ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2018; 14(4):406-417. doi:10.7150/ijbs.23542 This issue Cite
Research Paper
Simvastatin Suppresses Proliferation and Migration in Non-small Cell Lung Cancer via Pyroptosis
Fengjiao Wang1#, Wei Liu2#, Jinfeng Ning1, Junfeng Wang1, Yaoguo Lang1, Xiangyuan Jin1, Kaibin Zhu1, Xiuyun Wang3, Xiaoguang Li4, Fan Yang5, Jianqun Ma1 ![]() , Shidong Xu1
, Shidong Xu1 ![]()
1. Department of Thoracic Surgery, Harbin Medical University Cancer Hospital, Harbin, 150001, China
2. The Fourth Department of Medical Oncology, Harbin Medical University Cancer Hospital, Harbin, 150001, China
3. Department of Abdominal Ultrasound, First Hospital of Harbin Medical University, Harbin, 150001, China
4. Department of Cell Biology, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA
5. Department of Endocrinology, The Second Affiliated Hospital of Harbin Medical University, Harbin, 150001, China
#These authors contributed equally to this work and should be considered co-first authors
Abstract

Pyroptosis is a form of caspase-1-dependent programmed cell death with anti-tumor properties, but the underlying molecular mechanisms are not fully understood. The results of our study showed that the antihyperlipidemic drug simvastatin induced pyroptosis in non-small cell lung cancer (NSCLC) cell lines and a xenograft mouse model. Inhibition of pyroptosis attenuated the effects of simvastatin on tumor cell viability and migration. These data suggest that simvastatin may induce pyroptosis, thereby potentially serving as a novel therapeutic agent for NSCLC.
Keywords: Simvastatin, NSCLC, pyroptosis, caspase-1, cancer therapy
Introduction
Lung cancer is the leading cause of cancer-related death worldwide, with a dismal prognosis and a 5-year survival rate lower than 15% [1]. Non-small cell lung cancer (NSCLC), which includes adenocarcinoma, squamous cell carcinoma, and large-cell carcinoma, accounts for approximately 80% of all lung cancers [2]. Evidence has shown that, compared with small cell lung cancer, NSCLC is relatively insensitive to chemotherapy [3], and the effects of current therapeutic approaches have been less than optimal. Thus, novel strategies based on a better understanding of tumor biology may improve clinical survival rates.
Pyroptosis is a form of programmed cell death that is uniquely dependent on caspase-1 [4]. An important function of caspase-1 is to process inactive proforms of the inflammatory cytokines interleukin 1β (IL-1β) and IL-18 to generate their active forms [5]. The morphology of pyroptosis has characteristics of both apoptotic and necrotic cell death [6]. In monocytes, macrophages, dendritic cells, and other cell types, pyroptotic cells lose their cell membrane potential and undergo DNA fragmentation [7]. There is increasing evidence supporting the hypothesis that pyroptosis has anticancer properties, but the underlying molecular mechanisms are not fully understood.
Statins are prescribed for the treatment of elevated serum cholesterol levels in patients [8]. They reduce cholesterol levels by inhibiting the bottleneck enzyme of cholesterol synthesis, 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase [9]. In addition to their use in the treatment of lipid disorders, statins have been investigated for their anticarcinogenic effects in several models, including carcinoma of the colon and rectum, prostate, liver, breast, lung, and skin [10,11,12]. Simvastatin is one of the most widely used drugs and is considered to be very safe for long-term treatment [13].
We conducted this study to examine, in the context of NSCLC, the relationship between simvastatin and pyroptosis of the human lung cancer cells A549 and H1299. The results showed that simvastatin inhibited cell viability and mobility by inducing pyroptosis.
Materials and Methods
Lung cancer and adjacent non-tumor tissues
Samples of 5 paired NSCLC and adjacent non-tumor lung tissues (located > 5 cm away from the tumors) were obtained from patients treated at the Department of Thoracic Surgery, Harbin Medical University Cancer Hospital, China. The 5 patients, aged 45- 71 years, were all diagnosed with primary NSCLC and had received no other treatments before surgery. Both the tumor and non-tumor samples were confirmed as such by pathological examination. Samples were snap-frozen in liquid nitrogen after resection. Patients were excluded if they had recurrent NSCLC or had primary NSCLC treated with chemoradiotherapy before surgery. Written informed consent was obtained from each participant after a full explanation of the study was provided. This study was approved by the Ethics Committee of Harbin Medical University according to the Interdisciplinary Principles and Guidelines for the Use of Animals in Research, Testing, and Education (the New York Academy of Sciences, Ad Hoc Animal Research Committee).
Hematoxylin and eosin staining
Tumor and adjacent non-tumor tissues were fixed in 4% paraformaldehyde and then dehydrated. Processed samples were embedded in paraffin and cut into 5-μm thick sections using tissue-processing equipment. Sections were deparaffinized and stained with hematoxylin and eosin (HE) for histological analysis.
Cell culture
HLF-a human lung cells, A549 human adenocarcinoma cells, and H1299 NSCLC cells were a generous gift from Department of Genetics, Harbin Medical University. Cells were maintained in Dulbecco's modified Eagle medium (Gibco/ Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Gibco), penicillin (100 U/mL), and streptomycin (100 μg/mL) and incubated in a humidified atmosphere of 5% CO2 95% air at 37°C.
Immunohistochemical analysis
Paraffin sections were baked at 60°C, deparaffinized in xylene, rehydrated in graded ethanol, and microwaved for antigen retrieval. Slides were incubated with primary antibodies against caspase-1 (Cell Signaling Technologies, Beverly, MA) at 4°C overnight. The slides were then washed, further incubated with secondary antibodies for 2 h at room temperature, and stained with diaminobenzidine.
