ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2017; 13(7):888-900. doi:10.7150/ijbs.18468 This issue Cite
Research Paper
Crosstalk between Mitochondrial Fission and Oxidative Stress in Paraquat-Induced Apoptosis in Mouse Alveolar Type II Cells
Guangju Zhao1, 2*, Kaiqiang Cao1, 2*, Changqin Xu1, 2, Aifang Sun1, Wang Lu1, Yi Zheng3, 4, Haixiao Li1, 2, Guangliang Hong1, 2, Bing Wu1, Qiaomeng Qiu1, Zhongqiu Lu1, 2 ![]()
1. Emergency Department, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, 325000, China;
2. Wenzhou Municipal Key Laboratory of Emergency, Critical care, and Disaster Medicine, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, 325000, China;
3. Department of Microbiology and immunology, School of Laboratory Medicine, Wenzhou Medical University, Wenzhou 325000, China;
4. Key Lab of Laboratory Medicine, Ministry of Education of China, Wenzhou 325000, China.
* The first two authors contributed equally to this study
Abstract

Paraquat (PQ), as a highly effective and nonselective herbicide, induces cell apoptosis through generation of superoxide anions which forms reactive oxygen species (ROS). Mitochondria, as regulators for cellular redox signaling, have been proved to play an important role in PQ-induced cell apoptosis. This study aimed to evaluate whether and how mitochondrial fission interacts with oxidative stress in PQ-induced apoptosis in mouse alveolar type II (AT-II) cells. Firstly, we demonstrated that PQ promoted apoptosis and release of cytochrome-c (Cyt-c). Furthermore, we showed that PQ broke down mitochondrial network, enhanced the expression of fission-related proteins, increased Drp1 mitochondrial translocation while decreased the expression of fusion-related proteins in AT-II cells. Besides, inhibiting mitochondrial fission using mdivi-1, a selective inhibitor of Drp1, markedly attenuated PQ-induced apoptosis, release of Cyt-c and the generation of ROS. These results indicate that mitochondrial fission involves in PQ-induced apoptosis. Further study demonstrated that antioxidant ascorbic acid inhibited Drp1 mitochondrial translocation, mitochondrial fission and attenuated PQ-induced apoptosis. Overall, our findings suggest that mitochondrial fission interplays with ROS in PQ-induced apoptosis in mouse AT-II cells and mitochondrial fission could serve as a potential therapeutic target in PQ poisoning.
Keywords: paraquat, mitochondrial fission, apoptosis, oxidative stress, reactive oxygen species.
Introduction
Paraquat (1, 10-dimethyl-4, 40-bipyridinium dichloride; PQ), as a highly effective and nonselective herbicide, has been extensively used all over the world since the middle of the 20th century. Human exposure to PQ would lead to accumulation of PQ in various organs, especially in lungs, resulting in cell apoptosis and organ damage. The pulmonary toxicity of PQ is characterized by pulmonary hemorrhage and edema in the early phase, followed by proliferation of fibroblasts and deposition of collagen [1]. Since lung is the primary target organ, the exact mechanism of PQ toxicity remains largely elusive. Some studies revealed PQ undergoes redox cycling in multiple organs and consequently result in producing superoxide anions which causes direct cellular damage and forms other reactive oxygen species (ROS) [2, 3]. On the other hand, PQ was regarded as a resource of ROS production, promoting depletion of MMP and releasing cytochrome-c (Cyt-c) from mitochondria and cell death [4]. Our previous studies illustrated that activating an antioxidant pathway could protect mice from PQ-induced lung injury, suggesting the important role of oxidative stress in PQ intoxication [5]. Other studies showed PQ inhibits mitochondrial complex I and III activities [6, 7], implying an undefined role of PQ targeting on mitochondrial function.
Mitochondria play a crucial role in regulating cellular redox signaling and balancing cell survival and death [8]. Further, mitochondria also contribute to activation of apoptosis related molecules (e.g. Caspase 3, caspase 9 and PARP) and overproduction of ROS during PQ-induced cell apoptosis [6, 9, 10]. Interestingly, recent studies illustrated that mitochondria are dynamic organelles which change shape through two opposing processes of fusion and fission. It has been observed that rapid fragmentation of mitochondria with a concomitant increase in ROS generation after exposure to high glucose concentrations [11], suggesting that mitochondrial fission plays an indispensable role in the mitochondrial-mediated oxidative stress. Currently, more and more studies have revealed mitochondrial fission tends to manipulate apoptosis, autophagy and ROS production [12-16].
In mammalian cells, mitochondrial fusion is controlled by mitochondria-shaping proteins, including the large GTPase mitofusins2 (Mfn2) and optic atrophy protein 1 (OPA1) [13], whereas the mitochondrial fission 1 protein (Fis1) and dynamin-related protein 1 (Drp1) are the key mediators of mitochondrial fission [17-19]. Drp1 is crucial for embryonic development and synapse formation in mice [20], and impaired developmental competence and mitochondrial function of embryos and cells was observed when a selective mitochondrial fission inhibitor of Drp-1 was implemented [21]. However, upon induction of apoptosis, inhibiting Drp1 prevented the loss of the mitochondrial inner membrane potential (ΔΨm) as well as the release of Cyt-c [22], indicating Drp1 promotes mitochondrial-mediated cell damage in apoptosis. Besides, studies also illustrated that silencing Drp1 expression with siRNA alleviated mitochondrial fission, ROS production, nitric oxide synthase (NOS) activation, and cGMP generation [14]. Thus far, evidence for the occurrence of mitochondrial fission and its involvement in PQ-induced apoptosis is scare.
In the present study, we have demonstrated potent evidence for mitochondrial fission in PQ-induced apoptosis using mouse alveolar epithelial type II (AT- II) cells. The mitochondrial fission is activated by Drp1 and Fis1. Importantly, suppression of Drp1 abrogates mitochondrial damage, Cyt-c release, oxidative stress and apoptosis. Interestingly, antioxidant ascorbic acid not only alleviates oxidative stress and apoptosis but also inhibits mitochondrial fission and Cyt-c release. Regulation of mitochondrial fission may offer a novel strategy for treatment of PQ intoxication.
