ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2018; 14(2):165-177. doi:10.7150/ijbs.23477 This issue Cite
Research Paper
Loss of p21 promoted tumorigenesis in the background of telomere dysfunctions induced by TRF2 and Wrn deficiency
Xiaoyu Si1,2*, Chihao Shao1*, Jing Li1,3, Shuting Jia1, Wenru Tang1, Jihong Zhang1, Julun Yang3, Xiaoming Wu1 ![]() , Ying Luo1,2,4
, Ying Luo1,2,4 ![]()
1. Lab of Molecular Genetics of Aging & Tumor, Faculty of Medicine, Kunming University of Science & Technology, 727 South Jing Ming Road, Chenggong County, Kunming, Yunnan Province, China, 650500
2. College of Biological Engineering, Qilu University of Technology (Shandong Academy of Sciences), 3501 University Road, Changqing County, Jinan, Shandong Province, China, 250353
3. Faculty of Environmental Science and Engineering, Kunming University of Science & Technology, 727 South Jing Ming Road, Chenggong County, Kunming, Yunnan Province, China, 650500
4. Department of Pathology, Kunming General Hospital, 212 Daguan Road, Kunming, Yunnan Province, China, 650032
5. Yunnan Provincial Institute of Digestive Disease
*These authors contribute equally to this work.
Abstract

Werner syndrome (WS) is a rare autosomal recessive progeria disease with genetic instability/cancer predisposition, thus a good model in understanding aging related carcinogenesis. Telomere dysfunction induced cellular senescence is essential in the manifestation of the WS phenotype. Our previous data has shown that p21 (encoded by Cdkn1a gene) could induce cellular senescence and suppress cellular growth of ALT (alternative lengthening of telomere) tumors derived from WS, suggested that p21 might play a key role in maintaining senescence of WS cells.
To confirm the role of p21 in suppressing telomere dysfunction induced tumorigenesis, we overexpressed dominant negative protein TRF2ΔBΔM in p21-/- mouse embryonic fibroblasts (MEFs). To further stress the cell, we crossed Wrn-/- mice with p21-/- mice to obtained p21-/-Wrn-/- MEFs, and overexpressed TRF2ΔBΔM in these MEFs to induce telomere dysfunction similar to that in WS cells. Our data showed that, in the context of p21-/- TRF2ΔBΔM, loss of p21 function rescued cellular senescence, and induced p53 mutation, but did not induce tumorigenesis. However, in the set of p21-/-Wrn-/-TRF2ΔBΔM, loss of p21 function induced p53 mutation and tumorigenesis.
To further verify the role of p21 in suppressing telomere dysfunction related tumorigenesis, we knocked down p21 in non-tumorigenic immortalized cells derived from WS MEFs (mTerc-/-Wrn-/-), and found that loss of p21 could induce ALT tumorigenesis, which displayed typical smear pattern of telomere length and arc-shaped telomeric DNA. In another hand, recovering telomerase activity in these MEFs could also induce tumorigenesis without affecting p21 expression level. Together our data suggested that p21 controlled cell cycle regulation played an essential role in suppressing telomere dysfunction-related tumorigenesis. These data also suggested that the genetic context is essential in determining the role of p21 in cancer prevention. Therefore, targeting p21 in the treatment of human degenerative diseases would require a personalized genetic background screen.
Keywords: p21, Wrn, telomere dysfunction, senescence, tumorigenesis
Introduction
Werner syndrome protein (WRN in human, Wrn in mouse) is one member of the RecQ helicase family implicated in the maintenance of genome stability. Defects in WRN cause human pathologies linked with cancer predisposition and premature aging, such as Werner syndrome (WS)1-5. WS is a rare autosomal recessive genetic instability/cancer predisposition disorder that displays many symptoms of premature aging6. Therefore, WS is a good model in studying the crosstalk between aging and cancer. However, mice with Wrn deletion did not develop any obvious aging phenotypes7, 8. It is observed that fibroblasts and B-lymphoblastoid cell lines derived from WS patients exhibited accelerated rates of telomere shortening compared to age-matched control cells9, 10 and senescence of fibroblasts from WS patients could be rescued by the overexpression of telomerase11. These data suggested that telomere dysfunction might play an essential role in the manifestation of the WS phenotypes. Different from human, adult mouse somatic cells still maintain telomerase activity and chromosomes of mouse cells contain long telomere, which might prevent the progeria phenotypes in Wrn deficient mice12. Two laboratories independently generated mice with both telomerase and Wrn deficiency. The late generation (G4-6) of mTerc-/-Wrn-/-mice exhibited many of the clinical features observed in WS patients, including early onset of wound-healing defects, osteoporosis with skeletal fractures, hypogonadism, cataract formation, type II diabetes and short life span12, 13. While G3 mTerc-/-Wrn-/-mice with longer telomeres did not show these premature aging phenotypes, suggesting that loss of Wrn in the setting of short telomere is necessary for the manifestation of WS in mice. At cellular level, Wrn deficiency and telomere dysfunction cooperated to accelerate telomere shortening, resulting in accelerated cellular senescence. Elevated telomere-telomere recombinetion in these cells promoted escape from senescence and engagement of the ALT tumorigenesis14.
Our previous study has shown that all those ALT tumorigenic cell lines (SCID3B-1, SCID3B-2, SCID9B-1) derived from senescent WS MEFs gained the same point mutation in tumor suppressor gene Trp53, encoded a mutant p53 protein, p53N236S, which promoted the ALT tumorigenesis and abrogated the expression of p21Cip1/Waf1 (referred as p21 later) and p16Ink4a (referred as p16 later). While those non-tumorigenic immortalized cell lines (395-6B-1, 395-7A-1, 395-7A-2, 395-7A-3) derived from senescent WS MEFs maintained wild type p53, as well as p21 expression15. Our further study showed that overexpression of p21 into these ALT tumorigenic cell lines demethylated p16 promoters and restored cellular senescence16, suggesting p21 played an essential role in inhibiting ALT tumorigenesis.
The p21 protein encoded by cyclin-dependent kinase inhibitor 1A (Cdkn1a) is a negative regulator of the cell cycle and belongs to the Cip and Kip family of Cdk inhibitors. It functions as the mediator for p53 tumor suppression17. The p21 also plays important role in connecting the two key pathways for cellular senescence: p53-Rb and p16-Rb18, 19. After 20 years of study, growing evidence reveals that p21 expression can both promote and inhibit tumorigenesis, depending on the cellular context20. Thus, it is interesting to study the consequence of loss of p21 function in the background of telomere dysfunction.