Immunofluorescence staining
Cells were plated on coverslips in 24-well plates and allowed to reach 60% confluence. Then, the cells were treated with simvastatin or simvastatin+Ac-YVAD-CMK (inhibitor of caspase-1) for 48 h, fixed in 4% paraformaldehyde, permeabilized with 0.05% Triton X-100 in phosphate buffered saline (PBS), and blocked in 10% bovine serum albumin in PBS. The cells were incubated with primary antibodies against caspase-1 (Cell Signaling Technologies) at 4°C overnight, then washed 3 times in PBS and incubated for 1 h with the appropriate secondary antibodies (1:1,000). The stained cells were observed under a confocal microscope.
MTT assay
Cell viability was determined with an MTT assay according to the manufacturer's instructions. Briefly, cells (2×104 cells/well) were seeded in a 96-well plate and treated with simvastatin or simvastatin+Ac-YVAD-CMK. After treatment, 20 μL MTT solution (0.5 mg/mL; Sigma) were added to each well and incubated at 37°C for 48 h. After carefully removing the culture medium, 200 μL dimethyl sulfoxide was added to each well to dissolve the formazin, and the absorbance was measured at 490 nm using a microplate reader.
Qualitative polymerase chain reaction analysis
Caspase-1 mRNA levels were determined by qualitative polymerase chain reaction (qPCR). Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA) from tissues and cells. First-strand cDNA was synthesized using a reverse transcriptase kit (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions. qPCR was performed with the SYBR Green PCR Master Mix Kit (Applied Biosystems) and using a 7500 FAST Real-Time PCR System (Applied Biosystems). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control. The following primers were used. Caspase-1: Forward, 5'-ACACGTCTTGCCCTCATTATCT-3', Reverse, 5'-ATAACCTTGGGCTTGTCTTTCA-3'; GAPDH: Forward, 5'-ATCACTGCCACCCAGAAGAC3', Reverse, 5'-TTTCTAGACGGCAGGTCAGG-3'. IL-1β: forward 5' ATGATGGCTTATTACAGTGGCAA 3', reverse 5' GTCGGAGATTCGTAGCTGGA 3'.
Western blot analysis
Protein samples were extracted from tissues and cells for western blot analysis. The concentration of protein samples was determined using the BCA Protein Assay Kit (Bio-Rad, Mississauga, ON, Canada). Protein samples were resolved on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and electrotransferred onto polyvinylidene fluoride membranes. After blocking in 5% nonfat milk for 2 h at room temperature, the membranes were treated with antibodies against caspase-1 (Cell Signaling Technologies), IL-1β, IL-18, and GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C, followed by secondary antibodies at room temperature for 1 h. Western blot bands were quantified using Odyssey v1.2 software by measuring the band intensity (area × OD) for each group. GAPDH served as an internal control.
Wound healing assay
H1299 and A549 cells were incubated in a 6-well culture plate to achieve 90% confluence, and the cell monolayer was scratched in a straight line with a pipette tip. To remove the debris and smooth the edge of the scratch, the culture medium was removed and the wells were washed 3 times with medium. Then, cells were treated in different conditions, including 1 and 2 μm simvastatin. After 48 h, the width of the open area was immediately measured and the distance of wound closure was used to estimate motility.
Transwell assay
Cell migration and invasion were evaluated using 8-μm transwell filters (BD Biosciences, Franklin Lakes, NJ). Briefly, H1299 and A549 cells in different conditions were suspended in 0.5 mL serum-free media and added to the upper chamber with an uncoated or Matrigel-coated membrane; medium containing 10% FBS was added to the lower chamber. After incubation for 24 h, cells that migrated or invaded through and adhered to the bottom of the membrane were fixed and stained. Five random fields were captured for each membrane, and the migratory and invasive cells were counted and averaged.
Terminal deoxynucleotidyl transferase-mediated-digoxigenin-11-dUTP nick end labeling
DNA fragmentation in A549 and H1299 cells treated with simvastatin, or simvastatin+Ac-YVAD-CMK was detected by terminal deoxynucleotidyl transferase-mediated-digoxigenin-11-dUTP nick end labeling (TUNEL) according to the instructions of the TUNEL kit (Roche, Indianapolis, IN, USA).
Xenograft mouse model
Approval was obtained from the Ethics Committee of Harbin Medical University. All related procedures conformed to the Animal Protection Act of China. BALB/c-nude male mice (age 8 weeks, weight 18-20 g) were given free access to food and water and were maintained in a pathogen-free environment. Fifteen mice were randomly divided into three groups: normal group, tumor group, and tumor+simvastatin group. H1299 lung cancer cells (5×106 cells suspended in 100 μL PBS) or PBS as a control were injected via the tail vein, and the animals were fed by oral gavage with saline in the tumor group and simvastatin (5 mg·kg-1·d-1) in the tumor+simvastatin group. Mice were sacrificed after 4 weeks, and tumors were identified and isolated for further examination.
Statistical analysis
Data are expressed as the mean ± standard error of mean (SEM) and were analyzed with SPSS 13.0 software (IBM Corporation, Armonk, NY). Statistical differences between two groups were determined using Student's t-test and among multiple groups using analysis of variance. A two-tailed P < 0.05 was taken to indicate statistical significance.