Materials and Methods
Materials
Paraquat, 2′, 7′- dichlorodihydrofluorescein diacetate (DCFH-DA), Hoechst 33342 was purchased from Sigma-Aldrich (St Louis, MO, USA). Dulbecco's Modified Eagle Medium (DMEM) used for AT-II cell culture was obtained from Gibco (Carlsbad, CA, USA). Cell counting kit-8 was from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Annexin V-FITC/propidium iodide (PI) apoptosis detection kit was obtained from Nanjing KeyGen Biotech Co., Ltd. (Nanjing, China). Mitochondrial division inhibitor 1 (Mdivi-1) and anti-Fis1 mAb was purchased from Enzo Life Sciences (Farmingdale, NY, USA). Ascorbic Acid was purchased from Biogot Technology (Nanjing, China). MitoTracker Green FM were provided by Molecular probes (Eugene, OR, USA). Anti-Drp1 mAb, anti-Opa1 mAb, anti-Fis1 mAb, anti-Mfn2 mAb and anti-β-actin mAb used in this study were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-Cyt C antibody, anti-Cox IV antibody and Alexa Fluor 555 labeled secondary antibody were provided by Beyotime Co., Ltd. (Shanghai, China).
Cell culture
Mouse AT-II cells were from our own laboratory [23]. Cells were cultured in DMEM containing 10% fetal bovine serum and 100U/ml antibiotic (penicillin and streptomycin) solution in a humidified incubator with 5% CO2 atmosphere at 37℃. Medium was changed every other day.
Cell Counting Kit 8 assay
AT II cells (5×104 cells/ml) were inoculated to 96-well plates with 0.1 ml per well and incubated for 24 h until reaching about 70% confluency. Then, cells were treated with media that contain different concentrations (200, 400, 800 and 1600μM) of PQ. After 24 hours, 20μl CCK-8 agent was added to each well, and further incubated for 1.5 h. The microplate reader (Dynex Technologies, USA) was used for fluorescence detection at a wavelength of 450 nm. The cell survival rate (%) = (mean absorbance in four wells of PQ group/ mean absorbance in four wells of control group) × 100%).
Flow cytometry analysis
The cell apoptotic rate was detected using an Annexin V-FITC/propidium iodide (PI) apoptosis detection kit following the manufacturer's instructions. In Brief, following exposure to PQ, the cells were harvested and washed with ice-cold PBS twice. Then, the cells were re-suspended in 500 μl binding buffer containing 5 μl of Annexin V-FITC and 5 μl of PI at a concentration of 1×106 cells/ml, followed by incubation for another 15 min at room temperature in the dark. Finally, apoptosis was measured with a FACScan flow cytometer (Becton‑Dickinson, Franklin Lakes, NJ, USA) and analysis was performed using FlowJo software (Tree Star, San Carlos, CA).
Hoechst staining
The changes in apoptotic AT-II cells were detected by staining with Hoechst 33342. Briefly, after treatment, Hoechst 33342 staining solution (5μg/ml) was added to each well, and the cells incubated for 5min at 37℃ in darkness. The analysis were performed by fluorescence microscopy (Olympus, Japan).
ROS generation measurement
AT-II cells (5×104 cells/ml) were inoculated to 96 well plates with 0.1 ml per well and incubated for 24 h until reaching about 70% confluency. Then, the AT-II cells in different treatment conditions were incubated with 10μM DCFH-DA at 37℃ for 15min. The medium was aspirated carefully and 100 μl PBS was added into each well. ROS generation was determined by fluorescence intensity with setting of 485 and 525 nm for excitation and emission wavelengths, respectively.
Mitochondrial morphology
Mitochondrial morphology was examined in AT II cells stained with 200 nM MitoTracker Green FM by a fluorescence microscope (Olympus, Japan) or a confocal microscope (Nikon, Japan). Wholly fragmented or punctuated mitochondria are defined as fragmented mitochondria, mitochondria with clearly networks are defined as tabulated mitochondria, and the others are regarded as intermediate ones. Percentages of AT-II cells exhibiting indicated mitochondrial morphologies were determined as a percentage of the total number of cells counted (at least 100 cells per experiment).
Immunofluorescence
AT-II cells were incubated with MitoTracker Green FM for 1h at 37℃. After washing with DMEM three times, cells were fixed in 95% ethanol. Then, cells were incubated with primary anti-Cyt C antibody at 4℃ for 24 h followed by staining with Alexa Fluor 555 labeled secondary antibody for 1h at room temperature, respectively. Photographs were taken by using a confocal fluorescence microscope (Nikon, Japan). Cells were counted as having released Cyt-c if there was a loss of tubule staining. Cells were evaluated by 2 blinded observers. Results are expressed as cells releasing Cyt-c as the percentage of total cells counted.
Western blot analysis
Whole cell and mitochondrial lysates were papered by using lysis buffer (Beyotime Biotechnology, China) for 30 min on ice. Subsequently, the cell lysates were centrifuged at 12000g for 15 min at 4˚C. The supernatants were collected and the protein concentration was detected using the bicinchoninic acid (BCA) protein assay kit (Thermo Fisher, Waltham, MA, USA). Equal amounts of protein (40 μg/lane for whole cell protein, 20μg/lane for mitochondria protein) were loaded onto a polyacrymide gel and run at 80 V for 1 h, followed by a 120 V run for 30 min, and then transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA) by electroblotting. After blocking with 5% skimmed powdered milk for 2 h, the membranes were correspondingly incubated with anti-Mfn2 (1:1,000), anti-OPA1 (1:1,000), anti-Drp1 (1:1,000), anti-Fis1 (1:1,000) and β-actin (1:5,000) or Cox IV (1:1,000) overnight at 4˚C. The membranes were then washed in Tris‑buffered saline‑Tween‑20 (TBST) 3 times for 10 min each and further incubated with horseradish peroxidase (HRP)‑conjugated goat anti‑rabbit secondary antibodies (Biogot Technology, China) for 2 h at room temperature and detected using an chemiluminescence (ECL) detection system (Santa Cruz Biotechnology, Co., Ltd., USA).
Statistical analysis
Data were expressed as means ± SD and analyzed using SPSS 19.0 software (SPSS Inc., Chicago, IL, USA). GraphPad Prism 6 (GraphPad Software Inc., La Jolla, CA, USA) was used to prepare the figures. Difference between two groups were analyzed by two-tailed Student's test and one-way analysis of variance (ANOVA) was used for comparing three or more group means. P values< 0.05 were considered statistically significant.