It has been reported that in the background of Terc-/-mice, deletion of p21 prolonged the lifespan of telomerase-deficient mice with dysfunctional telomeres and did not accelerate chromosomal instability or cancer formation21. Similarly in the background of Wrn-/- mice, deletion of p21 did not show any acceleration of tumorigenesis during the first year of life22. However, as mentioned above, when generation of WS mouse model, telomere dysfunction is necessary for the manifestation of WS in mice and single Wrn null mice did not develop any obvious aging phenotypes. It will be interesting to know the effect of loss of p21 function in the background of both telomere dysfunction and Wrn null. As the essential components in shelterin complex, telomeric repeat binding factors TRF1 and TRF2 are present at telomeres throughout the cell cycle. TRF1 is an inhibitor of telomerase, acting in cis to negatively regulate telomere length, while TRF2 protects telomeres from DNA damage repair process and end-to-end chromosome fusions23-25. TRF2ΔBΔM is the dominant negative form of TRF2 that lacks both the basic and Myb domain and could sequester endogenous TRF2 away from telomere ends, thus induces telomere dysfunction23. We overexpressed TRF2ΔBΔM in Wrn-/- p21-/- MEFs to induce multiple telomere dysfunction to research the effect of loss of p21 function with multiple telomere dysfunction induced by TRF2ΔBΔM and Wrn-/-.
Materials and Methods
Cell Lines, Constructs and Antibodies
The immortalized cell lines 395-3B-1, 395-9B-1, 395-6B-1, 395-7A-1were established as described14. Transgenic MEFs were prepared and harvested from individual day 13.5 mouse embryos from designed parental mice with p21+/- or Wrn+/- genotypes (heterozygous knockouts of either p21 or WRN) and the harvested MEFs were cultured and genotyped. MEFs with wild type, p21-/-, Wrn-/- or p21-/-Wrn-/- genotypes were used for experiments. Wild-type MEFs were harvested and prepared from individual day 13.5 embryos obtained from wild-type C57/B6 mice. Wild-type MEFs were used for experiments prior to reaching passage 10, and the cells were passaged at a ratio of 1:2 or 1:3 so that they would not be diluted and become senescent quickly. Generally, for each genotype, more than three lines of MEFs were harvested for experiments and all cell lines were sequenced for p53 mutations. All cell lines were cultured in DMEM supplemented with 10% fetal bovine serum (Hyclone, CA) in a 3% oxygen, 5% CO2 incubator at 37°C.
The pBabe-TRF2ΔBΔM (with a cMyc tag) construct, a gift from Dr. de Lange's lab (as described in23), was introduced into MEFs. For transient transfection, cells were used 48h after transfection. For stable expression, antibiotic selection was applied for 1-2 months and expression of TRF2ΔBΔM was analyzed by both Western blot and immunostaining.
Antibodies used for western blot were anti-p16Ink4a (M-156) (1:500, Santa Cruz, CA, USA), anti-Chk2 (1:500, BD Transduction Laboratories, CA, USA), anti-phospho-p53 (Ser15) (1:500, Cell Signaling, MA, USA), anti-cMyc (9E10) (1:500, Santa Cruz, CA, USA), anti-p19Arf (1:1250, Upstate, NY, USA), anti-γ-H2AX (1:1000, Upstate, NY, USA), and anti-γ-tubulin (1:5000, Upstate, NY, USA).
Growth Curve Assay
A total of 2.5×104 cells were seeded per well in 12 well plates and triplicates were prepared for each sample. At each time point, cells were fixed for 20 min at room temperature in 10% buffered formalin and stained with 1 mL 0.1% crystal violet for 20 min at room temperature. After the plates were washed and dried, cells were treated with 2mL 10% acetic acid and the OD of the extracted dye was measured at 595nm. Statistical analysis was performed to compare the difference between cell lines (n=3).
Immunostaining Assay
Cells were fixed with 2% paraformaldehyde and 2% sucrose in 1× PBS (pH 7.6) for 10min and then permeabilized with 1% NP-40. After pre-incubation with 5% BSA/PBS, cells were incubated first with the primary antibody and then with the secondary antibody in 1% BSA/PBS for 1h at room temperature. After DAPI staining, the slides were mounted with ProLong mounting medium (Invitrogen, CA, USA). Fluorescence microscopy was performed using a Nikon Eclipse 90i fluorescence microscope and a Nikon Digital Sight CCD (controlled by NIS-Elements 3.0 software).
Transformation assay
1000 cells were seeded in a 10cm cell culture treated plate and cultured with DMEM supplemented with 10% fetal bovine serum. After cultured for 10-14 days, the plates were rinsed with PBS, and cells were fixed with 10% buffered formalin for 10min. The transformed cell colonies were stained with 0.01% crystal violet for 5min.
SA-β-Gal Staining
SA-β-Gal staining was performed as described previously26. Briefly, cultured cells were washed in 1×PBS and fixed for 3-5min (room temperature) in 2% formaldehyde, 0.2% glutaraldehyde. Fixed cells were stained with fresh stain solution for SA-β-galactosidase activity at 37°C for 4h. The percentage of cells positive for SA-β-Gal staining were quantified and statistically analyzed (n=3).
RNA interference of p21
The p21 sequence 5' CGGTGGAACTTTGACTTCG 3'27 was cloned into a pSUPER.retro vector (OligoEngine, WA). The shRNA construct was introduced into the non-tumorigenic, immortalized cell line 395-6B-1, 395-7A-1 and selected for stable expression. Cells with stable knock down of p21 were injected into SCID mice to observe tumorigenesis.
2D gel electrophoresis and terminal restriction fragment (TRF)-Southern blot
Telomeric circles were detected by neutral-neutral 2D gel electrophoresis as described28, 29. Briefly, 40 μg of genomic DNA was digested by Hinf I and Rsa I and resolved by size in the first dimension and by shape in the second dimension. In-gel hybridization was then performed using a TTAGGG telomeric probe. TRF-Southern blot was performed as described previously12.
Injection of cells into SCID mice and tumor cells harvest
1x 106 cells were injected subcutaneously into each site of SCID mice. When the size of the largest tumor reached 1 cm, the mice were sacrificed and the tumors were collected and digested in a tumor digesting cocktail (4 mg/ml collagenase D and 4 mg/ml dispase II). Isolated tumor cells were plated and cultured in DMEM supplemented with 10% fetal bovine serum. All mouse procedures were performed with the approval of the Animal Care and Use Committee of the Kunming University of Science & Technology.
Results
Loss of p21 function rescues cellular senescence induced by telomere dysfunction
Since p21 is a negative regulator of the cell cycle and play an important role in cellular senescence, loss of p21 function might change cellular growth potential and rescue cellular senescence induced by telomere dysfunction. To investigate the growth potential of MEFs with the genotypes p21-/-, Wrn-/- and p21-/-Wrn-/-, we performed the growth curve analysis using wild types (WT) MEFs as the control. We found that WT and Wrn-/- MEFs grew slower than p21-/- MEFs, while knock out of p21 function (p21-/-Wrn-/-) rescued the growth potential of Wrn-/- MEFs (Figure 1A). These data suggested that the growth barrier caused by dysfunction of Wrn deficiency could be overcome by the loss of p21 function. We further detected the proliferation cell nuclear antigen (PCNA) levels of MEFs with different genotypes in their early passages (passage number≤5) or late passages (passage number=10) by Western blot analysis. The results revealed that compared to their early passages, WT and Wrn-/- MEFs in late passages had dramatically decreased PCNA expression (Figure 1B, WT-p10, Wrn-/--p10), however p21-/-and p21-/-Wrn-/- MEFs in late passages still maintained similar level of PCNA expression as in early generation (Figure 1B, p21-/--p10 and p21-/-Wrn-/--p10). These results suggested that loss of p21 function facilitated celllular proliferation capacity. Our data also showed that compared to WT MEFs in late passage (Figure 1B, WT-p10), Wrn-/- MEFs in late passage showed elevated p19ARF (p19) and p16 expression (Figure 1B, Wrn-/--p10), suggesting that loss of Wrn function activated the expression of p19 and p16, which might cause the growth barrier of Wrn-/- MEFs due to negative regulation of the cell cycle.
