, Xu L, Xu YG. RNA interference-mediated downregulation of Beclin1 attenuates cerebral ischemic injury in rats. Acta Pharmacol Sin. 2009;30:919-27
41. Kurdi A, De Doncker M, Leloup A, Neels H, Timmermans JP, Lemmens K. et al. Continuous administration of the mTORC1 inhibitor everolimus induces tolerance and decreases autophagy in mice. Br J Pharmacol. 2016
Author contact
![]() Corresponding author: P. Andy Li, M.D., Ph.D. Department of Pharmaceutical Sciences, North Carolina Central University, 1801 Fayetteville Street, Durham, NC 27707. Fax: 919-530-6600; E-mail: pliedu
Corresponding author: P. Andy Li, M.D., Ph.D. Department of Pharmaceutical Sciences, North Carolina Central University, 1801 Fayetteville Street, Durham, NC 27707. Fax: 919-530-6600; E-mail: pliedu
Received 2016-12-29
Accepted 2017-1-28
Published 2017-7-7