Results
PQ inhibits cell viability and induces apoptosis in AT-II cells
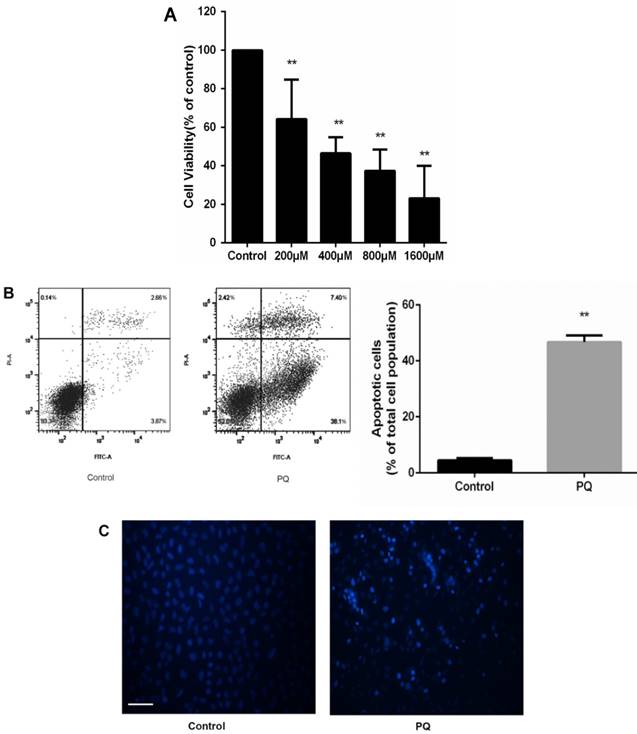
To investigate the cytotoxic effects of PQ on AT-II cells, we incubated the cells with PQ (200 - 1600μM) for 24 h. CCK-8 assay was used to assess cell viability and the mean inhibitory concentration (IC50) was calculated. As show in Fig 1A, cell viability was reduced to 61, 46, 37 and 23% after 200, 400, 800 and 1600μM PQ treatment for 24h. The IC50 value of PQ against AT-II cells was 675μM. Then, AT-II cells were treated with 600 μM PQ for 24 h and the early apoptotic cell was determined by Annexin V-FITC/PI staining. Additionally, Hoechst 33342 staining for double stranded DNA was used to observe nuclear fragmentation or apoptotic body. As shown in Fig. 1B, treatment of 600 μM for 24 h significantly enhanced early apoptosis of AT-II cells compared with control group (P<0.01). Similarly, the fluorescent microscopic images showed increased number of Hoechst 33342 positive cells after PQ treatment (Fig 1C).
PQ induces Cyt-c release in AT-II cells
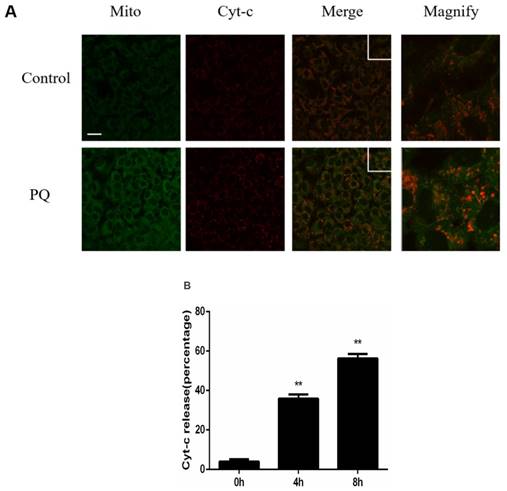
Previously study illustrated that mitochondria play a crucial role in PQ-induced cell apoptosis, and release of Cyt-c from mitochondria is a key initiative step in apoptotic process. Therefore, we investigated the effect of PQ stimulation on the release of Cyt-c. As shown in Figure 2A, Mitochondria and Cyt-c were co-stained in AT-II cells and PQ group released increased Cyt-c. Time course experiments (Figure 2B) revealed that Cyt-c released from mitochondrial in a time-dependent manner after PQ treatment (P < 0.01). These results suggested PQ induced Cyt-c release in AT-II cells.
PQ reduces cell viability and induces apoptosis of mouse AT-II cells (A) Mouse AT-II cells were treated with PQ (200, 400, 800, 1600 μM) for 24 h. The values are given as the means ± SD from 4 independent experiments. The IC50 value of PQ against AT-II cells was 675μM. ** P < 0.01 vs control group. (B) Mouse AT-II cells were treated with 600 μM PQ for 24 h. The apoptosis of the cells was detected by using a FACSalibur flow cytometer. The values are given as the means ± SD from 4 independent experiments. ** P < 0.01 vs control group. (C) Mouse AT-II cells were treated with 600 μM PQ for 24 h and then stained with Hoechst 33342. The cells were detected by a fluorescence microscope (Scale bar: 100 μm).
PQ induces release of Cyt-c in mouse AT-II cells. (A) Mouse AT-II cells were treated with 600 μM PQ for 8 h. Cyt-c was visualized by staining with anti-Cyt-c antibody, followed by secondary antibody. The mitochondria were visualized by Mito-Green staining and were observed under a confocal fluorescence microscope. (Images on the right side are magnified views of the boxed areas on its left side. Scale bar: 20 μm) (B) Mouse AT-II cells were treated with 600 μM PQ for 0, 4 and 8 h. The cells with released Cyt-c were counted. The values are represented the means ± SD of three independent experiments (at least 100 cells per experiment). ** P < 0.01, vs 0 h group.