We performed SA-β-Gal staining to detect cellular senescence in late passage of p21-/-, Wrn-/- and p21-/-Wrn-/- MEFs. The results indicated that abundant cellular senescence occurred in passage 13 of Wrn-/- MEFs (57.31±7.19% of cells were positive for SA-β-Gal staining), but not occurred in the same passage of p21-/- MEFs (1.31±0.56%of cells were positive for SA-β-Gal staining) or p21-/-Wrn-/- MEFs (2.55±0.27% of cells were positive for SA-β-Gal staining) (Figure 1C).
We then overexpressed the dominant negative protein TRF2ΔBΔM in these MEFs to disturb TRF2 function and introduced further telomere dysfunction. The SA-β-Gal staining data showed that even with telomere dysfunction caused by both Wrn and TRF2 dysfunction, loss of p21 function could still rescue the cellular senescence (Figure 1D).
Together these results suggested that loss of p21 function rescued cellular senescence induced by telomere dysfunction.
Surprisingly, we found that compared to the control WT MEFs, MEFs with p21 knocking out showed higher level of p16 proteins, suggested a compensatory overexpression of p16 induced by the loss of p21 function (Figure 1E), though those cells still underwent cell cycle progress. These results suggested an interactive play between p21 and p16 in progress of cellular senescence.
Loss of p21 function promoted chromosome doubling
Chromosome doubling is a frequent event occurred in the process of continuous cell propagation, and contributes to aneuploidy and tumorigenesis. Tetraploidization has been proposed as an intermediate step toward aneuploidy in human cancer, and tetraploidization was found in p53-deficient cells experiencing a prolonged DNA damage signal due to persistent telomere dysfunction30.
To understand more about the cellular consequences caused by loss of p21 function in the setting of telomere dysfunction, we analyzed karyotypes of WT, p21-/-, Wrn-/- and p21-/-Wrn-/- MEFs in their late passages (Table 1). We found that there were about 12.5% of cells in late passage of WT MEFs showed chromosome doubling, while the percentile is 65.9% in the late passage of p21-/- MEFs and 52.2% in the late passage of p21-/-Wrn-/- MEFs. We then overexpressed TRF2ΔBΔM in the late passages of p21-/- and p21-/-Wrn-/- MEFs and analyzed the karyotypes. The introduction of TRF2ΔBΔM in p21-/- MEFs increased the proportion of cells with chromosome doubling to 87.2%. Surprisingly, the proportion of cells with chromosome doubling in p21-/-Wrn-/- MEFs with overexpression of TRF2ΔBΔM is only 46.3%, suggested that overexpression of TRF2ΔBΔM in p21-/-Wrn-/- MEFs did not increase the chromosome doubling, even decreased a little if there was any (Table 1).
These results indicated that lack of p21 function increased the incidence of chromosome doubling, and loss of TRF2 function further promoted the chromosome doubling, while loss of Wrn function rendered the chromosome doubling.
The chromosome doubling induced by loss of p21 function might be one of the reasons that loss of p21 function helped to overcome cellular senescence induced by telomere dysfunction.
Dependent on the extent of telomere dysfunction, loss of p21 function could induce tumorigenic p53 mutation and tumorigenesis
It has been reported that the price of escaping from senescence by editing cellular proliferation might be tumorigenesis31. To test that whether loss of p21 function in the setting of telomere dysfunction could induce tumorigenesis, we kept passaging p21-/-+TRF2ΔBΔM MEFs escaping of senescence to observe whether these cells become tumorigenic. We detected expression of TRF2ΔBΔM in continuous passages by Western blot and fluorescent immunostaining, and found that expression of TRF2ΔBΔM was gradually reduced along with passaging time (Figure 2A, B). The percentage of TRF2ΔBΔM positive cells reduced from 73.1% in early stage to 24.2% in middle stage, and to almost zero in late stage (Figure 2E, F, G, H). Consistent with this, the expression of DNA damage response (DDR)-related factors, such as p53, p19 and γ-H2AX, were gradually reduced along with the loss of TRF2ΔBΔM in the early to middle-advanced stage of cell passages (Figure 2A, B, C, D). These results suggest that p21-/-+TRF2ΔBΔM MEFs may reduce DDR by eliminating exogenous TRF2ΔBΔM.
Loss of p21 function rescued cellular senescence induced by telomere dysfunction. A. Growth curves of WT, Wrn-/-, p21-/- and p21-/-Wrn-/- MEFs. The asterisk in day 4 showed the statistical significant difference between Wrn-/- and p21-/- MEFs (p= 0.02). The asterisk in day 6 showed the statistical significant difference between Wrn-/- and p21-/- MEFs (p= 0.01), and between Wrn-/- and p21-/-Wrn-/- MEFs (p=0.02). B. Analysis of PCNA, p16 and p19ARF (p19) expression levels in MEFs with different genotypes in their early passages (passage number≤5) or late passages (passage number=10) by Western blot. The quantification result was showed at the right side. C. SA-β-Gal staining of p21-/-, Wrn-/- and p21-/-Wrn-/- MEFs in late passage (passage number=13), showing that loss of p21 function could rescue the cellular senescence induced by Wrn-/-. The quantification result was showed at the right side. D. SA-β-Gal staining of p21-/-+TRF2ΔBΔM, Wrn-/+TRF2ΔBΔM and p21-/-Wrn-/-+TRF2ΔBΔM MEFs, showing that even with telomere dysfunction caused by both Wrn and TRF2 dysfunction, loss of p21 function could still rescue the cellular senescence. The quantification result was showed at the right side. E. Compared to the control WT MEFs, MEFs with p21 knocking out showed higher level of p16 expression.
