ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2018; 14(3):266-279. doi:10.7150/ijbs.23574 This issue Cite
Research Paper
Identification of key genes and pathways in human clear cell renal cell carcinoma (ccRCC) by co-expression analysis
Lushun Yuan1,2,#, Guang Zeng1,#, Liang Chen1, Gang Wang1,2,3, Xiaolong Wang4, Xinyue Cao3, Mengxin Lu1, Xuefeng Liu5, Guofeng Qian6, Yu Xiao1,3,7 ![]() , Xinghuan Wang1,2
, Xinghuan Wang1,2 ![]()
1. Department of Urology, Zhongnan Hospital of Wuhan University, Wuhan, China
2. Medical Research Institute, School of Medicine, Wuhan University, Wuhan, China.
3. Department of Biological Repositories, Zhongnan Hospital of Wuhan University, Wuhan, China
4. Department of Urology, Ludwig-Maximilians-University Munich, Munich, Germany
5. Department of Pathology, Lombardi Comprehensive Cancer Center, Georgetown University Medical School, Washington DC, USA
6. Department of Endocrinology, The First Affiliated Hospital of Zhejiang University, Hangzhou, China
7. Laboratory of Precision Medicine, Zhongnan Hospital of Wuhan University, Wuhan, China
#These authors contributed equally to this work.
Abstract

Human clear cell renal cell carcinoma (ccRCC) is the most common solid lesion within kidney, and its prognostic is influenced by the progression covering a complex network of gene interactions. In our study, we screened differential expressed genes, and constructed protein-protein interaction (PPI) network and a weighted gene co-expression network to identify key genes and pathways associated with the progression of ccRCC (n = 56). Functional and pathway enrichment analysis demonstrated that upregulated differentially expressed genes (DEGs) were significantly enriched in response to wounding, positive regulation of immune system process, leukocyte activation, immune response and cell activation. Downregulated DEGs were significantly enriched in oxidation reduction, monovalent inorganic cation transport, ion transport, excretion and anion transport. In the PPI network, top 10 hub genes were identified (TOP2A, MYC, ALB, CDK1, VEGFA, MMP9, PTPRC, CASR, EGFR and PTGS2). In co-expression network, 6 ccRCC-related modules were identified. They were associated with immune response, metabolic process, cell cycle regulation, angiogenesis and ion transport. In conclusion, our study illustrated the hub genes and pathways involved in the progress of ccRCC, and further molecular biological experiments are needed to confirm the function of the candidate biomarkers in human ccRCC.
Keywords: clear cell renal cell carcinoma (ccRCC), differentially expressed genes (DEGs), biomarker, weighted gene co-expression network analysis (WGCNA), protein-protein interaction (PPI)
Introduction
Renal cell carcinoma (RCC), which makes up approximately 3% of all adult malignancies and 90-95% of adult kidney neoplasm, is being diagnosed with increasing incidence and mortality rates worldwide [1]. Clear cell renal cell carcinoma (ccRCC) is the most common tumor in kidney. However, its prognostic is influenced by the progression covering a complex network of gene interactions. As the most common pathological type of renal cancer, apart from surgery, it is both radiotherapy and chemotherapy resistant. At present, biomarkers for early detection and follow-up of the disease are not available. Therefore, targeted therapies are the best choices of non-surgical treatment for ccRCC [2]. Many studies revealed that targeted therapies, such as multipotase inhibitors, anti-VEGF antibodies and mTOR, had been approved for clinical use [3]. However, identification of novel therapeutic targets or biomarkers for prognostic, diagnostic or predictive use remains a priority.
Currently, gene expression profiles have been used to identify genes associated with progression of renal cancer [4-7]. Meanwhile, some researchers also used integrated approach to screen changes in renal carcinogenesis [8, 9]. However, most studies just focused on the screening of differentially expressed genes and ignored the high degree of interconnection between genes, although genes with similar expression patterns may be functionally related. Weighted gene expression network analysis (WGCNA) is a systems biology method for describing the correlation patterns among genes across microarray or RNA Sequence data, and it is an algorithm used for finding clusters (modules) of highly correlated genes as well as identifying phenotype related modules or genes clusters [10]. Nowadays, there are various studies revealing the phenotypes-related genes via WGCNA method, especially in cancers [11, 12]. For example, Zhou et al. found that TOP2A could be used as the potential prognostic and progression biomarker for pancreatic ductal adenocarcinoma [12]. Wang et al. revealed that ASPM could cause cirrhosis and eventually led to hepatocellular carcinoma [13]. In this study, we attempt to firstly screen differential expressed genes, construct protein-protein interaction networks and a co-expression network of relationships between genes through a systematic biology method based on WGCNA and to identify key genes and pathways participating in the carcinogenesis of ccRCC [13, 14].
Materials and Methods
Data collection
Gene expression profile was downloaded from Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). Dataset GSE53000 performed on Affymetrix Human Gene 1.0 ST Array [transcript (gene) version] (Affymetrix, Santa Clara, CA, USA) was used to screen differential expressed genes, construct protein-protein interaction networks and co-expression networks and identify hub genes and pathways in this study. This dataset included 56 clear cell renal cell carcinoma samples (including 2 lymph node metastasis samples and 1 venous thrombus samples) and 6 normal kidney samples.