Effect of PQ on mitochondrial networks in AT-II cells
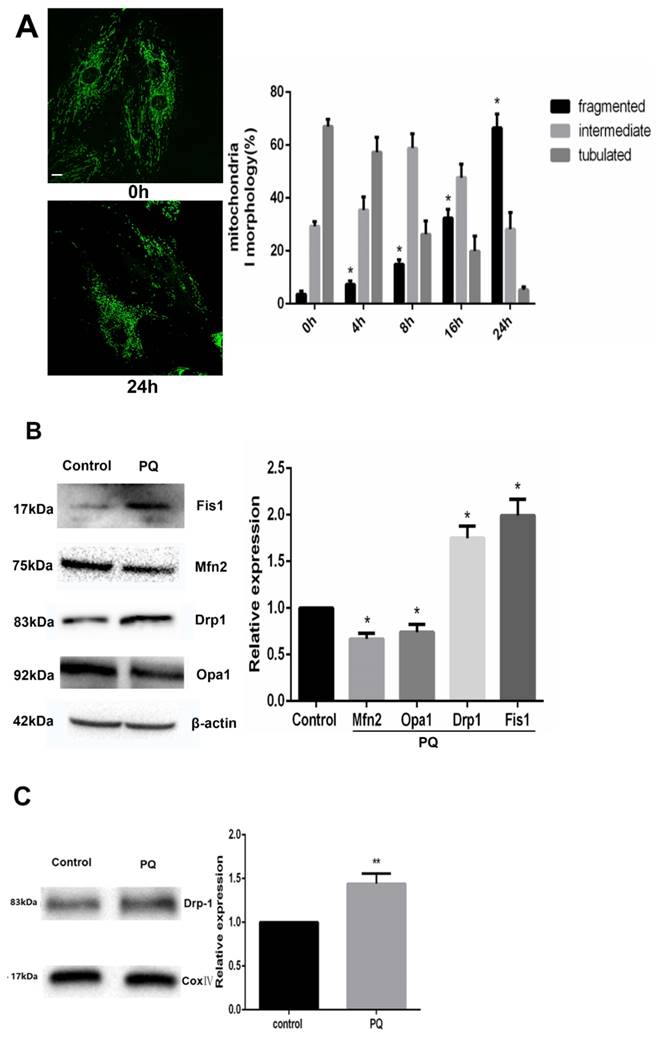
As PQ was proved to induce cells to release increased Cyt-c, we examined the alteration of mitochondrial dynamics in AT-II cells with or without PQ stimulation using fluorescence microscopy in the present study. As shown in Fig 3A, the mitochondria in control cells were filamentous or of long tubular like appearance. However, following exposure to PQ, normal tubular mitochondria network broke down and became short and small within 24 h which indicating mitochondrial fragmentation. Further, the percentage of fragmented mitochondria increased with culture time following exposure to PQ while the percentage of tubulated mitochondria decreased.
Effect of PQ on mitochondria-shaping protein expression and Drp1 mitochondrial translocation in AT-II cells
Mitochondrial morphology is controlled by mitochondria-shaping proteins. As shown in Fig 3B. Exposure to PQ increased protein levels of the fission protein Fis1 as well as Drp1 (P<0.05). In contrast, the fusion proteins Mfn2 and OPA1 were significantly decreased after PQ stimulation (P<0.05). As Drp1 mitochondrial translocation plays a crucial role in mitochondria fission, we further investigated the protein levels of Drp1 in mitochondria. As shown in Fig 3C, the protein levels of Drp1 in mitochondria was higher in PQ group compared with control group (P<0.01).
Inhibition of mitochondrial fission prevents PQ-induced apoptosis and Cyt-c release
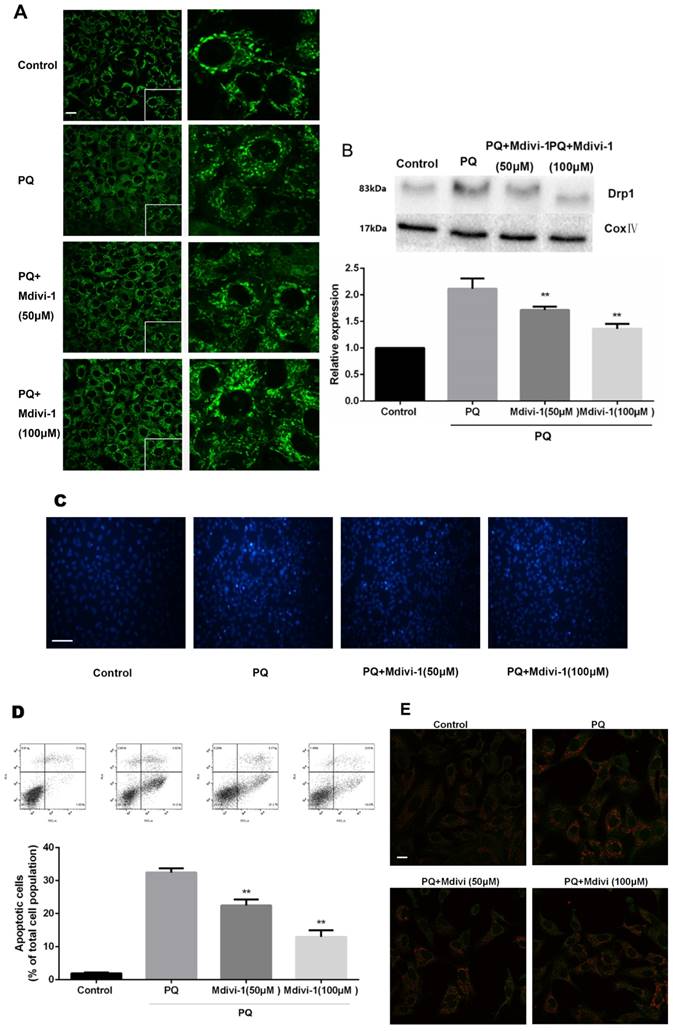
To investigate the role of the change of mitochondrial morphology in PQ-induced cell apoptosis, Mdivi-1, an effective inhibitor for mitochondrial division [24], was chosen for experiment. As shown in Fig 4A-B, under treatment of PQ, Mdivi-1 at 50 and 100μM significantly decreased fragmentation of the mitochondria network and mitochondrial Drp1 protein levels (P<0.01). Besides, among cells stained with Hoechst 33342, cells at 50 and 100μM of Mdivi-1 group were accompanied with much less apoptotic body than PQ group (Fig 4C). Meanwhile, the early apoptotic cells markedly decreased after 50 and 100μM Mdivi-1 stimulation in PQ group (P<0.01) (Fig 4D). In line with above observation, Cyt-c released from mitochondrial strongly decreased after stimulation of Mdivi-1 (Fig 4E).