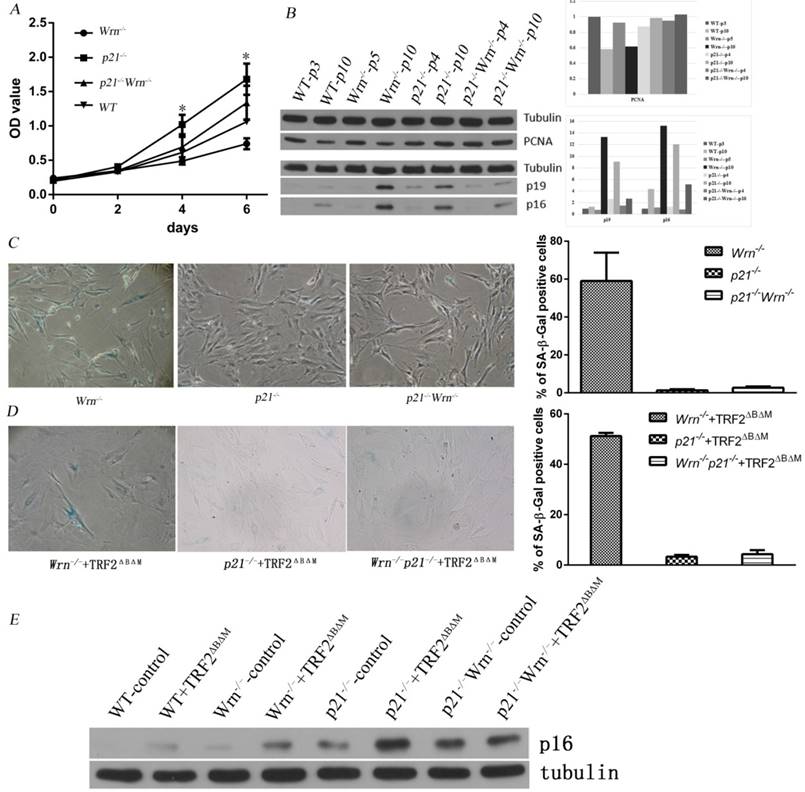
p21-/-+TRF2ΔBΔM MEFs reduced DNA damage response by eliminating exogenous TRF2ΔBΔM. A-B. Analysis of TRF2ΔBΔM levels and the expression of DNA damage response related factors, p53, p19 in p21-/-+TRF2ΔBΔM MEFs in different passages by Western blot. The statistic analysis indicated that p19 expression level in middle-advanced stage was significantly down-regulated compared to early stage (**p<0.01) and was significantly up-regulated in late stage compared to middle-advanced stage (**p<0.01). C-D. Analysis of γ-H2AX level in p21-/-+TRF2ΔBΔM MEFs in different passages by Western blot. The statistic analysis indicated that γ-H2AX expressed much less in middle stage compared to early stage (**p<0.01) and was highly expressed in late stage (**p<0.01). E-H. Immunostaining of TRF2ΔBΔM in p21-/-+TRF2ΔBΔM MEFs in different passages. The percentage of TRF2ΔBΔM positive cells reduced from 73.1% in early stage to 24.2% in middle stage, and to almost zero in late stage (**p<0.01).
The chromosome aberrations occurred in MEFs or tumor cells with different genotype.
| MEFs' GMEFs' Genotype | metaphases counted | fusion | breakage | double minutes | diploid 2N=40 | Aneuploid | Proportion of chromosome doubling | ||
|---|---|---|---|---|---|---|---|---|---|
| 40<2N≤80 | 2N>80 | 2N<40 | |||||||
| WT-p10 | 40 | 0 | 1 | 0 | 32 | 5 | 0 | 3 | 12.5% |
| Wrn-/- -p10 | 22 | 0 | 0 | 0 | 13 | 7 | 0 | 2 | 31.8% |
| p21-/- -p11 | 44 | 0 | 1 | 0 | 4 | 28 | 1 | 11 | 65.9% |
| Wrn-/- p21-/- -p11 | 46 | 1 | 0 | 0 | 15 | 21 | 3 | 7 | 52.2% |
| p21-/-+TRF2ΔBΔM-p11 | 39 | 3 | 0 | 0 | 5 | 32 | 2 | 0 | 87.2% |
| Wrn-/-p21-/-+TRF2ΔBΔM-p11 | 41 | 1 | 0 | 0 | 21 | 17 | 2 | 1 | 46.3% |
| p21-/-+TRF2ΔBΔM-p59 | 38 | 4 | 0 | 1 | 0 | 15 | 15 | 8 | 78.9% |
| Wrn-/-p21-/-+TRF2ΔBΔM-p21 | 41 | 13 | 0 | 0 | 0 | 32 | 9 | 0 | 100% |
| Wrn-/-p21-/-+TRF2ΔBΔM-p59 | 41 | 15 | 6 | 3 | 0 | 18 | 18 | 5 | 87.8% |
| Wrn-/-p21-/-+TRF2ΔBΔM tumor cells | 58 | 7 | 0 | 0 | 0 | 40 | 13 | 5 | 91.4% |
Interestingly, we observed a sudden increase of p19 and γ-H2AX in late stage of p21-/-+TRF2ΔBΔM MEFs (Figure 2A, B, C, D), suggesting that the DNA damage surveillant gene was mutated. Therefore, we sequenced p53 gene in late stage of p21-/-+TRF2ΔBΔM MEFs and found that p53 was mutated at C138W. Since mutation of p53 usually induces genomic instability, we detected karyotypes of these MEFs with p53 mutation at C138W. The data showed that cells with polyploidy and aneuploidy karyotypes increased greatly, and cells with chromosome number more than 80 reached 39.5% of total cells, this percentage was only 5.1% in early stage cells (Table 1). These results suggest that p53 mutation increased genomic instability.
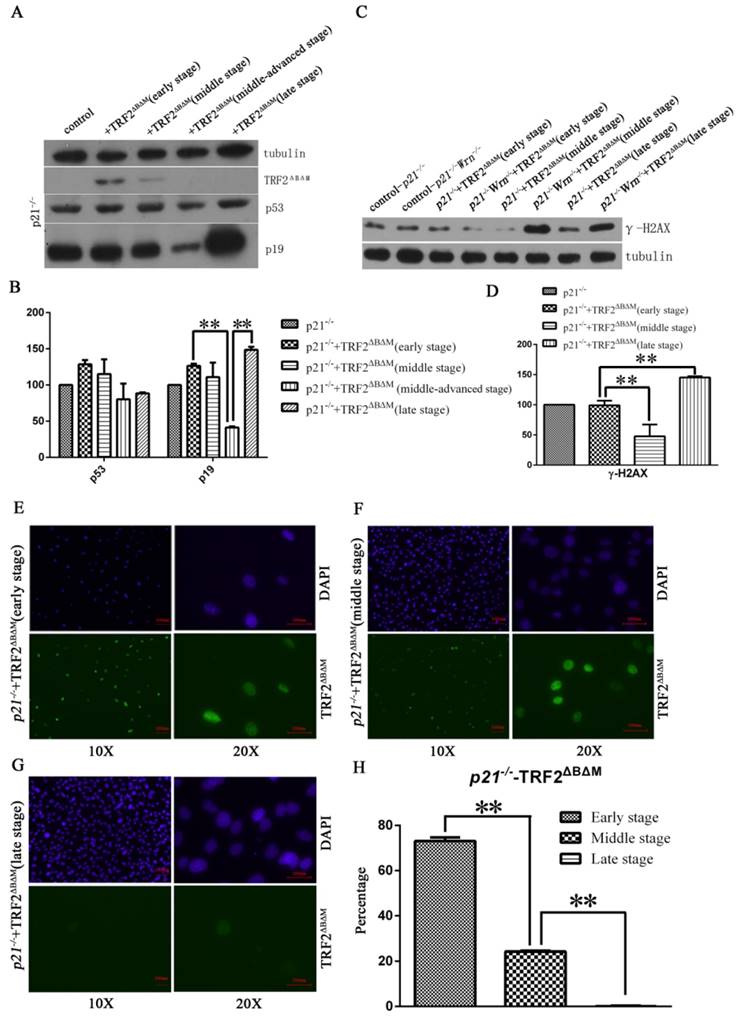
To further test that whether these cells with p53 mutation became cancerous, we performed transformation assay and xenograft assay. We found that compared to p21-/-+TRF2ΔBΔM MEFs without p53 mutation, p21-/-+TRF2ΔBΔM MEFs with p53 mutation obviously formed more colonies (Figure 3A, upper panel). The data suggested that the p53 mutation cells gained a strong ability of colony formation. Further we subcutaneously injected p53 mutation cells to SCID mice and nude mice. We did not observe tumor formation at injected points after 5 months. These results suggested that p53 mutation cells were not cancerous although they had a strong ability of colony formation. Together these data showed that loss of p21 function could not induce cells to form tumor with telomere dysfunction induced by TRF2ΔBΔM alone.