Study design and data preprocessing
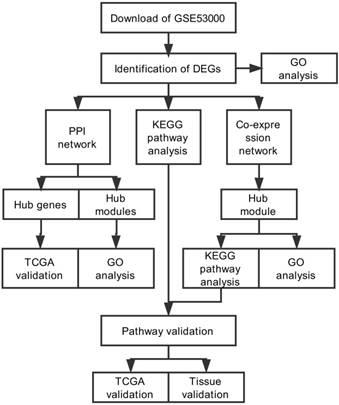
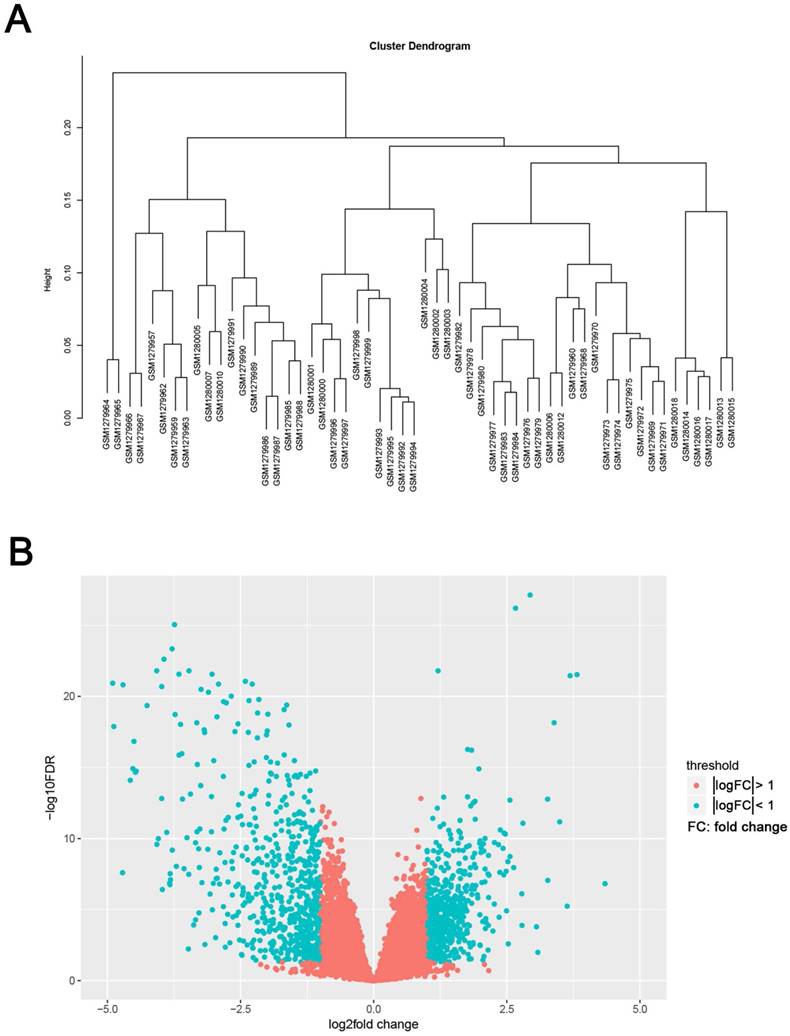
The study design was performed in a flow diagram (Fig. 1). Raw expression data were calculated following the pre-processing procedures: RMA background correction, log2transformation, quantile normalization and median polish algorithm summarization using the “affy” R package. Probes were annotated by the Affymetrix annotation files. Microarray quality was assessed by sample clustering according to the distance between different samples in Pearson's correlation matrices. No samples were removed from subsequent analysis in the test dataset (Fig. 2A).
Flow diagram of study. Data preparing, processing and analysis was shown in the flow diagram.
Differentially expressed genes (DEGs) screening
We use the “limma” R package to screen the DEGs between ccRCC samples and normal kidney samples. The FDR (false discovery rate) < 0.05 and |log2fold change (FC)| > 1 were chosen as the cut-off criteria (Fig. 2B). Furthermore, we chose the same condition and screened differentially expressed genes between ccRCC samples and metastasis samples.
Functional and pathway enrichment analysis
The Database for Annotation, Visualization and Integrated Discovery (DAVID) (http://david.abcc.ncifcrf.gov/) is an online program providing a comprehensive set of functional annotation tools for investigators to understand biological meaning behind large list of genes [15]. Enriched biological themes of DEGs, particularly GO terms and visualization of those on KEGG pathway maps were performed using DAVID database, p < 0.05 was set as the cut-off criterion.
Samples clustering and identification of differentially expressed genes (DEGs) in ccRCC tissues. (A) Samples clustering of GSE53000 to detect outliers. (B) The volcano plot of all DEGs.
PPI network construction
We used Search Tool for the Retrieval of Interacting Genes (STRING) Database (STRING) (http://www.string-db.org/) to assess protein-protein interaction (PPI) information [16]. In addition, to evaluate the interrelationships of DEGs, we used STRING database for analysis and Cytoscape software for visualization [17]. Confidence score > 0.4 was set as significant.
Hub module selection and functional analysis
We used plug-in Molecular Complex Detection (MCODE) to select hub modules of PPI network in Cytoscape [18]. Meanwhile, degree = 5, node score = 0.2, k-core = 2, and max. depth = 100 were used as cut-off criteria. Then we uploaded the genes in hub module to DAVID database to perform functional analysis.
Co-expression network construction and module functional analysis
Firstly, expression data profile of DEGs was tested to check if they were the good samples and good genes. Then, we used the “WGCNA” package in R to construct co-expression network for the DEGs [19, 20]. At first, the Pearson's correlation matrices were both performed for all pair-wise genes. Then, a weighted adjacency matrix was constructed using a power function amn = |cmn|β (cmn = Pearson's correlation between gene m and gene n; amn = adjacency between gene m and gene n). β was a soft-thresholding parameter that could emphasize strong correlations between genes and penalize weak correlations. Next, the adjacency was transformed into topological overlap matrix (TOM), which could measure the network connectivity of a gene defined as the sum of its adjacency with all other genes for network generation. To classify genes with similar expression profiles into gene modules, average linkage hierarchical clustering was conducted according to the TOM-based dissimilarity measure with a minimum size (gene group) of 50 for the genes dendrogram. To further analyze the module, we calculated the dissimilarity of module eigengenes, chose a cut line for module dendrogram and merged some modules, after which we performed GO enrichment analysis and KEGG pathway enrichment analysis on gene modules to characterize modules related to ccRCC.
Hub genes and pathway validation
Here, we chose genes with the most connectivities as hub genes and used TCGA KIRC data to perform validation using GEPIA database [21]. Common pathways in all modules were chosen to perform validation on both TCGA data and patients tissues.
Preparation for human ccRCC samples
The ccRCC and paracancerous tissues samples were collected from patients after surgery at Zhongnan Hospital of Wuhan University. The histology diagnosis was confirmed by two pathologists independently. The ccRCC and paracancerous tissues were immediately frozen and stored in liquid nitrogen or fixed in 4 % PFA after collection. The study using ccRCC and paracancerous tissue samples for total RNA isolation and qRT-PCR analysis was approved by the Ethics Committee at Zhongnan Hospital of Wuhan University (approval number: 2015029). Informed consent was obtained from all subjects.
Total RNA isolation
Total RNA from ccRCC tissues were isolated using RNeasy Mini Kit (Cat. #74101, Qiagen, Germany) according to the manufacturer's instruction. DNase I digestion (Cat. #79254, Qiagen, Germany) was used in each RNA preparation to remove genomic DNA. After that, total RNA quantity was measured using NanoPhotometer (Cat. #N60, Implen, Germany).
Quantitative real time PCR (qRT-PCR)
The cDNA was synthesized using 1 µg of total RNA isolated from patients tissues by ReverTra Ace qPCR RT Kit (Toyobo, China) and qRT-PCR was performed using 400 ng cDNA per 25 μl reaction. Each reaction was conducted with iQTM SYBR® Green Supermix (Bio-Rad, China) using 400 or 500 ng of cDNA in a final volume of 25 µl. Primers sequences and annealing temperature were summarized in Supplementary Tab. S1.