PQ induces mitochondrial fission in AT-II cells. (A) Mouse AT-II cells were treated with 600 μM PQ for 0, 4, 8, 16, 24 h. Cells were expressing mito-GREEN and were observed under a fluorescence microscope. The values are expressed as the means ± SD from 5 independent experiments. * P < 0.05, vs 0 h group. (Scale bar: 10 μm) (B) Protein expression of Mfn2, OPA1, Drp1, Fis1 in mouse AT-II cells following exposure to 600 μM PQ for 24 h. The protein expression of Mfn2, OPA1, Drp1 and Fis1 was measured by western blot analysis. The values are given as the means ± SD from 5 independent experiments. * P < 0.05, vs control group. (C) The mitochondrial location of Drp1 was detected using western blotting. The Cox IV was used as the loading control to normalize Drp1 levels. The values are given as the means ± SD from 4 independent experiments. ** P < 0.01, vs control group
The mitochondrial morphology, apoptosis and the release of Cyt-c of mouse AT-II cells after co-incubated with PQ and Mdivi-1. Mouse AT-II cells were treated with 600 μM PQ and Mdivi-1 (50 and 100 μM) for 24 h. (A) Cells were expressing mito-GREEN and were observed under a fluorescence microscope. (Images in the right row are magnified views of the boxed areas in left row. Scale bar: 20 μm) (B) The mitochondrial location of Drp1 was determined by using western blot. The Cox IV was used as the loading control. The values are expressed as the means ± SD from 4 independent experiments. ** P < 0.01, vs PQ group. (C) Cells were stained with Hoechst 33342 and observed by fluorescence microscope. (Scale bar: 100 μm) (D) The apoptosis of the cells was detected by using a FACSalibur flow cytometer. The values are expressed as the means ± SD from 5 independent experiments. ** P < 0.01, vs PQ-treated group. (E) Cyt-c was visualized by staining with anti-Cyt-c antibody, followed by secondary antibody. Cells were observed under a fluorescence microscope. (Scale bar: 20 μm).
Inhibition of mitochondrial fission prevents PQ-induced ROS production
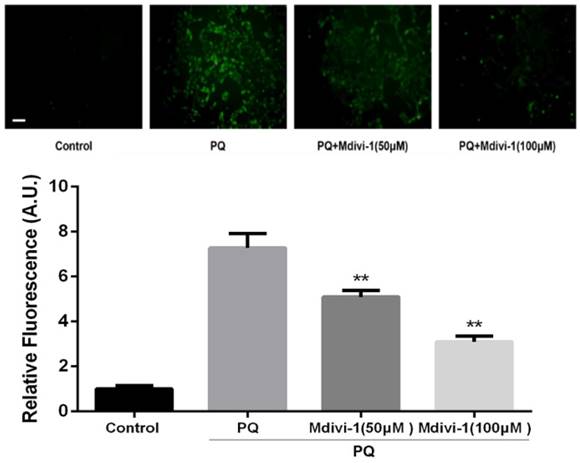
ROS has been reported as an important mediator in PQ-induced cell apoptosis. DCFH-DA, a ROS-sensitive fluorometric probe, was used to measure ROS generation in present study. As shown in Fig.5, PQ treatment significantly enhanced ROS production in AT-II cells. Interestingly, inhibiting mitochondria fission by Mdivi-1 decreased the ROS production after PQ stimulation compared with PQ group (P<0.01).
Ascorbic Acid alleviates PQ-induced mitochondrial fragmentation, Drp1 mitochondrial translocation and cell apoptosis
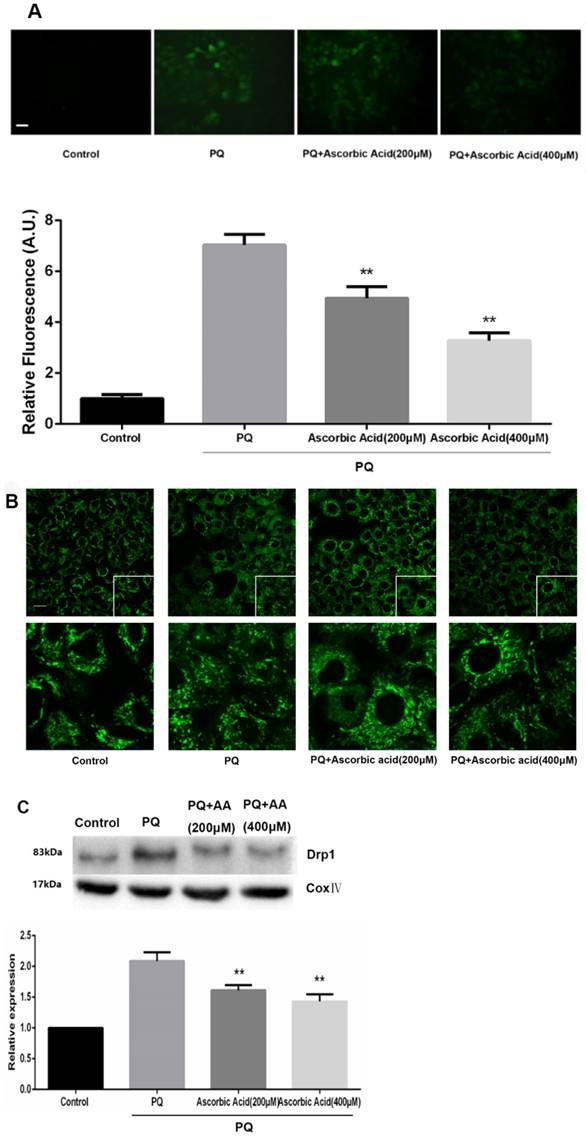
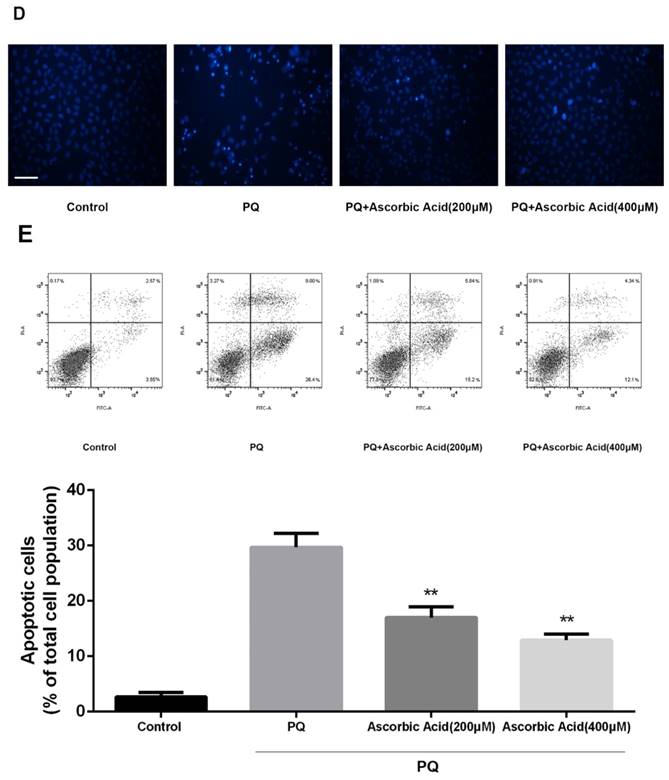
To further examine whether antioxidant attenuated mitochondrial fission upon PQ treatment, we imaged mitochondrial morphology change in the presence of ascorbic acid. As shown in Fig.6A, treatment with ascorbic acid significantly down-regulated the generation of ROS after PQ stimulation (P<0.01) (Fig.6A). Interestingly, ascorbic acid-treated cells performed enhanced tubular mitochondria compared with untreated groups following exposed to PQ (Fig.6B). Drp1 mitochondrial translocation in PQ-treated cells was also alleviated by ascorbic acid stimulation (P<0.01) (Fig.6C). Besides, we also tested the apoptosis after co-treatment with ascorbic acid and PQ in AT-II cells. As shown in Fig 6D, these PQ-treated groups with ascorbic acid presented decreased apoptotic body, indicating that ascorbic acid inhibited apoptosis. Furthermore, with the concentration of ascorbic acid increased, the effect of anti-apoptosis arose accordingly. Meanwhile, the early apoptotic cells decreased significantly after 200 and 400μM ascorbic acid stimulation in PQ group (P<0.01) (Fig 6E).