In WS mouse model, defects in both telomerase and Wrn caused telomere dysfunction and induced aging and tumor formation. To mimic this genetic background and test the p21 function in tumorigenesis, we kept on passaging p21-/-Wrn-/-+TRF2ΔBΔM MEFs to observe the fate of cells. We also observed that the expression of TRF2ΔBΔM was gradually reduced along with passaging (Figure 3B). However, unlike in p21-/-+TRF2ΔBΔM MEFs, DDR factors, such as phosphorylated p53, p19, p16 and γ-H2AX, were all increased along with passaging in p21-/-Wrn-/-+TRF2ΔBΔM MEFs (Figure 3B, Figure 2C), suggesting the continuous existence of DNA damages. These results implied that when challenged with multiple telomere dysfunctions, cells suffered more serious telomere DNA damages. Surprisingly, cells with intense activated DDR signals did not senescence. On the contrast, these cells proliferated more rapidly, as showed by increased cellular proliferation factor PCNA (Figure 3B). We also observed a sudden burst of cell proliferation in the middle stage of cell passaging, suggesting p53 mutation at this time point.
Therefore we sequenced p53 gene in the middle stage of cell passaging and found that p53 was mutated at R270L. We also found a few cells that still maintained wild type p53 at 270 at this stage. Then we further sequenced p53 genes in late stage cells, and found p53 was mutated at R270L in all cells, suggesting a positive selection for the cells bearing mutant p53 and thus higher proliferation potential.
The p53 mutation of R270L in mice is R273L in human, which is one of the most frequent p53 mutations in human cancer. IARC TP53 database showed that mutation R273L lose transactivation capacities of WAF1, MDM2, BAX, h1433s, AIP1, GADD45, NOXA, P53R2 etc. R273L has been reported as a somatic mutation in 155 tumor samples and as a germline mutation in 2 families with Li-Fraumeni syndromes (IARC TP53 database, R18, April 201632).
We further detected tumorigenic capacity of p21-/-Wrn-/-+TRF2ΔBΔM MEFs with p53R270L mutation by transformation assay and xenograft assay. We found that p21-/-Wrn-/-+TRF2ΔBΔM MEFs with p53 mutation formed more and bigger colonies than the cells without p53R270L mutation (Figure 3A, lower panel). Then we injected the cells to SCID mice and nude mice subcutaneously. We found that tumors formed at injected points in SCID mice (Figure 3C) and in nude mice (Figure 3D). The hematoxylin-eosin (HE) staining showed that the tumors were sarcomas (Figure 3E). Thus, we verified that p21-/-Wrn-/-+TRF2ΔBΔM MEFs in late stage with p53 R270L mutation were cancerous. We sequenced p53 of the tumors and also found R270L mutation in all tumor cells. These results suggested that p21 deletion together with multiple telomere dysfunctions promoted tumorigenic p53 mutation.
Loss of p21 function could induce cells to form tumor with telomere dysfunction induced by TRF2ΔBΔM and Wrn defect. A. Transformation assay showed that in either p21-/-+TRF2ΔBΔM or p21-/-Wrn-/-+TRF2ΔBΔM MEFs, late stage MEFs with p53 mutation formed more colonies than early stage MEFs without p53 mutation. B. Analysis of expression of DDR factors in passages. The TRF2ΔBΔM was gradually reduced along with passaging. However, the expression of p53, p19, p16, PCNA increased along with passaging in p21-/-Wrn-/-+TRF2ΔBΔM MEFs. C-D. Xenograft assay showed that p21-/-Wrn-/-+TRF2ΔBΔM MEFs with p53 mutation could generate tumors in SCID mice(C) and nude mice (D). 50 days in nude mice. E. The hematoxylin-eosin (HE) staining results of tumor tissue showed that the tumors were sarcomas. F. Western blot showed that compared to normal muscle tissue (left lane), p19 and PCNA were highly expressed in tumor tissue (right lane).
By Western blot assay, we found that compared to normal tissue, p19 and PCNA were highly expressed in tumor tissue and cultured tumor cells (Figure 3B, F). Different from late stage cells and tumor cells, tumor tissue did not express high levels of p53, p16 and phosphorylated p53 (Figure 3B). And no TRF2ΔBΔM was detected in tumor tissue and tumor cells (Figure 3B, F), suggesting that although it initiated the chromosome aneuploidy, TRF2ΔBΔM was eliminated by established tumors for maintaining better telomere function.
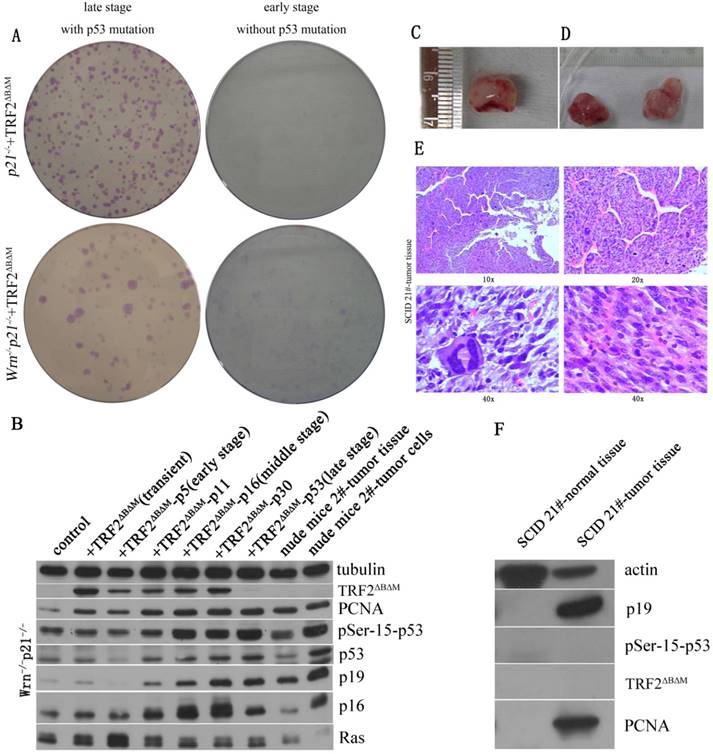
Then we detected karyotypes in MEFs with p53 mutation at R270L in middle stage and late stage. We found that all cells had increased polyploidy and aneuploidy compared to early stage cells (Table 1). The ration of chromosome fusion increased to 31.7% and cells with chromosome number more than 80 increased to 22% in middle stage (Figure 4A, Table 1). These results suggested that p53 mutation increased genomic instability. In late stage cells, genomic instability further enhanced, displaying high ration of chromosome fusion (36.6%), chromosome breakage (14.6%), cells with chromosome number more than 80 increased to 43.9% and double minutes were observed (Figure 4B, Table 1).