Statistical analyses
All analyses were performed three times and represent data from three individual experiments. Two-tailed Student's t-test was used for significance of differences between subgroups. Statistical analyses were performed with SPSS 16.0. Statistical significance was set at probability values of p < 0.05.
Results
Identification of DEGs in ccRCC tissues
The gene expression profiling of GSE53000 including 56 ccRCC tissues and 6 normal kidney tissues were analysed. Using “limma” package of R software, selecting p < 0.05 and |log2fold change (FC)| > 1 as the cut-off criteria, 1175 DEGs were identified, of which, 533 were upregulated and 642 were downregulated (Supplementary Tab. S2). The volcano plot of all DEGs is shown in Fig. 2B. Meanwhile, we screened 11 genes differentially expressed in metastasis samples compared with primary ccRCC (Supplementary Tab. S3).
Functional enrichment analysis of DEGs
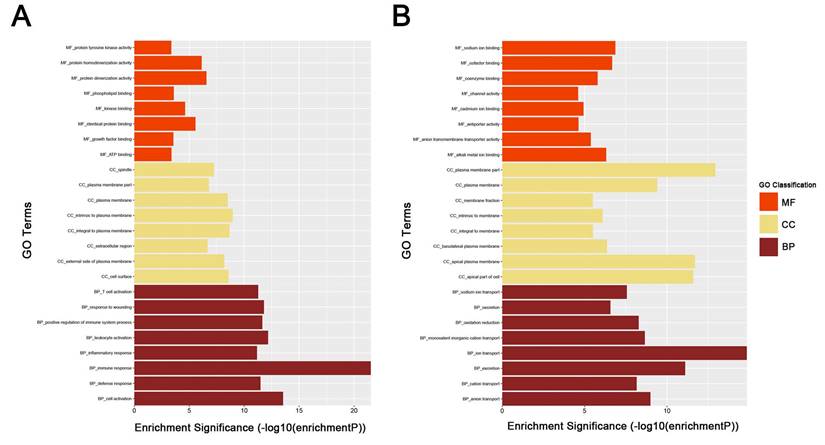
To obtain further insight into the function of DEGs of ccRCC, the up- and downregulated DEGs were respectively uploaded to the DAVID database. GO analysis results showed as to biological process (BP), the upregulated DEGs significantly enriched in response to wounding, positive regulation of immune system process, leukocyte activation, immune response and cell activation, and the downregulated DEGs significantly enriched in oxidation reduction, monovalent inorganic cation transport, ion transport, excretion and anion transport. For molecular function (MF), the upregulated DEGs significantly enriched in protein homodimerization activity, protein dimerizetion activity, phospholipid binding, kinase binding and identical protein binding, and downregulated DEGs significantly enriched in sodium ion binding, cofactor binding, alkali metal ion binding, coenzyme binding and anion transmembrane transporter activity. About cellular component, the upregulated DEGs significantly enriched in intrinsic to plasma membrane, integral to plasma membrane, cell surface, plasma membrane and external side of plasma membrane, and downregulated DEGs significantly enriched in plasma membrane part, apical plasma membrane, apical part of cell, plasma membrane and basolateral plasma membrane (Fig. 3A-B). These significantly enriched GO terms could help us a lot to further understand the role of DEGs played in ccRCC development and progression.
KEGG pathway enrichment analysis of DEGs
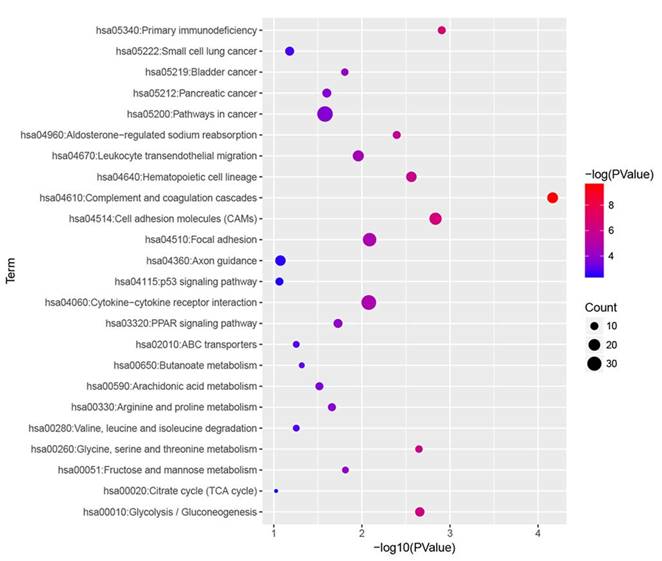
KEGG pathway analysis was performed for all DEGs, and we found that complement and coagulation cascades, primary immunodeficiency, cell adhesion molecules (CAMs), glycolysis / gluconeogenesis and glycine, serine and threonine metabolism were most significantly enriched (Fig. 4).
PPI network construction
Based on the string profile obtained from STRING analysis tool (Supplementary Tab. S3), the PPI network of DEGs consisted of 949 nodes and 4774 edges, including 367 upregulated genes and 414 downregulated genes. We considered the top 10 DEGs with high degree of connectivity as the hub genes of ccRCC (TOP2A, MYC, ALB, CDK1, VEGFA, MMP9, PTPRC, CASR, EGFR and PTGS2), which may play a critical role in tumorogenesis and proliferation.
Hub module selection and validation
Degree cut-off = 5, node score cut-off = 0.2, k-core = 2, and max. depth = 100 as the criterion, top 3 significant modules were selected by using plug-in MCODE. Gene Ontology analysis of each module was performed in DAVID database (Fig. 5).
GO analysis of DEGs. (A) upregulated DEGs with fold change > 2. (B) downregulated DEGs with fold change < -2. BP: biological process, CC: cellular component, MF: molecular function.
KEGG enrichment analysis of all DEGs with |fold change| > 2. All differentially expressed genes (DEGs) were analysed by KEGG enrichment. Fold change > 2 was set as cut-off value.