Results
Caspase-1, IL-1β, and IL-18 were downregulated in NSCLC tumor tissues
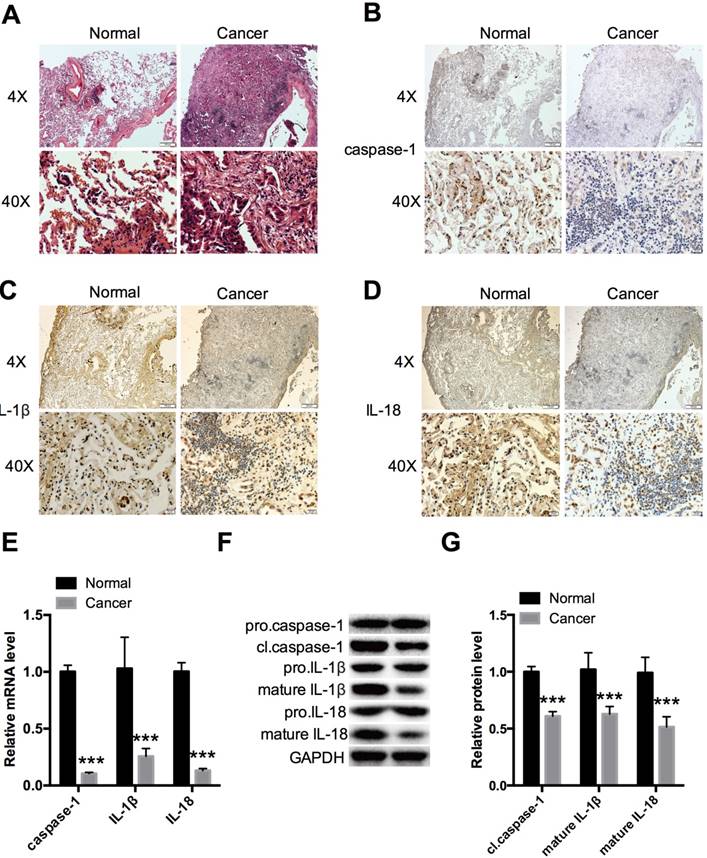
Tumor and adjacent non-tumor tissues were compared in NSCLC tissues for expression of caspase-1 (Fig. 1A). Immunohistochemistry revealed remarkably decreased expression in tumor tissues compared with normal tissues (Fig. 1B). The cleavage of pro-IL-1β and pro-IL-18 by caspase-1 is a principle requirement for the release of IL-1β and IL-18 [5]; thus, IL-1β and IL-18 are markers of caspase-1 activation. We therefore evaluated IL-1β and IL-18 expression and found that these were downregulated in tumor tissues (Fig. 1C, 1D). Caspases are synthesized as inactive enzymogens (pro-caspases) that are activated only with an appropriate stimulus. Similar to other caspases, caspase-1 is proteolytically activated from a proenzyme to produce a tetramer of its two active subunits, p20 and p10 [4]. We detected the protein expression of the active p20 subunit of caspase-1, which reflected the activity of caspase-1. The expression level of caspase-1 was also monitored by qPCR (Fig. 1E) and western blot analysis (Fig. 1F, G) in the 5 paired NSCLC and adjacent non-tumor lung tissue samples. The results demonstrated that caspase-1 was downregulated in the tumor group. The same trends were seen for IL-1β and IL-18 by qPCR, and for the mature forms of IL-1β and IL-18 by western blotting.
Caspase-1 expression was low in H1299 and A549 NSCLC cells
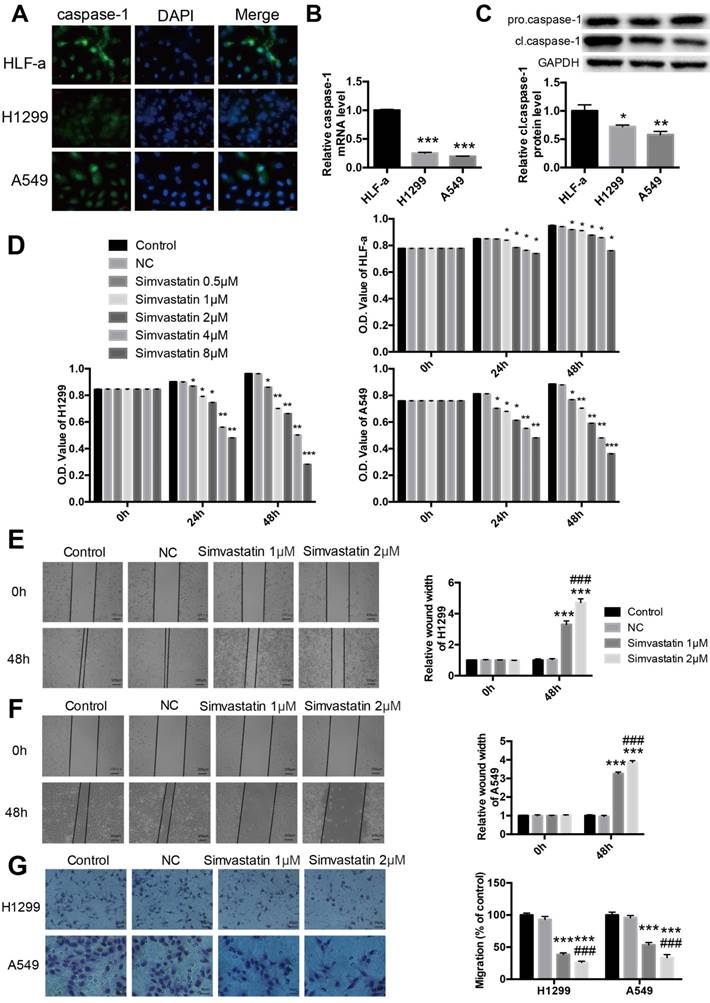
To further explore the expression of caspase-1 in NSCLC cell lines, immunofluorescence staining was used to confirm the difference between normal lung and NSCLC cell lines. As shown in Fig. 2A, compared with normal HLF-a lung cells, the expression of caspase-1 was remarkably decreased in H1299 and A549 NSCLC cells. Similar results were observed at both the mRNA level of caspase-1 and protein level of cleaved caspase-1 (cl.caspase-1) in the tumor groups (Fig. 2B, C).