Discussion
Previous studies have been illustrated that the apoptosis of AT II cells through the mitochondrial pathway contributes to PQ-induced pulmonary fibrosis [1, 25, 26, 27]. PQ exerted oxidative damage and cell death through impairing mitochondrial function and membrane permeability [10, 28], indicating mitochondria have a crucial role in PQ- induced apoptosis. Nevertheless, the relationship between mitochondria and oxidative stress during PQ intoxication remains largely elusive. In this study, it has been expected that PQ promotes cell apoptosis and induces Cyt-c release, correspond with most studies. Besides, we found that mitochondria become fragmented after incubated with PQ in AT-II cells. Importantly, prevention of mitochondrial fission alleviates Cyt-c release, cell apoptosis as well as ROS production. Interestingly, antioxidant ascorbic acid not only inhibits oxidative stress and cell apoptosis but also abrogates mitochondrial fragmentation.
ROS production in mouse AT-II cells after co-incubated with PQ and Mdivi-1. Mouse AT-II cells were treated with 600 μM PQ and Mdivi-1 (50 and 100 μM) for 24 h. Then cells were stained with DCFH-DA and observed by a fluorescence microscope. The values are expressed as the means ± SD from 6 independent experiments. ** P < 0.01, vs PQ-treated group. (Scale bar: 50 μm)
Mitochondrial fragmentation, apoptosis and ROS production in mouse AT-II cells after co-incubated with PQ and Ascorbic Acid. Mouse AT-II cells were treated with 600 μM PQ and Ascorbic Acid (200 and 400 μM) for 24 h. (A) Cells were stained with DCFH-DA and observed by a fluorescence microscope. The values are expressed as the means ± SD from 6 independent experiments. ** P < 0.01, vs PQ-treated group. (Scale bar: 50 μm). (B) Cells were observed under a fluorescence microscope. (Images in the second row are magnified views of the boxed areas in first row. Scale bar: 20 μm). (C) The mitochondrial location of Drp1 was detected using western blotting. The Cox IV was used as the loading control to normalize Drp1 levels. The values are expressed as the means ± SD from 4 independent experiments. ** P < 0.01, vs PQ group. (D) Cells were stained with Hoechst 33342 and observed by a fluorescence microscope. (Scale bar: 100μm). (E) The apoptosis of the cells was detected by using a FACSalibur flow cytometer. The values are expressed as the means ± SD from 4 independent experiments. ** P < 0.01, vs PQ-treated group.
Mitochondrial morphology possessing two opposing processes: fusion and fission. Imbalance of mitochondrial fusion and fission is thought to play a crucial role in pathological conditions [29]. For example, Jheng et al. showed mitochondrial fragmentation contributes to insulin resistance in skeletal muscle [30]. Besides, previous study has illustrated the relationship between mitochondrial dynamics and cell apoptosis [31]. Mizumura et al. proved that mitochondrial fission inhibitor Mdivi-1 protected against cigarette smoke (CS)-induced pulmonary epithelial cell death and mitochondrial dysfunction in vitro [32]. Dong et al. showed evidences that Drp1 mitochondrial location and increased mitochondria fission contribute to cardiomyocyte death [33]. However, some studies showed that mitochondrial fission may be not involved in the process of apoptosis [34, 35]. For example, Philippe et al. showed that alleviating mitochondrial fragmentation does not decrease cell death induced by Bax/Bak-dependent apoptosis stimuli indicating mitochondria fission is a dispensable event in this process [36]. The latest work by Lefevre et al. showed that deletion of Fis1 did not cause lifespan extension in yeast peroxisome-deficient cells [37]. In this study, we demonstrated convincing evidence that PQ induced a dramatic increases in fragment mitochondria compared with untreated cells. Besides, by detecting the release of Cyt-c, we found PQ aggrandized Cyt-c release from mitochondria. Of note, in the study of Western blotting, we found that Drp1 and Fis1 which recruitment mitochondrial fission showed a significant increase while Opa1 and Mfn2, both playing a leading role in mitochondrial fusion, declined apparently in PQ-treated group. Additionally, the redistribution of Drp1 to mitochondria increased significantly in response to PQ. These results suggested that PQ facilitated mitochondrial fission through upregulation of fission-related proteins.