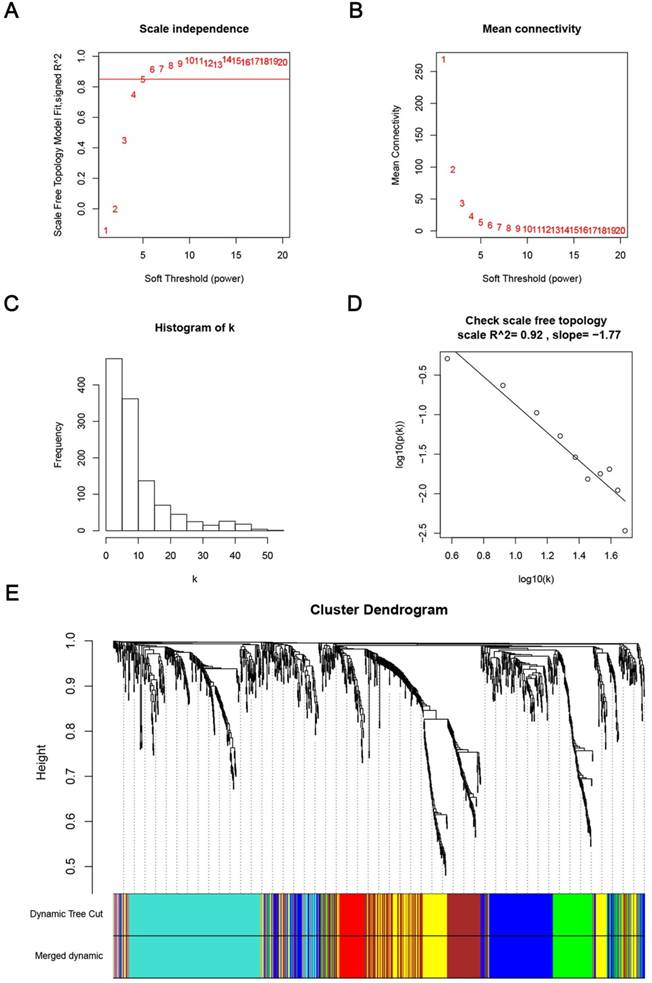
Weighted co-expression network construction and analysis
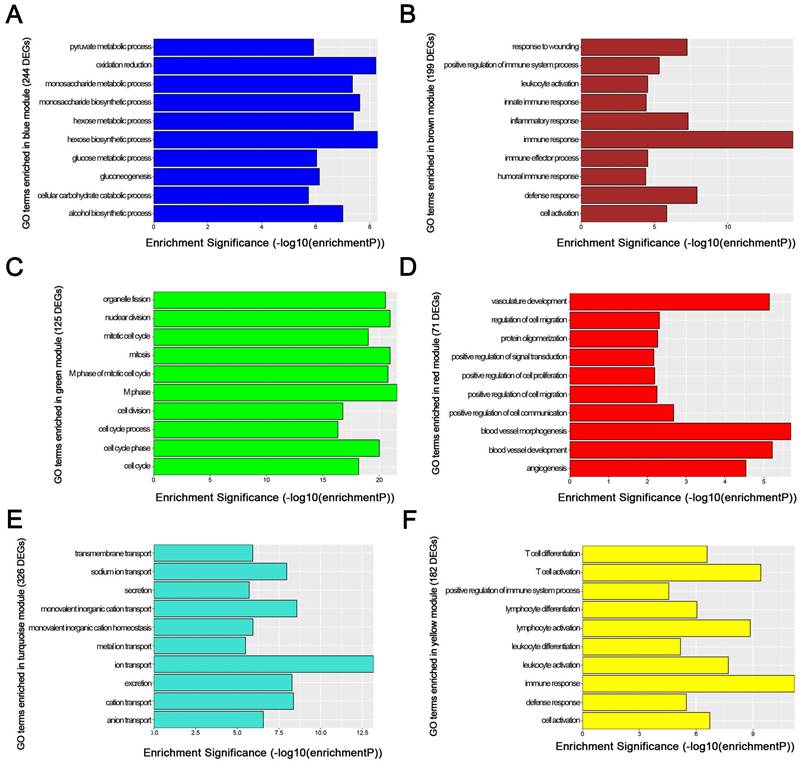
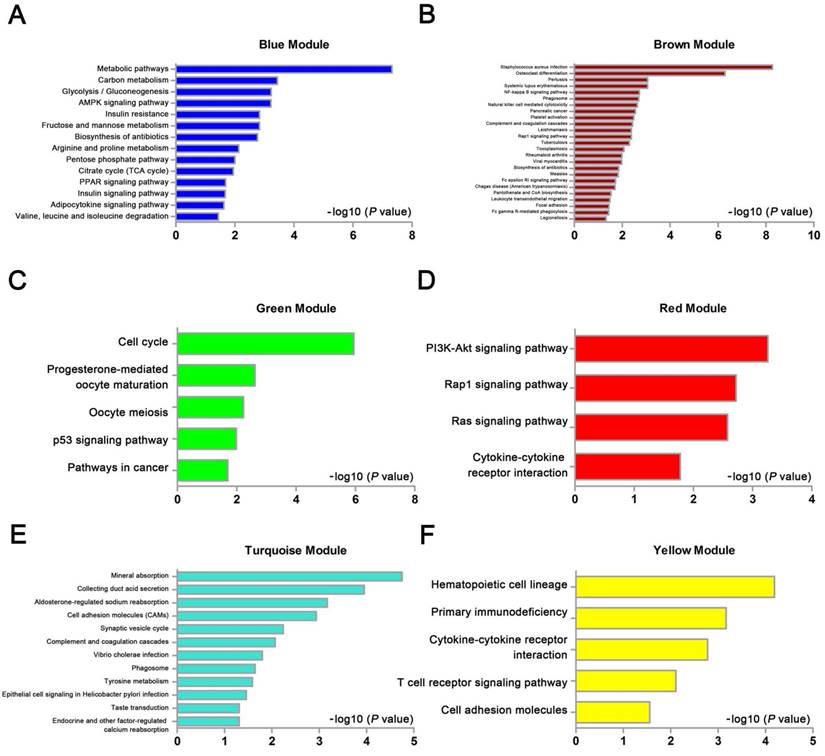
We used “WGCNA” package in R, after the quality assessment for expression data matrix of GSE53000, select the power of β = 6 (scale free R2 = 0.92) to ensure a scalefree network (Fig. 6A-D). After putting the DEGs with similar expression patterns into modules by average linkage clustering, a total of 7 modules were identified (Fig. 6E). Except grey module, Gene Ontology was performed for other 6 modules so as to explore the underlying biological process correlated to ccRCC. In Fig. 7, we could find that DEGs in blue module significantly enriched in hexose biosynthetic process, oxidation reduction and monosaccharide biosynthetic process. In brown module, DEGs were enriched in immune response, defense response and inflammatory response. In green module, DEGs were enriched in M phase, nuclear division and mitosis. In red module, DEGs were enriched in blood vessel morphogenesis, blood vessel development and vasculature development. In turquoise module, DEGs were enriched in ion transport, monovalent inorganic cation transport and cation transport. In yellow module, DEGs were enriched in immune response, T cell activation and lymphocyte activation. In Fig. 8, we found the common pathways related to renal carcinoma: PI3K/AKT pathway, Rap1 pathway, NF-kappa B pathway and insulin associated pathways.