Expression levels of caspase-1, IL-1β, and IL-18 in NSCLC tissues (A)HE staining showing histopathological changes in tumors at 4× and 40× magnification. Immunohistochemical analysis revealed expression of caspase-1 (B), IL-1β (C), and IL-18 (D) in 5 paired NSCLC and adjacent non-tumor lung tissue samples at 4× and 40× magnification. (E) Relative expression of caspase-1, IL-1β, and IL-18 was measured by qPCR. (F, G) Western blot analysis of caspase-1, IL-1β, and IL-18 expression in different forms in two groups. ***P < 0.001 compared with the normal group. Normal: adjacent non-tumor tissue; Cancer: tumor tissue. Data are expressed as the mean ± SEM, n=3.
Expression of caspase-1 in NSCLC cell lines and effects of simvastatin on NSCLC cell proliferation and mobility Expression of caspase-1 in normal lung HLF-a cells and H1299 and A549 NSCLC cells. (A) Immunofluorescence staining was used to reveal expression of caspase-1 (Green). (B) Caspase-1 mRNA expression detected by qPCR. (C) pro-caspase-1 and cl-caspase-1 protein expression detected by western blotting. (D) HLF-a, H1299, and A549 cells were incubated with 0.5, 1, 2, 4, and 8 μM simvastatin for 24 or 48 h. Control cells remained untreated. The proportion of surviving cells was determined by MTT assay. Effects of simvastatin (1 and 2 μM) on cell migration were evaluated by wound healing assay (E, F) and transwell assay (G). GAPDH served as an internal control. Data are expressed as the mean ± SEM, n=3. *P < 0.05 versus HLF-a; **P < 0.01 versus HLF-a; ***P < 0.001 versus HLF-a. ###P < 0.001 versus 1 μM simvastatin.
Simvastatin reduced the viability and motility of H1299 and A549 cells
Cell proliferation, migration, and invasion are important characteristics of cancer cells and indicators of malignancy. As demonstrated by the MTT assay, simvastatin significantly reduced H1299 and A549 cell viability in a dose-dependent manner (Fig. 2D). Treatment with 8 μM simvastatin for 48 h led to the strong inhibition of tumor cell viability. As shown in the wound healing assay, treatment with 1 or 2 μM simvastatin resulted in a significant reduction in the migration of NSCLC cells compared with the control (Fig. 2E, F). Similar results were noted in the transwell migration assay (Fig. 2G).
Simvastatin induced pyroptosis in H1299 and A549 cells by activating NLRP3 -caspase-1- IL-1β and IL-18 pathways
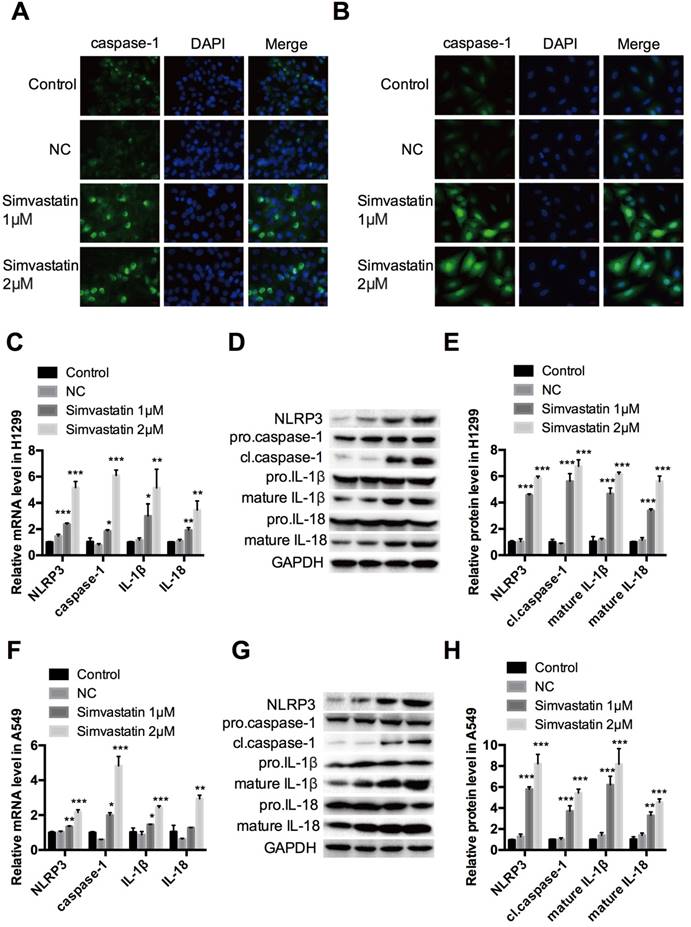
Treatment with simvastatin for 48 h in A549 and H1299 cancer cells increased growth inhibition in a concentration-dependent manner. Interestingly, the same dosage of simvastatin had less or even no suppressive effects on the proliferation of HLF-a cells. To explore the underlying mechanism, we examined caspase-1 expression by immunofluorescence staining, qPCR, and western blot analysis. The results showed that caspase-1 immunofluorescence staining (Fig. 3A, B), as well as mRNA (Fig. 3C, F) and cl.caspase-1 protein (Fig. 3D, E, G, H) expression were all upregulated in H1299 and A549 cells in a concentration-dependent manner after simvastatin treatment. To confirm this, caspase-1 upstream markers (nucleotide-binding domain and leucine-rich repeat-containing (NLR) pyrin domain 3 [NLRP3]) and downstream markers (mature IL-1β and IL-18) were also analyzed. They all had remarkably higher mRNA and protein expression than the control group. Taken together, these data show that simvastatin induced caspase-1 expression and activation, leading to pyroptosis in NSCLC cells.