Furthermore, we detected karyotype of cultured tumor cells derived from xenograft tumors. We found that compared to the p21-/-Wrn-/-+TRF2ΔBΔM MEFs that were injected for tumor xenograft assay, genomic instability in stabilized tumor cells was improved, displaying as less chromosomal fusion (12.1%), no chromosomal breakages or double minutes, about 69% of cells have chromosomal number between 40 and 80 (Figure 4C, Table 1). The characteristic was similar to p21-/-Wrn-/-+TRF2ΔBΔM MEFs in middle stage. The result suggests that tumor cells in vivo were changed to survive better.
The karyotype of MEFs and cultured tumor cells derived from xenograft tumors. A. The karyotype of p21-/-Wrn-/-+TRF2ΔBΔM MEFs in their middle stage. The percentage of cell with more than 80 chromosomes increased to 22%. Arrows showed chromosome fusion. B. The karyotype of p21-/-Wrn-/-+TRF2ΔBΔM MEFs in their late stage. The percentage of cell with more than 80 chromosomes increased to 43.9% and double minute chromosomes were observed. Arrows showed chromosome fusion, breakage and double minutes. C. The karyotype of cultured tumor cells derived from a xenograft tumor. Arrows showed chromosome fusion.
Loss of p21 function is necessary for ALT tumorigenesis but not for telomerase positive tumorigenesis. A. Knock down of p21 expression in 395-6B-1 and 395-7A-1 cells by shRNA. B. The xenograft assay showed that either knocking down p21 in 395-6B-1 or rescuing telomerase activity by putting back Terc (mTR) could facilitate the tumorigenesis in the SCID mouse. The arrows pointed to shoulders showed the tumors derived from 395-6B-1+p21-shRNA, and the arrows pointed to hips showed the tumors derived from 395-6B-1+mTR. C. Western blot analysis of the protein expression level in telomerase negative tumors or telomerase positive tumors with or without p53N236S mutation. It showed that p21 level is low in telomerase negative tumors derived from 395-6B-1+p21-shRNA, while telomerase positive SCID6B-1+mTR tumor maintained normal level of p21 expression. The mTR promoted the expression of mutant p53N236S. D. TRF-Southern assay confirmed that like SCID3B-1 and SCID9B-1 tumors, the SCID6B-1+p21-shRNA tumor showed smear pattern of telomere length, thus is ALT tumor. However, SCID6B-1+mTR tumor showed homogenous telomere length. E. 2D gel electrophoresis detected the typical ALT feature of telomeric circles in SCID6B-1+p21-shRNA tumor, further confirmed its ALT characteristics. Arrows showed the arc-shaped telomeric circles.
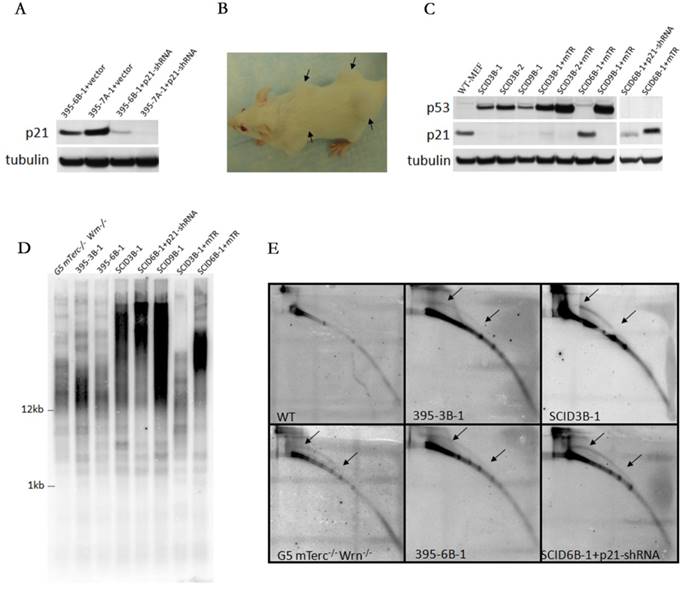
Loss of p21 function is necessary for ALT tumorigenesis but not for telomerase positive tumorigenesis
The above data showed that in the setting of multiple telomere dysfunctions, loss of p21 could induce p53 mutation and tumorigenesis. However, we could not tell the tumorigenesis is due to p53R270L mutation or due to the loss of p21 function.
Previously we have reported two series of cell lines derived from senescent WS MEFs (G5 mTerc-/-Wrn-/-). Both series of cell lines escaped senescent and form immortalized colonies, however, xenograft assays using immortalized cell lines revealed two distinct categories: one set of immortalized cell lines formed tumors (tumorigenic cell lines SCID3B-1, SCID3B-2, SCID9B-1), whereas the second set of cell lines did not form tumors within 6 months (non-tumorigenic immortalized cell lines 395-6B-1, 395-7A-1, 395-7A-2, 395-7A-3)14, 16. When we look into the molecular difference of these two series, we found that all three tumorigenic cell lines obtained the same p53 mutation, p53N236S, while p53 kept wild type in all four non-tumorigenic immortalized cell lines 395-6B-1, 395-7A-1, 395-7A-2 and 395-7A-3. However, Ink4a locus surrounding chromosome deletion occurred in these four non-tumorigenic immortalized cell lines16. Thus we used these cell lines to test the function of p21 in inhibiting tumorigenesis in the setting of multiple telomere dysfunctions and wild type p53 function.
We used shRNA to knock down p21 expression in non-tumorigenic immortalized cell lines 395-6B-1, 395-7A-1 and established stabilized p21 knocking down cell lines (Figure 5A), and then subcutaneously injected the cells into SCID mice. We observed tumorigenesis derived from 395-6B-1 cells (Figure 5B, pointed by arrows), but not from 395-7A-1 cells. Due to the knockout of telomerase template RNA (Terc) in these cells, the tumors derived from 395-6B-1 were ALT tumors. These data suggested that p21 play a key role in suppression of ALT tumor in WS MEFs with negative telomerase activity.