Hub genes and pathway validation
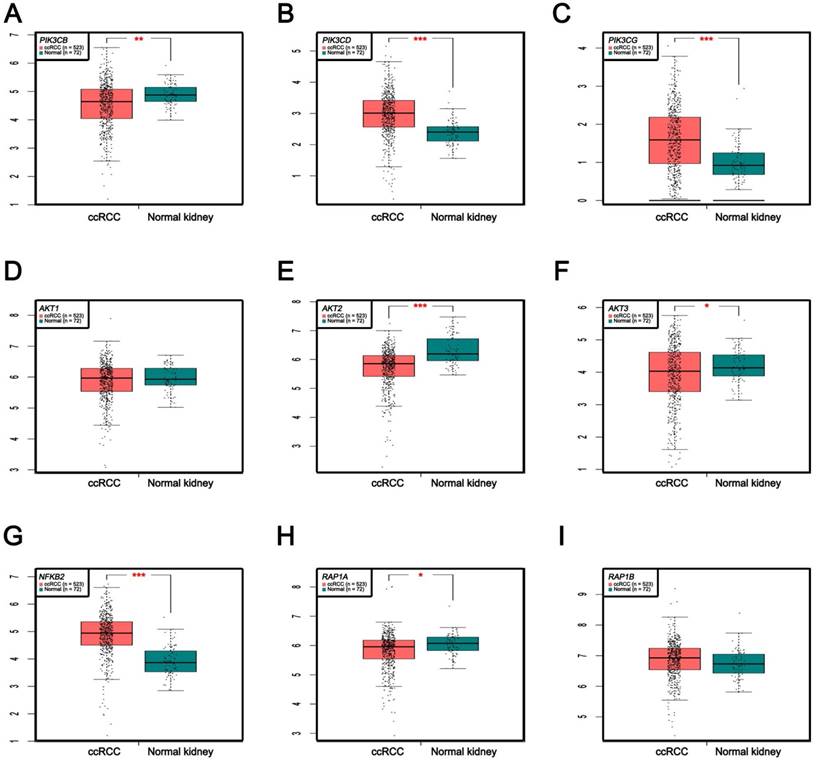
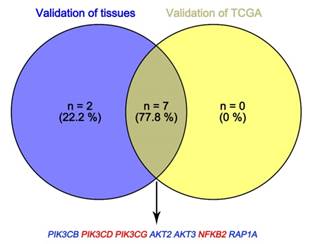
In Supplementary Fig. S1-2, we performed validation of hub genes using TCGA KIRC data and microarray data (GSE53000). We found that CASR and PTGS2 were tumor suppressors in both microarray and RNA-Seq data; CDK1, EGFR, MMP9, MYC, PTPRC, TOP2A and VEGFA were oncogenes in the common expression data. To perform the pathway validation, we chose the common pathway related to renal carcinogenesis and validated the key molecules using qRT-PCR. We found PIK3CD, PIK3CG and NFKB2 were upregulated in tumor samples and PIK3CB, AKT1, AKT2, AKT3, RAP1A and RAP1B were downregulated in tumor samples (Fig. 9). To further validate the pathway, we use TCGA KIRC data to perform the validation as well (Fig. 10). And we found that there were 7 genes deregulated in common validation sets (PIK3CB, PIK3CD, PIK3CG, AKT2, AKT3, NFKB2, RAP1A, Fig. 11).
Discussion
Clear cell renal cell carcinoma is biologically heterogeneous and has variable clinical courses, therefore, it is essential to understand the molecular mechanism for better diagnosis and treatment of ccRCC. In this study, we investigated the gene expression profile of GSE53000, including 56 clear cell renal cell carcinoma samples (including 2 lymph node metastasis samples and 1 venous thrombus samples) and 6 normal kidney samples to explore the molecular mechanism of ccRCC and find some biomarkers, which might be helpful therapeutic targets by using bioinformatics analysis.
In this study, results showed that expressions of total 1175 genes were altered between normal kidney tissues and ccRCC tissues at FDR < 0.05. Among the 1175 DEGs, 533 were upregulated and 642 were downregulated. PPI network analysis and WGCNA analysis were performed to identify protein-protein interactions and gene co-expression modules related with the clinical features of ccRCC. In addition, functional and pathway analysis were also performed to find ccRCC-related biological process and pathways.
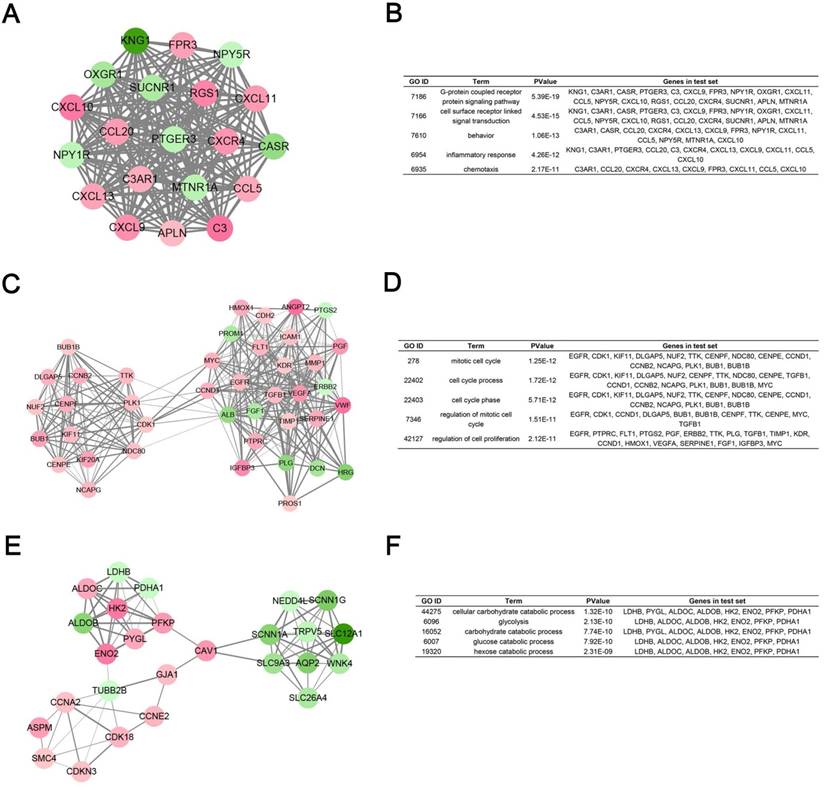
Module analysis of PPI network. (A) Module rank 1. (B) GO enrichment analysis of module rank 1. (C) Module rank 2. (D) GO enrichment analysis of module rank 2. (E) Module rank 3. (F) GO enrichment analysis of module rank 3.