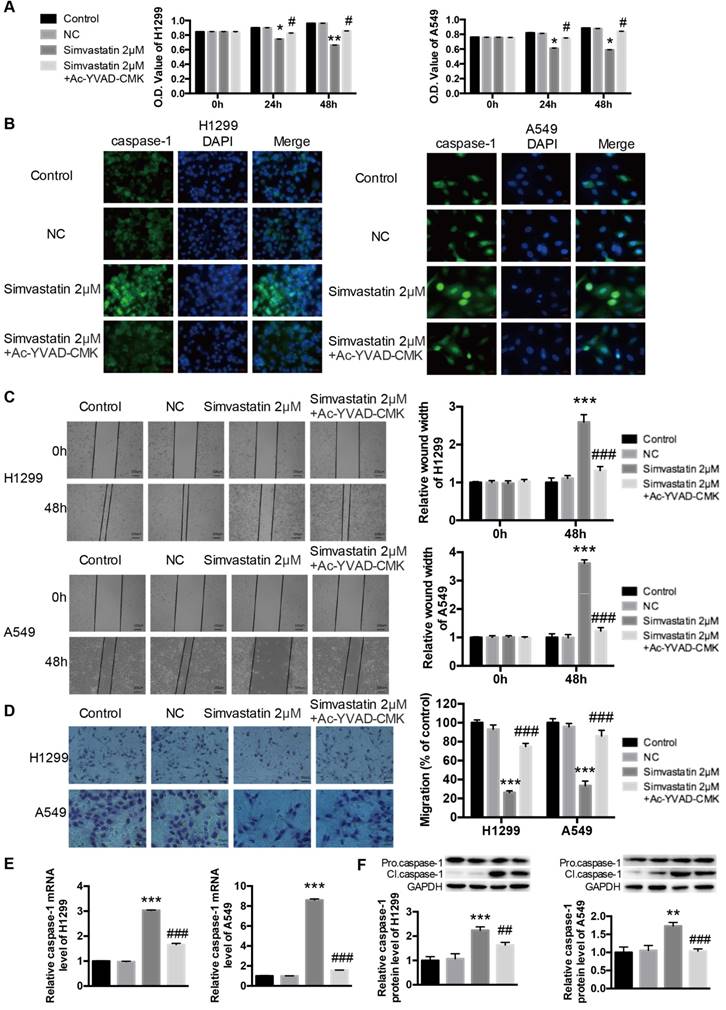
Ac-YVAD-CMK attenuated the effects of simvastatin on tumor cell viability, motility, and caspase-1 expression
Ac-YVAD-CMK is a specific caspase-1 inhibitor that can inhibit caspase-1 activation, caspase-1 expression, and pyroptotic cell death. Simvastatin significantly reduced the number of viable H1299 and A549 cancer cells (Fig. 4A). However, co-treatment with Ac-YVAD-CMK (100 μM) decreased simvastatin-mediated growth inhibition. Caspase-1 activation was measured using immunofluorescence staining. As shown in Figure 4B, there was a marked increase in activated caspase-1 in H1299 and A549 cells after treatment with simvastatin, whereas Ac-YVAD-CMK decreased this effect. Wound healing and transwell assays (Fig. 4C, D) showed that Ac-YVAD-CMK also attenuated the inhibitory effects of simvastatin on the motility of H1299 and A549 cells. As for caspase-1 expression, the results were coincident, and Ac-YVAD-CMK decreased simvastatin-induced caspase-1 expression at both the mRNA and protein levels (Fig. 4E, F).
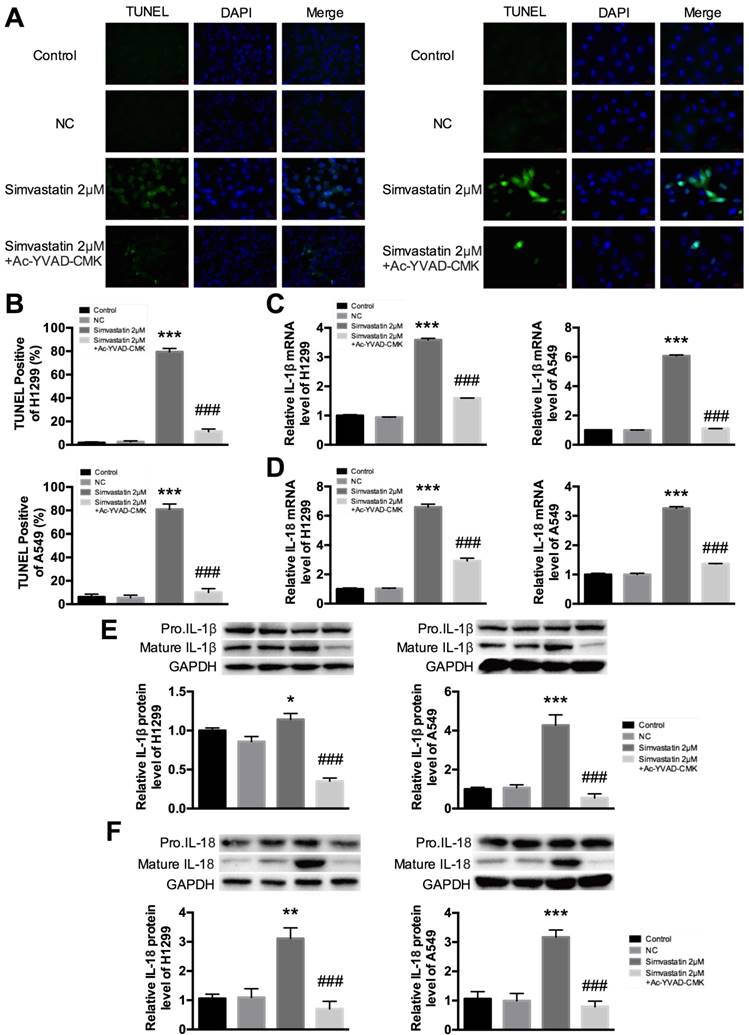
Ac-YVAD-CMK decreased simvastatin-induced pyroptosis in H1299 and A549 cells
Figure 5A and B show fluorescent signals of TUNEL and DAPI staining of H1299 and A549 cells; TUNEL staining was used to monitor DNA damage, and DAPI staining was used to monitor morphological changes of nuclei. In the untreated group, there was a small number of dead cells. Compared with this group, cell death rate was significantly increased at 48 h after treatment with simvastatin. However, the increase was markedly reduced in the groups treated with Ac-YVAD-CMK. These results suggest that treatment with simvastatin led to cell death by inducing pyroptosis. To ensure that pyroptosis was involved in this process, we examined the change in expression of IL-1β and IL-18. Cleavage of pro-IL-1β and pro-IL-18 by caspase-1 is a principle requirement for the release of IL-1β and IL-18 [5]. We evaluated the expression of IL-1β and IL-18 in A549 and H1299 cells treated with simvastatin or simvastatin+Ac-YVAD-CMK. As expected, mRNA expression of IL-1β and IL-18 dramatically increased after treatment with simvastatin. However, co-treatment with Ac-YVAD-CMK suppressed IL-1β and IL-18 mRNA expression (Fig. 5C, D). Similar results were observed for mature IL-1β and IL-18 protein levels. These results suggest that simvastatin-induced inhibitory effects on cell viability were mediated by caspase-1-induced pyroptosis.