To further verify the role of p21 in telomerase negative tumor, we transfected the Terc (mTR) in 395-6B-1 cells and try to rescue the telomerase activity. The ALT tumorigenic cell lines 395-3B-1, 3B-2 and 395-9B-1 were also transfected with Terc and used as positive controls. We successfully manifested telomerase positive tumorigenesis in 395-6B-1 cells by putting back Terc (Figure 5C, D).
When we compared the p21 protein expression level in telomerase negative tumor SCID6B-1+p21-shRNA with telomerase positive tumor SCID6B-1+mTR, we found that SCID6B-1+mTR tumor maintained normal level of p21 expression (Figure 5C). Considered that 395-6B-1 cells (bearing wild type p53) could not generate tumor, while after knocking down p21, 395-6B-1-+p21-shRNA could form tumor. Together these data suggests that p21 function is the essential barrier for ALT tumorigenesis. However, when telomerase is available, even with functional p53 and p21, due to loss of Ink4a locus, tumorigenesis is still possible. The ALT tumorigenic cell lines 395-3B-1, 3B-2 and 395-9B-1 (bearing mutated p53: p53N236S) lost p21 expression due to p53 mutation. Overexpressing Terc (mTR) in these cells accelerated the tumorigenesis, and interestingly promoted the mutant p53 expression (Figure 5C).
By TRF-southern and two-dimensional (2D) gel electrophoresis, we showed that tumor SCID6B-1-p21-shRNA formed typical smear pattern of telomere length like ALT tumors SCID9B-1 and SCID3B-1 (Figure 5D), and also formed arc-shaped circular extra-chromosomal telomeric DNA (t-circles) in 2D assay (Figure 5E). However, telomerase positive tumor SCID6B-1+mTR showed homogenous telomere length (Figure 5D). These data confirmed that the tumor SCID6B-1-p21-shRNA generated by knocking down p21 is ALT tumor, and overexpressing Terc (mTR) rescued the telomerase activity in SCID6B-1+mTR tumor.
Discussion
It has been documented that deletion of p21 prolongs the lifespan of mice with dysfunctional telomeres induced by knocking out the RNA component of telomerase (Terc)21. It was found that p21 deletion improved hematolymphopoiesis and the maintenance of intestinal epithelia without rescuing telomere function. Moreover, apoptotic responses remained intact and p21 deletion did not accelerate chromosomal instability or cancer formation in these mice. These data showed that knocking out of the p21 gene could improve stem cell function and the lifespan of mice with dysfunctional telomeres without accelerating cancer formation21. In the background of Wrn deficient mice, deletion of p21 did not show any acceleration of tumorigenesis during the first year of life22. Thus, in the presence of functional wild-type p53, therapeutically targeting p21 may help to improve regeneration and organ homeostasis in chronic diseases and aging-related disorders associated with telomere dysfunction33.
In consistent with these data, we found that loss of p21 function could rescue the cellular senescence induced by telomere dysfunction caused by either Wrn deficiency or TRF2ΔBΔM alone, without increasing cellular tumorigenesis. Interestingly, in these cells rescued from senescence by p21 deletion, p16 expression was up-regulated, suggesting that p21 and p16 interacted with each other in regulating cellular senescence. It also implied that overexpression of p16 alone might not be enough to maintain cellular senescence status in the condition of p21 deletion. Our result agrees with a study in human cells in which telomere shortening triggered senescence of human cells through a pathway involving ATM, p53, and p21, but not p1634. These results suggest that p21 play a primary role in cellular senescence induced by telomere dysfunction in mice.
We also found that chromosome doubling increased in p21 null MEFs suffered from telomere dysfunction induced by TRF2ΔBΔM. Previous study has proposed that the lack of p53 function and prolonged DNA damage due to persistent telomere dysfunction were essential for induction of tetraploidy. The lack of p53 function could release cells from a G1/S block and enter the S phase in response to DNA damage. The prolonged DNA damage could arrest cells in G2 and re-entered a second S phase, which resulting in whole-genome reduplication and tetraploidy30. Since p21 is the key downstream effector of p53 in G1/S checkpoint, this proposal might also be applicable to p21 deficient cells. Our data revealed that the loss of p21 function did the same as the lack of p53 that released MEFs from a G1/S checkpoint, and overexpression of TRF2ΔBΔM induced prolonged DNA damages and arrested MEFs in G2/M phase, displayed as increased chromosome doubling in p21 null MEFs with telomere dysfunction induced by TRF2ΔBΔM. On the other hand, Wrn deletion significantly induced G1/S arrest35, which was adverse to the role of loss of p21 function. Therefore compared to p21-/- MEFs, p21-/-Wrn-/- MEFs has lower proportion of chromosome doubling.
However, our data showed that if we added up telomere dysfunctions by combining Wrn deficiency and TRF2 dysfunction, loss of p21 function could induce cells to form tumor. This data demonstrated that p21 plays an important role in suppressing ALT tumorigenesis in the setting of multiple telomere dysfuntions induced by TRF2ΔBΔM and Wrn-/-. It is interesting that we also found that in the late stage of MEFs with exogenous TRF2ΔBΔM or tumors derived from these MEFs, cells tend to eliminate exogenous TRF2ΔBΔM expression. This could be caused by the senescence of cells with high level of TRF2ΔBΔM expression. Theoretically, loss of p21 should endow the cells with tolerance to TRF2ΔBΔM expression, which was consistent with the data that loss of p21 postponed the cellular senescence induced by TRF2ΔBΔM. However, it is also possible that loss of p21 regulated the expression level of TRF2ΔBΔM.
Furthermore, knocking down p21 in the non-tumorigenic cell line 395-6B-1 resulted in ALT tumorigenesis despite the retention of wild-type p53 status in these cells. However, some cell lines did not undergo ALT tumorigenesis after knock down of p21 (395-7A-1, 395-7A-2, 395-7A-3), which implies that p21 knockdown on this genetic background might not be sufficient to promote ALT tumorigenesis. The immortalized cell line 395-6B-1 could be predisposed to ALT tumorigenesis for unknown reasons that occurred during the escape from senescence. Second, when we restored telomerase activity in 395-6B-1 cells, we were able to engineer tumors with regular p21 expression. It is interesting to see that either p21 knock down or restoration of telomerase function was able to induce tumorigenesis in 395-6B-1 cells with wild type p53 function. Further study into the genetic background of 395-6B-1 cells may reveal additional correlations between p21 and telomere function.
Together, our data showed that the p21 function is essential for suppressing ALT tumorigenesis and that p21 might play a key role in maintenance the senescence status induced by telomere dysfunction. An in vitro study found that restoring p53 function in ALT cells resulted in p21Cip1/Waf1 up-regulation and growth arrest/senescence, suggesting a preventive role for p21 in ALT tumorigenesis36.