Determination of soft-thresholding power in the weighted gene co-expression network analysis (WGCNA). (A) Analysis of the scale-free fit index for various soft-thresholding powers (β). (B) Analysis of the mean connectivity for various soft-thresholding powers. (C) Histogram of connectivity distribution when β = 6. (D) Checking the scale free topology when β = 6. (E) Dendrogram of all differentially expressed genes clustered based on a dissimilarity measure (1-TOM).
According to the GO analysis of DEGs, we found that upregulated DEGs were significantly enriched in immune response, cell activation, leukocyte activation and positive regulation of immune system process; downregulated DEGs were significantly enriched in ion transport, monovalent inorganic cation transport, cation transport and anion transport. Giraldo NA et al. investigated that the infiltration and the localization of DC, and the expression of immune checkpoints (PD-1, LAG-3, PD-L1, and PD-L2) in relation with prognosis in the tumor microenvironment modulated the clinical impact of CD8(+) T cells in ccRCC [22]. In addition, Balan M et al. also reported that c-Met can promote increased survival of renal cancer cells through the regulation of HO-1 and PD-L1 [23]. Ciarimboli G et al. found that OCT2 played a decisive role in the renal secretion of creatinine and the process could be inhibited by OCT2 substrates [24]. Pochini L et al. also discovered that OCTN cation transporters were associated with several pathologies [25]. Meanwhile, KEGG pathway analysis revealed those glycolysis/gluconeogenesis and glycine, serine and threonine metabolisms were significantly enriched. Many studies illustrated that carcinogenesis could have very closely correlation with metabolism [26-28]. As to renal carcinoma, significant progress had been made to understand the metabolic derangements present, which had been derived through translational, in vitro, and in vivo studies. So far, von Hippel-Lindau (VHL) loss was the well-characterized metabolic features linked to renal cancer [29]. And several metabolic pathways were altered, including glycolysis and oxidative phosphorylation due to VHL loss and the influence caused by increasing expression of hypoxia-inducible factor [30-35].
GO enrichment analysis of 6 genes modules. (A) blue module, (B) brown module, (C) green module, (D) red module, (E) turquoise module and (F) yellow module.
KEGG pathway enrichment analysis of 6 genes modules. (A) blue module, (B) brown module, (C) green module, (D) red module, (E) turquoise module and (F) yellow module.
Furthermore, protein-protein interaction network analysis demonstrated that TOP2A, MYC, ALB, CDK1, VEGFA, MMP9, PTPRC, CASR, EGFR and PTGS2 had the highest degree of connectivity among DEGs. TOP2A and CDK1 played an important role in regulation of cell cycle, which might modulate the tumor proliferation [36-38]. MYC, as an oncogene, encoded a transcription factor, triggered selective gene expression amplification to promote cell growth and proliferation [39]. Shroff EH et al. demonstrated that MYC overexpression causes RCC and pointed to the inhibition of glutamine metabolism [40]. ALB was reported to be an independent prognostic factor for patients with mRCC treated with angiogenesis-targeted therapy [41]. VEGFA, as a member of PDGF/VEGF growth factor family, induced proliferation and migration of vascular endothelial cells, and was essential for both physiological and pathological angiogenesis, which played a vital role in renal carcinogenesis [42, 43]. MMP9 (matrix metalloproteinase-9) was reported to have a strong correlation with tumor invasion and migration [44-46]. PTPRC, as an essential regulator of T- and B-cell antigen receptor signaling, might influence tumor proliferation via immune regulation [47]. CASR was a robust promoter of differentiation in colonic epithelial cells and functions as a tumor suppressor in colon cancer [48]. EGFR, upregulated in ccRCC, was a receptor tyrosine kinase involved in many important aspects of cell biology that are related to tumorigenesis [49-51]. PTGS2 was reported to be an prognostic biomarker for poor clinical outcome of upper tract urothelial cancer [52].
WGCNA analysis found six modules with highly relevant expression pattern. Then Gene Ontology enrichment analysis was performed to explore the biological process of each module. Blue module including 244 DEGs was involved in glycometabolic process such as hexose biosynthetic process, monosaccharide biosynthetic process and hexose metabolic process. Many studies had mentioned the close relationship between metabolism and renal carcinoma [53, 54]. Brown module including 199 DEGs was associated with immune response. Meanwhile, yellow module including 182 DEGs also participated in immune response, which revealed the correlation between renal cancer and immune regulation. 125 DEGs in green module were enriched in cell cycle regulation, playing a critical role in tumor proliferation. Red module including 71 DEGs was closely related to angiogenesis and vascular repair, relating to the prognosis of ccRCC. Turquoise module including 326 DEGs has a strong correlation to ion transport, which might participated in the tumor migration and invasion.
Pathway validation using qRT-PCR analysis. (A) PIK3CB, (B) PIK3CD, (C) PIK3CG, (D) AKT1, (E) AKT2, (F) AKT3, (G) NFKB2, (H) RAP1A and (I) RAP1B.
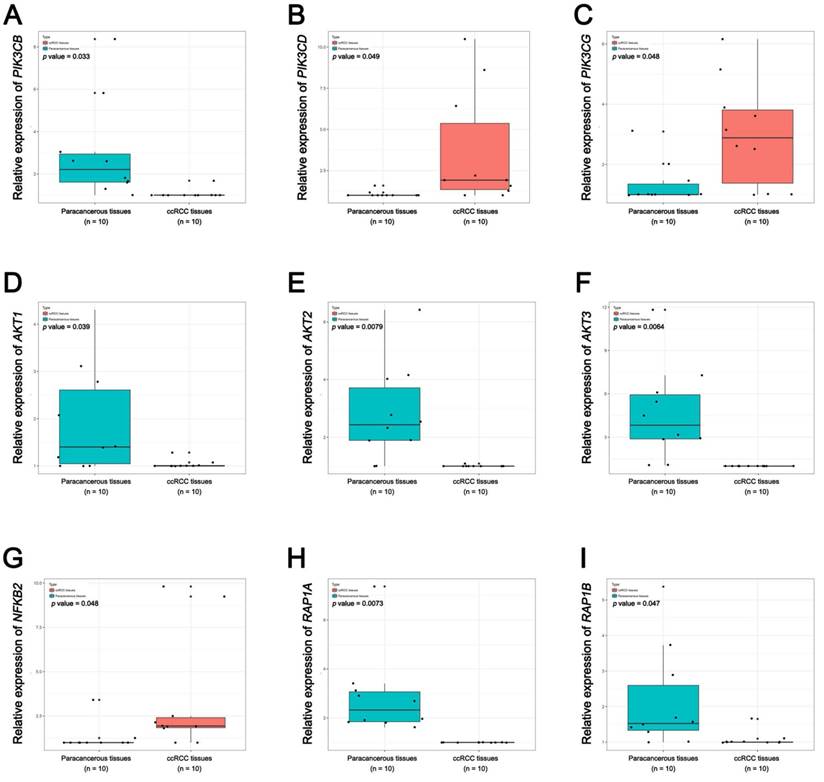
Pathway validation using TCGA KIRC data. (A) PIK3CB, (B) PIK3CD, (C) PIK3CG, (D) AKT1, (E) AKT2, (F) AKT3, (G) NFKB2, (H) RAP1A and (I) RAP1B (* p < 0.05; ** p < 0.01; *** p <0.001).