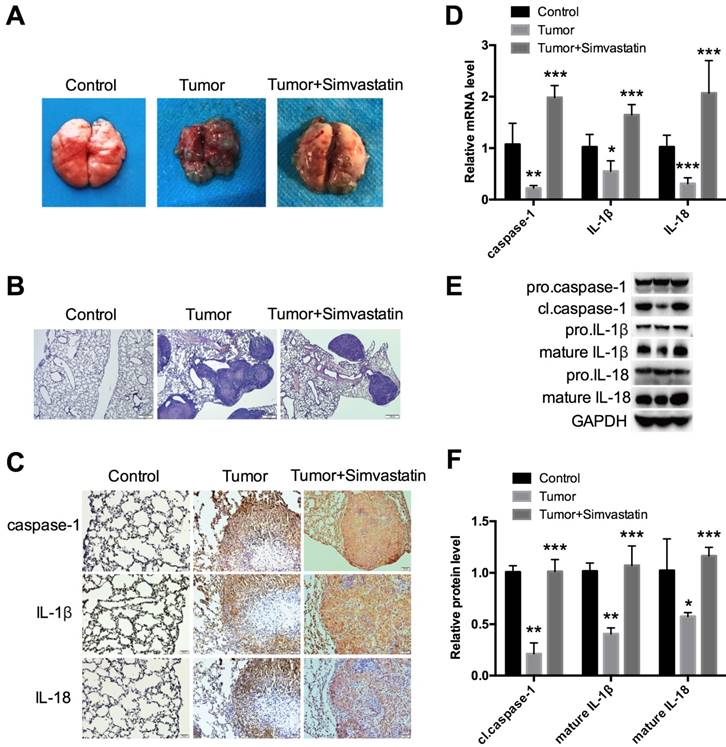
Simvastatin suppressed NSCLC tumor growth in a xenograft mouse model by activating caspase-1-induced pyroptosis
To further explore the effects of simvastatin in vivo, a xenograft mouse model was established. H1299 cells were injected into nude mice via the tail vein. The mice were then fed saline or simvastatin daily for 4 weeks until sacrifice to evaluate the lung tissue. As expected, simvastatin strongly suppressed tumor growth as shown in Figure 6A. From HE staining, each tumor focus had a similar size, but the total number was clearly lower in the simvastatin treatment group (Fig. 6B), suggesting that simvastatin had stronger inhibitory effects on cell invasion and migration than on cell growth. Interestingly, expression of caspase-1, IL-1β, and IL-18 was low, especially in the center of the tumor, but it was homogeneously high after treatment with simvastatin (Fig. 6C). mRNA expression (Fig. 6D) and protein expression (Fig. 6E, F) of caspase-1, IL-1β, and IL-18 in different forms were all analyzed. Similar to the results from the in vitro experiments, caspase-1, IL-1β, and IL-18 expression was suppressed in the tumors and could be reversed by simvastatin. These data suggest that simvastatin suppressed NSCLC tumor growth in mice by activating caspase-1-mediated pyroptosis.
Effects of simvastatin treatment on caspase-1 expression H1299 and A549 lung cancer cells were incubated with 1 or 2 μM simvastatin for 24 h. Immunofluorescence staining revealed the expression of caspase-1 (green) in (A) H1299 and (B) A549 cells. (C, F) qPCR was performed to detect the expression of caspase-1 and its upstream (NLRP3) and downstream (IL-1β, IL-18) markers. NLRP3, cl-caspase-1, pro-IL-1β, mature IL-1β, pro-IL-18, and mature IL-18 protein expression was (D, G) evaluated by western blotting and (E, H) was quantified. *P < 0.05 versus control; **P < 0.01 versus control; ***P < 0.001 versus control.
Ac-YVAD-CMK decreased effects of simvastatin on cells and caspase-1 expression H1299 and A549 cells were treated with 2 μM simvastatin or simvastatin+Ac-YVAD-CMK (100 μM) for 48 h. (A) The number of viable cells was determined by MTT assay. Caspase-1 activation and expression were analyzed by (B) immunofluorescence staining, (E) qPCR, and (F) western blotting. Cell mobility was measured by (C) wound healing assay and (D) transwell assay. *P < 0.05 versus control; **P < 0.01 versus control; ***P < 0.001 versus control; #P < 0.05 versus 2 μm simvastatin; #P < 0.01 versus 2 μm simvastatin; ###P < 0.001 versus 2 μM simvastatin.