These data also suggested that the genetic context is essential in determining the role of p21 in cancer prevention. Therefore, targeting p21 in the treatment of human degenerative diseases would require a personalized genetic background screen. These results shed light on the p21 important roles in cellular senescence and tumor suppression.
Supplementary Material
Supplementary figure S1.
Acknowledgements
This work was supported by NSFC grants to Y.L. (30771194, 81460224). We thank Dr. Sandy Chang from Yale University and Dr. Ronald A Depinho from MD Anderson Cancer Center for kindly providing the WS mice, the related cell lines, and the mTR construct. We thank Dr. Tyler Jacks from MIT for kindly provided the p21 knock out mice. We thank Dr. Titia de Lange from Rockefeller University for kindly providing the pBabe-TRF2ΔBΔM construct.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bohr VA. Rising from the RecQ-age: the role of human RecQ helicases in genome maintenance. Trends in biochemical sciences. 2008;33(12):609-20
2. Brosh RM Jr, Bohr VA. Human premature aging, DNA repair and RecQ helicases. Nucleic acids research. 2007;35(22):7527-44
3. Chu WK, Hickson ID. RecQ helicases: multifunctional genome caretakers. Nature reviews Cancer. 2009;9(9):644-54
4. Hanada K, Hickson ID. Molecular genetics of RecQ helicase disorders. Cellular and molecular life sciences: CMLS. 2007;64(17):2306-22
5. Singh DK, Ahn B, Bohr VA. Roles of RECQ helicases in recombination based DNA repair, genomic stability and aging. Biogerontology. 2009;10(3):235-52
6. Ozgenc A, Loeb LA. Werner Syndrome, aging and cancer. Genome dynamics. 2006;1:206-17
7. Lebel M, Leder P. A deletion within the murine Werner syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of cellular proliferative capacity. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(22):13097-102
8. Lombard DB, Beard C, Johnson B, Marciniak RA, Dausman J, Bronson R. et al. Mutations in the WRN gene in mice accelerate mortality in a p53-null background. Molecular and cellular biology. 2000;20(9):3286-91
9. Schulz VP, Zakian VA, Ogburn CE, McKay J, Jarzebowicz AA, Edland SD. et al. Accelerated loss of telomeric repeats may not explain accelerated replicative decline of Werner syndrome cells. Human genetics. 1996;97(6):750-4
10. Tahara H, Tokutake Y, Maeda S, Kataoka H, Watanabe T, Satoh M. et al. Abnormal telomere dynamics of B-lymphoblastoid cell strains from Werner's syndrome patients transformed by Epstein-Barr virus. Oncogene. 1997;15(16):1911-20
11. Wyllie FS, Jones CJ, Skinner JW, Haughton MF, Wallis C, Wynford-Thomas D. et al. Telomerase prevents the accelerated cell ageing of Werner syndrome fibroblasts. Nature genetics. 2000;24(1):16-7
12. Chang S, Multani AS, Cabrera NG, Naylor ML, Laud P, Lombard D. et al. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nature genetics. 2004;36(8):877-82
13. Du X, Shen J, Kugan N, Furth EE, Lombard DB, Cheung C. et al. Telomere shortening exposes functions for the mouse Werner and Bloom syndrome genes. Molecular and cellular biology. 2004;24(19):8437-46
14. Laud PR, Multani AS, Bailey SM, Wu L, Ma J, Kingsley C. et al. Elevated telomere-telomere recombination in WRN-deficient, telomere dysfunctional cells promotes escape from senescence and engagement of the ALT pathway. Genes & development. 2005;19(21):2560-70
15. Jia S, Zhao L, Tang W, Luo Y. The gain of function of p53 mutant p53S in promoting tumorigenesis by cross-talking with H-RasV12. International journal of biological sciences. 2012;8(5):596-605
16. Wu X, Jia S, Zhang X, Si X, Tang W, Luo Y. Two mechanisms underlying the loss of p16(Ink4a) function are associated with distinct tumorigenic consequences for WS MEFs escaping from senescence. Mechanisms of ageing and development. 2012;133(8):549-55
17. el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM. et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75(4):817-25
18. Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. The international journal of biochemistry & cell biology. 2005;37(5):961-76
19. Pantoja C, Serrano M. Murine fibroblasts lacking p21 undergo senescence and are resistant to transformation by oncogenic Ras. Oncogene. 1999;18(35):4974-82
20. Warfel NA, El-Deiry WS. p21WAF1 and tumourigenesis: 20 years after. Current opinion in oncology. 2013;25(1):52-8
21. Choudhury AR, Ju Z, Djojosubroto MW, Schienke A, Lechel A, Schaetzlein S. et al. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nature genetics. 2007;39(1):99-105
22. Lebel M, Cardiff RD, Leder P. Tumorigenic effect of nonfunctional p53 or p21 in mice mutant in the Werner syndrome helicase. Cancer research. 2001;61(5):1816-9
23. van Steensel B, Smogorzewska A, de Lange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92(3):401-13
24. de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes & development. 2005;19(18):2100-10
25. Broccoli D, Smogorzewska A, Chong L, de Lange T. Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nature genetics. 1997;17(2):231-5
26. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C. et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(20):9363-7
27. Zou W, Zeng J, Zhuo M, Xu W, Sun L, Wang J. et al. Involvement of caspase-3 and p38 mitogen-activated protein kinase in cobalt chloride-induced apoptosis in PC12 cells. J Neurosci Res. 2002;67(6):837-43
28. Cesare AJ, Griffith JD. Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Molecular and cellular biology. 2004;24(22):9948-57
29. Wang RC, Smogorzewska A, de Lange T. Homologous recombination generates T-loop-sized deletions at human telomeres. Cell. 2004;119(3):355-68
30. Davoli T, Denchi EL, de Lange T. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell. 2010;141(1):81-93
31. Ding Z, Wu CJ, Jaskelioff M, Ivanova E, Kost-Alimova M, Protopopov A. et al. Telomerase reactivation following telomere dysfunction yields murine prostate tumors with bone metastases. Cell. 2012;148(5):896-907
32. Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J. et al. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Human mutation. 2016;37(9):865-76
33. Bell JF, Sharpless NE. Telomeres, p21 and the cancer-aging hypothesis. Nature genetics. 2007;39(1):11-2
34. Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Molecular cell. 2004;14(4):501-13
35. Sidorova JM, Li N, Folch A, Monnat RJ Jr. The RecQ helicase WRN is required for normal replication fork progression after DNA damage or replication fork arrest. Cell Cycle. 2008;7(6):796-807
36. Jiang WQ, Zhong ZH, Nguyen A, Henson JD, Toouli CD, Braithwaite AW. et al. Induction of alternative lengthening of telomeres-associated PML bodies by p53/p21 requires HP1 proteins. The Journal of cell biology. 2009;185(5):797-810
Author contact
![]() Corresponding authors: Email: yingluoedu.cn, xiao_tracyedu.cn
Corresponding authors: Email: yingluoedu.cn, xiao_tracyedu.cn
Received 2017-10-24
Accepted 2017-12-10
Published 2018-1-14