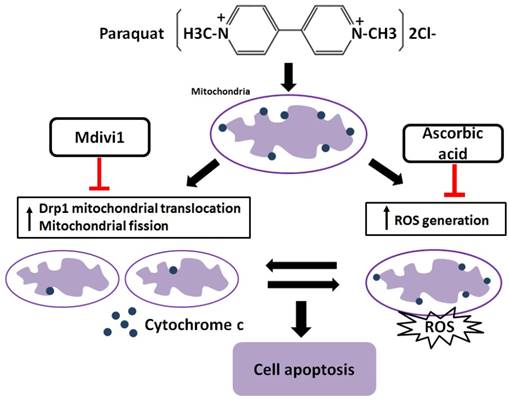
It has been reported that increased generation of ROS requires dynamic change of mitochondrial fission [38], and Drp1 play a major role in regulating ROS production in apoptosis [39]. Previous studies showed that the induction of the peroxidase activity of Cyt-c is a key event in early apoptosis [40], while the production of ROS, one of the primary factor of oxidative stress, is determined by Cyt-c-oxidase [41]. In this study, during AT-II cells of PQ intoxication, we detected the ROS production and release of Cyt-c and found that suppression of Drp1 by Mdivi-1 can block ROS generation and Cyt-c release. These results suggest that oxidative stress may be activated by mitochondrial fission and contributes to cell apoptosis. To further detect the potential relationship between oxidative stress and mitochondrial fission, ascorbic acid, an antioxidant, was employed in PQ-induced apoptosis system. Of note, co-treatment with ascorbic acid not only significantly reduced apoptosis and the generation of ROS, but also attenuated mitochondrial fragmentation, suggesting inhibiting generation of ROS may suppress the mitochondrial fission. Similarly, other studies indicated that mitochondrial fission contributes to the generation of ROS, while oxidative stress causes an imbalance in mitochondrial fission-fusion which leading to mitochondrial fragmentation [42]. Zhang and his colleagues proved ROS effects pulmonary artery smooth muscle cells (PASMCs) apoptosis through regulating Drp1 [43] and Zang et al. discovered that sepsis induces mitochondrial fission through ROS in heart [44]. In addition, recent evidence showed ROS promotes to progressive reduction of the number of mtDNA copies which contributes to impairment of the electron transport chain, mitochondrial uncoupling and bioenergetics dysfunction of mitochondria [45], a mechanism that would contribute to explaining the inherent relationship between oxidative stress and mitochondrial fission. Taken together, we illustrate a molecular apoptotic model in which PQ induces oxidative stress and mitochondrial fission in mouse AT-II cells, whereas inhibits either of them could suppress the other (Fig 7).
Crosstalk between mitochondrial fission and oxidative stress in paraquat-induced apoptosis in mouse alveolar type II (AT-II) cells. Stimulation of PQ leading to increased ROS generation and mitochondrial fission in AT-II cells from mouse. Inhibiting Drp1 mitochondrial translocation and mitochondrial fission by Mdivi-1 suppresses oxidative stress, Cyt-c release and cell apoptosis. Additionally, downregulation of ROS generation by ascorbic acid inhibits Drp1 mitochondrial translocation, mitochondrial fission as well as cell apoptosis. --|, inhibit.
In conclusion, the present study demonstrates that PQ induces oxidative stress and apoptosis through mitochondrial fission, and elucidates the underlying internal connection between mitochondrial fission and oxidative stress in PQ-induced apoptosis, hence providing further understanding of the biochemical mechanisms in PQ-induced apoptosis. Taken together, mitochondrial fission plays an important role in regulating PQ-induced cytotoxicity, supporting mitochondrial fission as a potential therapeutic target in PQ poisoning.
Abbreviations
PQ: paraquat; ROS: reactive oxygen species; Cyt-c: cytochrome-c; Mfn2: mitofusin 2; OPA1: optic atrophy protein 1; Fis1: fission 1; Drp1: dynamin-related protein 1; Mdivi-1: mitochondrial division inhibitor 1; NOS: nitric oxide synthase; PASMCs: pulmonary artery smooth muscle cells.
Acknowledgements
This work was supported, in part, by grants from Medical and Health Platform Backbone Personnel Plan of Zhejiang Province (2016RCA021) and grants from Wenzhou Municipal Science and Technology Project (Y20140673, Y20140244).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Dinis-Oliveira RJ, Duarte JA, Sanchez-Navarro A, Remiao F, Bastos ML, Carvalho F. Paraquat poisonings: mechanisms of lung toxicity, clinical features, and treatment. Critical reviews in toxicology. 2008;38:13-71
2. Rio MJ, Velez-Pardo C. Paraquat induces apoptosis in human lymphocytes: protective and rescue effects of glucose, cannabinoids and insulin-like growth factor-1. Growth factors. 2008;26:49-60
3. Chang X, Lu W, Dou T, Wang X, Lou D, Sun X. et al. Paraquat inhibits cell viability via enhanced oxidative stress and apoptosis in human neural progenitor cells. Chemico-biological interactions. 2013;206:248-55
4. Rincheval V, Bergeaud M, Mathieu L, Leroy J, Guillaume A, Mignotte B. et al. Differential effects of Bcl-2 and caspases on mitochondrial permeabilization during endogenous or exogenous reactive oxygen species-induced cell death: a comparative study of H(2)O(2), paraquat, t-BHP, etoposide and TNF-alpha-induced cell death. Cell biology and toxicology. 2012;28:239-53
5. Li S, Zhao G, Chen L, Ding Y, Lian J, Hong G. et al. Resveratrol protects mice from paraquat-induced lung injury: The important role of SIRT1 and NRF2 antioxidant pathways. Mol Med Rep. 2016;13:1833-8
6. Castello PR, Drechsel DA, Patel M. Mitochondria are a major source of paraquat-induced reactive oxygen species production in the brain. The Journal of biological chemistry. 2007;282:14186-93
7. Cocheme HM, Murphy MP. Complex I is the major site of mitochondrial superoxide production by paraquat. The Journal of biological chemistry. 2008;283:1786-98
8. J OU, Ryu SY, Jhun BS, Hurst S, Sheu SS. Mitochondrial ion channels/transporters as sensors and regulators of cellular redox signaling. Antioxidants & redox signaling. 2014;21:987-1006
9. Hong GL, Liu JM, Zhao GJ, Wang L, Liang G, Wu B. et al. The reversal of paraquat-induced mitochondria-mediated apoptosis by cycloartenyl ferulate, the important role of Nrf2 pathway. Experimental cell research. 2013;319:2845-55
10. Huang CL, Chao CC, Lee YC, Lu MK, Cheng JJ, Yang YC. et al. Paraquat Induces Cell Death Through Impairing Mitochondrial Membrane Permeability. Molecular neurobiology. 2016;53:2169-88
11. Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:2653-8