Venn plot of common deregulated genes. Blue: downregulated genes; red: upregulated genes.
In conclusion, by using a series of bioinformatics analysis, we have illustrated the hub genes and pathways which may be involved in the progress of ccRCC, based on differentially expressed genes between ccRCC samples and normal kidneys. However, further molecular biological experiments are needed to confirm the function of the candidate biomarkers in ccRCC.
Abbreviations
ccRCC: Clear Cell Renal Cell Carcinoma; WGCNA: Weighted Gene Co-expression Network Analysis; TCGA: The Cancer Genome Atlas; DEGs: Differentially Expressed Genes; TOM: Topological Overlap Matrix; STRING: Search Tool for the Retrieval of Interacting Genes; PPI: Protein-Protein Interaction; DAVID: Database for Annotation, Visualization and Integrated Discovery.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The excellent technical assistance of Yuan Zhu, Shanshan Zhang and Danni Shan at Zhongnan Hospital of Wuhan University is gratefully acknowledged. This study was supported in part by grants from Zhongnan Hospital of Wuhan University Science, Technology and Innovation Seed Fund (grant number: cxpy20160010 and cxpy20160031), Wuhan Clinical Cancer Research Center of Urology and Male Reproduction (grant number 303-230100055) and National Natural Science Foundation of China (grant number 81772730). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7-30
2. Vera-Badillo FE, Templeton AJ, Duran I. et al. Systemic therapy for non-clear cell renal cell carcinomas: a systematic review and meta-analysis. Eur Urol. 2015;67:740-9
3. Heng DY, Mackenzie MJ, Vaishampayan UN. et al. Primary anti-vascular endothelial growth factor (VEGF)-refractory metastatic renal cell carcinoma: clinical characteristics, risk factors, and subsequent therapy. Ann Oncol. 2012;23:1549-55
4. Dahinden C, Ingold B, Wild P. et al. Mining tissue microarray data to uncover combinations of biomarker expression patterns that improve intermediate staging and grading of clear cell renal cell cancer. Clin Cancer Res. 2010;16:88-98
5. Gerlinger M, Horswell S, Larkin J. et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014;46:225-33
6. Yang W, Yoshigoe K, Qin X. et al. Identification of genes and pathways involved in kidney renal clear cell carcinoma. BMC Bioinformatics. 2014;15(Suppl 17):S2
7. Yuan L, Chen L, Qian K, Qian G, Wu CL, Wang X, Xiao Y. Co-expression network analysis identified six hub genes in association with progression and prognosis in human clear cell renal cell carcinoma (ccRCC). Genom Data. 2017;14:132-40
8. White NM, Newsted DW, Masui O, Romaschin AD, Siu KW, Yousef GM. Identification and validation of dysregulated metabolic pathways in metastatic renal cell carcinoma. Tumour Biol. 2014;35:1833-46
9. Furge KA, Tan MH, Dykema K, Kort E, Stadler W, Yao X, Zhou M, Teh BT. Identification of deregulated oncogenic pathways in renal cell carcinoma: an integrated oncogenomic approach based on gene expression profiling. Oncogene. 2007;26:1346-50
10. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559
11. Yuan L, Shu B, Chen L. et al. Overexpression of COL3A1 confers a poor prognosis in human bladder cancer identified by co-expression analysis. Oncotarget. 2017;8:70508-20
12. Zhou Z, Liu S, Zhang M, Zhou R, Liu J, Chang Y, Zhao Q. Overexpression of Topoisomerase 2-Alpha Confers a Poor Prognosis in Pancreatic Adenocarcinoma Identified by Co-Expression Analysis. Dig Dis Sci. 2017;62(10):2790-2800
13. Wang F, Chang Y, Li J, Wang H, Zhou R, Qi J, Liu J, Zhao Q. Strong correlation between ASPM gene expression and HCV cirrhosis progression identified by co-expression analysis. Dig Liver Dis. 2017;49:70-6
14. Clarke C, Madden SF, Doolan P. et al. Correlating transcriptional networks to breast cancer survival: a large-scale coexpression analysis. Carcinogenesis. 2013;34:2300-8
15. Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3
16. Szklarczyk D, Franceschini A, Wyder S. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447-52
17. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498-504
18. Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4:2
19. Mason MJ, Fan G, Plath K, Zhou Q, Horvath S. Signed weighted gene co-expression network analysis of transcriptional regulation in murine embryonic stem cells. BMC Genomics. 2009;10:327
20. Horvath S, Dong J. Geometric interpretation of gene coexpression network analysis. PLoS Comput Biol. 2008;4:e1000117
21. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017 doi: 10.1093/nar/gkx247. [Epub ahead of print]
22. Giraldo NA, Becht E, Pages F. et al. Orchestration and Prognostic Significance of Immune Checkpoints in the Microenvironment of Primary and Metastatic Renal Cell Cancer. Clin Cancer Res. 2015;21:3031-40
23. Balan M, Mier y Teran E, Waaga-Gasser AM, Gasser M, Choueiri TK, Freeman G, Pal S. Novel roles of c-Met in the survival of renal cancer cells through the regulation of HO-1 and PD-L1 expression. J Biol Chem. 2015;290:8110-20
24. Sprowl JA, Lancaster CS, Pabla N. et al. Cisplatin-induced renal injury is independently mediated by OCT2 and p53. Clin Cancer Res. 2014;20:4026-35
25. Pochini L, Scalise M, Galluccio M, Indiveri C. OCTN cation transporters in health and disease: role as drug targets and assay development. J Biomol Screen. 2013;18:851-67
26. Yang M, Soga T, Pollard PJ. Oncometabolites: linking altered metabolism with cancer. J Clin Invest. 2013;123:3652-8