In vitro evidence for simvastatin-mediated pyroptosis (A) Representative photomicrographs showing TUNEL staining. H1299 and A549 cells were treated with 2 μM simvastatin or 2 μM simvastatin+100 μM Ac-YVAD-CMK. (B) Pyroptotic cells were induced after 48 h of simvastatin and were quantified. Cell death significantly increased in the simvastatin group compared with the untreated group. Treatment with Ac-YVAD-CMK led to decreased apoptotic death. TUNEL-positive cells, green; nuclei, blue. IL-1β and IL-18 activation and expression were analyzed by (C, D) qPCR and (E, F) western blotting. *P < 0.05, **P < 0.01, ***P < 0.001 versus control group; ###P < 0.001 versus 2 μM Berberine; results are representative of three separate independent experiments.
Simvastatin suppressed NSCLC tumor growth in a mouse model by activating caspase-1 H1299 cells were first injected into BALB/c-nude male mice through the tail vein. (A) Analysis of tumor after 4 weeks of continuous treatment with PBS or simvastatin. (B) Representative HE staining of the lung. (C) Immunohistochemical staining and (D) qPCR were used to detect expression of caspase-1, IL-1β, and IL-18. (E, F) Western blot was used to detect cl.caspase-1, mature IL-1β, and mature-IL-18 protein expression. *P < 0.05, **P < 0.01, ***P < 0.001 versus control group; results are representative of three separate independent experiments.
Discussion
Statins are beneficial to cardiovascular health and are used to prevent coronary heart disease [8]. Recently, however, their anticancer properties have received increasing attention. Cardwell et al. [13] provided some evidence that using statins reduced lung cancer-specific mortality whether used before or after diagnosis. A similar phase II trial showed improvement in survival in a group that received simvastatin plus gefitinib compared with gefitinib only [14]. Both studies proved that statin usage had effects in improving survival in lung cancer patients. Some basic studies have shown reduced proliferation and migration and increased apoptosis in several types of cancer, including lung adenocarcinoma [12,15]. Similarly, we showed that simvastatin inhibited proliferation and migration of A549 and H1299 lung cancer cells in a concentration-dependent manner, but these effects were not observed in normal HLF-a lung cells. The anticancer properties of simvastatin have the potential to increase chemoprevention and treatment efficacy in NSCLC, which could enable the dose of other agents to be reduced to minimize possible detrimental side effects [9]. Moreover, simvastatin was not toxic to normal lung cells. This makes it an ideal adjuvant therapy for NSCLC patients, although its underlying molecular mechanisms are not fully understood.
Simvastatin can serve as an inhibitor of HMG-CoA reductase by interfering with the metabolism of the mevalonate pathway. [9] However, it has been suggested that its anticancer mechanisms may be HMG-CoA-independent. [9] In lung cancer, Lee et al. [16] reported that simvastatin upregulated BIM expression to promote apoptosis. Li et al. [17] suggested that simvastatin enhanced oxidative stress and upregulated the expression of superoxide dismutase 2, inhibiting the proliferation of A549 lung cells in a concentration- and time-dependent manner. However, the present study is the first to indicate that pyroptosis is closely involved in the anticancer effects of simvastatin.
Pyroptosis is a unique inflammatory form of programmed cell death, induced by caspase-1 in the canonical pathway and by murine caspase-11 (human caspase-4/-5) in the noncanonical pathway [5,18]. Both ways of inducing pyroptosis require two-step activation. The first step is identical, in which inflammasomes are primed and the expression of pro-IL1β and pro-IL18 is increased by activating pattern-recognition receptors. In the second step, canonical and noncanonical pyroptosis differ. Activated caspase-1, activated by inflammasomes, catalyzes the proteolytic maturation of cytokine substrates pro-IL-1β and pro-IL-18 into their active forms and also executes programmed cell death, known as pyroptosis, in the canonical pathway [5]. In contrast, activated caspase-11 induces pyroptosis directly, but requires activated caspase-1 indirectly to produce the active forms of IL-1β and IL-18 in the noncanonical pathway [18]. Thus, activated caspase-1 is key in both pathways. In our study, pyroptosis induced by simvastatin by activating NLRP3, caspase-1, and mature forms of IL-1β and IL-18. Importantly, the inhibitory effects of simvastatin on migration could be attenuated by Ac-YVAD-CMK, a specific caspase-1 inhibitor, suggesting that simvastatin suppresses proliferation and migration NSCLC via pyroptosis.
The ability to escape programmed death, such as apoptosis, is the hallmark of cancer [19], and an immortal status is essential for cancer development. Therefore, it is reasonable that pyroptosis has anticancer properties [20]. Hu et al. [21, 22] reported that caspase-1 regulated colonic epithelial cell proliferation and programmed cell death directly and profoundly. In this colitis-associated colorectal cancer model, caspase-1-deficient colon epithelial cells promoted tumor formation in the early stage and exhibited increased tumor load compared with wild type. Chang et al. [23] showed that a cytochrome P450 1B1 inhibitor impaired the proliferation and migration of prostate cancer cells by upregulating caspase-1 expression and that reducing activated caspase-1 weakened its anticancer effects. However, little has been reported on the role of caspase-1-dependent pyroptosis in lung cancer. We demonstrated that caspase-1 was downregulated in NSCLC tumor tissues and cell lines H1299 and A549, which raises the possibility that pyroptosis participates in cancer suppression and that caspase-1 is a potential cancer therapeutic target.