12. Thomas KJ, Jacobson MR. Defects in mitochondrial fission protein dynamin-related protein 1 are linked to apoptotic resistance and autophagy in a lung cancer model. PloS one. 2012;7:e45319
13. Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:10190-5
14. Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE. et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation. 2011;124:444-53
15. Huang Q, Zhan L, Cao H, Li J, Lyu Y, Guo X. et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy. 2016;12:999-1014
16. Twig G, Elorza A, Molina AJA, Mohamed H, Wikstrom JD, Walzer G. et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. Embo J. 2008;27:433-46
17. Loson OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Molecular biology of the cell. 2013;24:659-67
18. Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q. et al. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nature cell biology. 1999;1:298-304
19. Loson OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Molecular biology of the cell. 2013;24:659-67
20. Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K. et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nature cell biology. 2009;11:958-66
21. Yeon JY, Min SH, Park HJ, Kim JW, Lee YH, Park SY. et al. Mdivi-1, mitochondrial fission inhibitor, impairs developmental competence and mitochondrial function of embryos and cells in pigs. J Reprod Develop. 2015;61:81-9
22. Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F. et al. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Developmental cell. 2001;1:515-25
23. Ding YW, Zhao GJ, Li XL, Hong GL, Li MF, Qiu QM. et al. SIRT1 exerts protective effects against paraquat-induced injury in mouse type II alveolar epithelial cells by deacetylating NRF2 in vitro. International journal of molecular medicine. 2016;37:1049-58
24. Kim H, Lee JY, Park KJ, Kim W-H, Roh GS. A mitochondrial division inhibitor, Mdivi-1, inhibits mitochondrial fragmentation and attenuates kainic acid-induced hippocampal cell death. BMC Neuroscience. 2016:17
25. Liu Z, Zhao H, Liu W, Li T, Wang Y, Zhao M. NLRP3 inflammasome activation is essential for paraquat-induced acute lung injury. Inflammation. 2015;38:433-44
26. He Y, Zou L, Zhou Y, Hu H, Yao R, Jiang Y. et al. Adiponectin ameliorates the apoptotic effects of paraquat on alveolar type cells via improvements in mitochondrial function. Mol Med Rep. 2016;14:746-52
27. Li G, Yuzhen L, Yi C, Xiaoxiang C, Wei Z, Changqing Z. et al. DNaseI protects against Paraquat-induced acute lung injury and pulmonary fibrosis mediated by mitochondrial DNA. BioMed research international. 2015;2015:386952
28. Huang CL, Lee YC, Yang YC, Kuo TY, Huang NK. Minocycline prevents paraquat-induced cell death through attenuating endoplasmic reticulum stress and mitochondrial dysfunction. Toxicology letters. 2012;209:203-10
29. Rosdah AA, Holien J, Delbridge LMD, Dusting GJ, Lim SY. Mitochondrial fission - a drug target for cytoprotection or cytodestruction?. Pharmacology Research & Perspectives. 2016;4:e00235
30. Jheng HF, Tsai PJ, Guo SM, Kuo LH, Chang CS, Su IJ. et al. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Molecular and cellular biology. 2012;32:309-19
31. Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev. 2008;22:1577-90
32. Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T. et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. 2014;124:3987-4003
33. Dong Y, Undyala VVR, Przyklenk K. Inhibition of mitochondrial fission as a molecular target for cardioprotection: critical importance of the timing of treatment. Basic Res Cardiol. 2016:111
34. James DI, Martinou JC. Mitochondrial dynamics and apoptosis: A painful separation. Developmental cell. 2008;15:341-3
35. Perfettini JL, Roumier T, Kroemer G. Mitochondrial fusion and fission in the control of apoptosis. Trends Cell Biol. 2005;15:179-83
36. Parone PA, James DI, Da Cruz S, Mattenberger Y, Donze O, Barja F. et al. Inhibiting the mitochondrial fission machinery does not prevent Bax/Bak-dependent apoptosis. Molecular and cellular biology. 2006;26:7397-408
37. Lefevre SD, Kumar S, van der Klei IJ. Inhibition of peroxisome fission, but not mitochondrial fission, increases yeast chronological lifespan. Cell cycle. 2015;14:1698-703
38. Yu TZ, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:2653-8
39. Zhang L, Ma C, Zhang C, Ma M, Zhang F, Zhang L. et al. Reactive oxygen species effect PASMCs apoptosis via regulation of dynamin-related protein 1 in hypoxic pulmonary hypertension. Histochemistry and cell biology. 2016;146:71-84
40. Abe M, Niibayashi R, Koubori S, Moriyama I, Miyoshi H. Molecular mechanisms for the induction of peroxidase activity of the cytochrome c-cardiolipin complex. Biochemistry. 2011;50:8383-91
41. Vogt S, Rhiel A, Weber P, Ramzan R. Revisiting Kadenbach: Electron flux rate through cytochrome c-oxidase determines the ATP-inhibitory effect and subsequent production of ROS. BioEssays: news and reviews in molecular, cellular and developmental biology. 2016;38:556-67
42. Wu S, Zhou F, Zhang Z, Xing D. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. The FEBS journal. 2011;278:941-54
43. Zhang LX, Ma C, Zhang C, Ma MF, Zhang FY, Zhang LL. et al. Reactive oxygen species effect PASMCs apoptosis via regulation of dynamin-related protein 1 in hypoxic pulmonary hypertension. Histochemistry and cell biology. 2016;146:71-84
44. Zang QS, Yao X, Wolf SE, Minei J. Sepsis Induces Mitochondrial Ros-Dependent Mitochondrial Fission In the Heart. Shock. 2015;43:54-5
45. Ikeda Y, Sciarretta S, Nagarajan N, Rubattu S, Volpe M, Frati G. et al. New insights into the role of mitochondrial dynamics and autophagy during oxidative stress and aging in the heart. Oxidative medicine and cellular longevity. 2014;2014:210934
Author contact
![]() Corresponding author: Zhongqiu LU, Emergency Department, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou 325000, P.R. China; Tel: (+86) 57755578563; E-mail: lzq_640815com
Corresponding author: Zhongqiu LU, Emergency Department, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou 325000, P.R. China; Tel: (+86) 57755578563; E-mail: lzq_640815com
Received 2016-11-23
Accepted 2017-3-10
Published 2017-7-7