27. Hirschey MD, DeBerardinis RJ, Diehl AM. et al. Dysregulated metabolism contributes to oncogenesis. Semin Cancer Biol. 2015;35(Suppl):S129-50
28. Robertson-Tessi M, Gillies RJ, Gatenby RA, Anderson AR. Impact of metabolic heterogeneity on tumor growth, invasion, and treatment outcomes. Cancer Res. 2015;75:1567-79
29. Sudarshan S, Karam JA, Brugarolas J. et al. Metabolism of kidney cancer: from the lab to clinical practice. Eur Urol. 2013;63:244-51
30. Yang F, Zhang H, Mei Y, Wu M. Reciprocal regulation of HIF-1alpha and lincRNA-p21 modulates the Warburg effect. Mol Cell. 2014;53:88-100
31. Guo Y, Meng X, Ma J, Zheng Y, Wang Q, Wang Y, Shang H. Human papillomavirus 16 E6 contributes HIF-1alpha induced Warburg effect by attenuating the VHL-HIF-1alpha interaction. Int J Mol Sci. 2014;15:7974-86
32. Malec V, Coulson JM, Urbe S, Clague MJ. Combined Analyses of the VHL and Hypoxia Signaling Axes in an Isogenic Pairing of Renal Clear Cell Carcinoma Cells. J Proteome Res. 2015;14:5263-72
33. Hervouet E, Cizkova A, Demont J. et al. HIF and reactive oxygen species regulate oxidative phosphorylation in cancer. Carcinogenesis. 2008;29:1528-37
34. Hervouet E, Demont J, Pecina P, Vojtiskova A, Houstek J, Simonnet H, Godinot C. A new role for the von Hippel-Lindau tumor suppressor protein: stimulation of mitochondrial oxidative phosphorylation complex biogenesis. Carcinogenesis. 2005;26:531-9
35. Mikhaylova O, Ignacak ML, Barankiewicz TJ. et al. The von Hippel-Lindau tumor suppressor protein and Egl-9-Type proline hydroxylases regulate the large subunit of RNA polymerase II in response to oxidative stress. Mol Cell Biol. 2008;28:2701-17
36. Hongo F, Takaha N, Oishi M. et al. CDK1 and CDK2 activity is a strong predictor of renal cell carcinoma recurrence. Urol Oncol. 2014;32:1240-6
37. Xi Q, Huang M, Wang Y. et al. The expression of CDK1 is associated with proliferation and can be a prognostic factor in epithelial ovarian cancer. Tumour Biol. 2015;36:4939-48
38. Kang X, Song C, Du X. et al. PTEN stabilizes TOP2A and regulates the DNA decatenation. Sci Rep. 2015;5:17873
39. Qu A, Jiang C, Cai Y, Kim JH, Tanaka N, Ward JM, Shah YM, Gonzalez FJ. Role of Myc in hepatocellular proliferation and hepatocarcinogenesis. J Hepatol. 2014;60:331-8
40. Shroff EH, Eberlin LS, Dang VM. et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc Natl Acad Sci U S A. 2015;112:6539-44
41. Stenman M, Laurell A, Lindskog M. Prognostic significance of serum albumin in patients with metastatic renal cell carcinoma. Med Oncol. 2014;31:841
42. Ma X, Shen D, Li H. et al. MicroRNA-185 inhibits cell proliferation and induces cell apoptosis by targeting VEGFA directly in von Hippel-Lindau-inactivated clear cell renal cell carcinoma. Urol Oncol. 2015;33:169 e1-11
43. Song W, Yeh CR, He D, Wang Y, Xie H, Pang ST, Chang LS, Li L, Yeh S. Infiltrating neutrophils promote renal cell carcinoma progression via VEGFa/HIF2alpha and estrogen receptor beta signals. Oncotarget. 2015;6:19290-304
44. Cheng X, Yang Y, Fan Z. et al. MKL1 potentiates lung cancer cell migration and invasion by epigenetically activating MMP9 transcription. Oncogene. 2015;34:5570-81
45. Lin H, Pan JC, Zhang FM. et al. Matrix metalloproteinase-9 is required for vasculogenic mimicry by clear cell renal carcinoma cells. Urol Oncol. 2015;33:168 e9-16
46. Baldwin RM, Haghandish N, Daneshmand M, Amin S, Paris G, Falls TJ, Bell JC, Islam S, Cote J. Protein arginine methyltransferase 7 promotes breast cancer cell invasion through the induction of MMP9 expression. Oncotarget. 2015;6:3013-32
47. Minarik I, Lastovicka J, Budinsky V, Kayserova J, Spisek R, Jarolim L, Fialova A, Babjuk M, Bartunkova J. Regulatory T cells, dendritic cells and neutrophils in patients with renal cell carcinoma. Immunol Lett. 2013;152:144-50
48. Singh N, Promkan M, Liu G, Varani J, Chakrabarty S. Role of calcium sensing receptor (CaSR) in tumorigenesis. Best Pract Res Clin Endocrinol Metab. 2013;27:455-63
49. Matusan-Ilijas K, Damante G, Fabbro D. et al. EGFR expression is linked to osteopontin and Nf-kappaB signaling in clear cell renal cell carcinoma. Clin Transl Oncol. 2013;15:65-71
50. Cossu-Rocca P, Muroni MR, Sanges F. et al. EGFR kinase-dependent and kinase-independent roles in clear cell renal cell carcinoma. Am J Cancer Res. 2016;6:71-83
51. Mirza Z, Schulten HJ, Farsi HM, Al-Maghrabi JA, Gari MA, Chaudhary AG, Abuzenadah AM, Al-Qahtani MH, Karim S. Molecular interaction of a kinase inhibitor midostaurin with anticancer drug targets, S100A8 and EGFR: transcriptional profiling and molecular docking study for kidney cancer therapeutics. PLoS One. 2015;10:e0119765
52. Ke HL, Tu HP, Lin HH. et al. Cyclooxygenase-2 (COX-2) up-regulation is a prognostic marker for poor clinical outcome of upper tract urothelial cancer. Anticancer Res. 2012;32:4111-6
53. Li B, Qiu B, Lee DS. et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature. 2014;513:251-5
54. Suhre K, Shin SY, Petersen AK. et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature. 2011;477:54-60
Author contact
![]() Corresponding authors: Dr. Xinghuan Wang, Email: wangxinghuanedu.cn, Tel. +86-27-6781-3104, Fax: +86-27-6781-2892, and Dr. Yu Xiao, Email: yu.xiaoedu.cn, Tel. +86-27-6781-2689, Fax: +86-27-6781-2892.
Corresponding authors: Dr. Xinghuan Wang, Email: wangxinghuanedu.cn, Tel. +86-27-6781-3104, Fax: +86-27-6781-2892, and Dr. Yu Xiao, Email: yu.xiaoedu.cn, Tel. +86-27-6781-2689, Fax: +86-27-6781-2892.
Received 2017-10-31
Accepted 2018-1-11
Published 2018-2-12