Other studies have reported that simvastatin augmented apoptosis in NSCLC cells [12, 14, 24]. Pyroptosis can be distinguished from apoptosis by three characteristics. First, pyroptosis is accompanied by inflammatory cytokine release such as IL-1β and IL-18. Second, pyroptosis is induced by caspase-1 and caspase-4/-5 in humans, but capase-2/-8/-9/-10 are required to activate the downstream caspases caspase-3/-6/-7 in apoptosis [20]. Third, similar to apoptosis, pyroptosis also results in nuclear fragmentation, but it is not as prominent as that observed in apoptosis [7]. However, TUNEL assay is useful for identification [7]. We detected activated capase-1, L-1β, and IL-18 by western blotting and DNA fragmentation by TUNEL assay, confirming that pyroptosis had occurred. Pyroptosis, apoptosis, and necrosis are different types of cell death; they are not mutually exclusive but are related [25]. Thus, pyroptosis, as a new molecular mechanism underlying the anticancer properties of simvastatin, warrants further investigation.
In summary, this study demonstrated that pyroptosis induced by simvastatin occurred in A549 and H1299 lung cancer cells and in a xenograft mouse model, indicating that simvastatin could be a potential drug for the clinical treatment of NSCLC without causing toxicity to normal lung cells. The findings of this study also shed some light on the anticancer effects of pyroptosis with NLRP3-caspase-1-L-1β and IL-18 axis activation, which should be evaluated further in additional studies.
Abbreviations
HE: hematoxylin and eosin; HMG-CoA: 3-hydroxy- 3-methylglutaryl-CoA; NSCLC: non-small cell lung cancer; PBS: phosphate buffered saline; PRR: pattern-recognition receptor; SEM: standard error of mean; TUNEL: Terminal deoxynucleotidyl transferase-mediated-digoxigenin-11-dUTP nick end labeling.
Acknowledgements
This work was supported by the Hai Yan Foundation (JJQN2016-05, JJMS2014-04), Health Department of Heilongjiang Province of China (2017-116), Harbin Bureau Science and Technology (2015RAQYJ101), Education Department of Heilongjiang Province of China (12541317), Wu Jieping Medical Foundation (320.6799.15046), and Heilongjiang Medial Science Institute (201606).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Shaw AT, Kim DW, Mehra R. et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370(13):1189-97
2. Travis WD, Travis LB, Devesa SS. Lung cancer. Cancer. 1995;75(1 Suppl):191-202
3. Brahmer J, Reckamp KL, Baas P. et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. New Engl J Med. 2015;373(2):123-35
4. Doitsh G, Galloway NLK, Geng X. et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. 2014;505(7484):509-14
5. Kolb R, Liu GH, Janowski AM. et al. Inflammasomes in cancer: a double-edged sword. Protein Cell. 2014;5(1):12-20
6. Miao EA, Rajan JV, Aderem A. Caspase-1-induced pyroptotic cell death. Immunol Rev. 2011;243(1):206-14
7. Brennan MA, Cookson BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol. 2000;38(1):31-40
8. Boudreau DM, Yu O, Johnson J. Statin use and cancer risk: a comprehensive review. Expert Opin Drug Saf. 2010;9(4):603-21
9. Chan KKW, Oza AM, Siu LL. The statins as anticancer agents. Clin Cancer Res. 2003;9(1):10-9
10. Wang ST, Ho HJ, Lin JT. et al. Simvastatin-induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells. Cell Death Dis. 2017;8(2):e2626
11. Liang Z, Li W. et al. Simvastatin suppresses the DNA replication licensing factor MCM7 and inhibits the growth of tamoxifen-resistant breast cancer cells. Sci Rep. 2017;7:41776
12. Alizadeh J, Zeki AA, Mirzaei N. et al. Mevalonate cascade inhibition by simvastatin induces the intrinsic apoptosis pathway via depletion of isoprenoids in tumor cells. Sci Rep. 2017;7:44841
13. Cardwell CR, McMenamin Ú, Hughes CM. et al. Statin use and survival from lung cancer: a population-based cohort study. Cancer Epidemiol Biomarkers Prev. 2015;24(5):833-841
14. Han JY, Lee SH, Yoo NJ. et al. A randomized phase II study of gefitinib plus simvastatin versus gefitinib alone in previously treated patients with advanced non-small cell lung cancer. Clin Cancer Res. 2011;17:1553-60
15. Yu X, Pan Y, Ma H. et al. Simvastatin inhibits proliferation and induces apoptosis in human lung cancer cells. Oncol Res. 2013;20(8):351-57
16. Lee HY, Kim IK, Lee HI. et al. The apoptotic effect of simvastatin via the upregulation of BIM in nonsmall cell lung cancer cells. Exp Lung Res. 2016;42(1):14-23
17. Li Y, Fu J, Yuan X. et al. Simvastatin inhibits the proliferation of A549 lung cancer cells through oxidative stress and up-regulation of SOD2. Pharmazie. 2014;69(8):610-14
18. Stowe I, Lee B, Kayagaki N. Caspase-11: arming the guards against bacterial infection. Immunol Rev. 2015;265(1):75-84
19. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-74
20. Lin C, Zhang J. Inflammasomes in inflammation-induced cancer. Front Immunol. 2017;8:271
21. Hu B, Elinav E, Flavell RA. Inflammasome-mediated suppression of inflammation-induced colorectal cancer progression is mediated by direct regulation of epithelial cell proliferation. Cell Cycle. 2011;10(12):1936-39
22. Hu B, Elinav E, Huber S. et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci U.S.A. 2010;107(50):21635-40
23. Chang I, Mitsui Y, Kim SK. et al. Cytochrome P450 1B1 inhibition suppresses tumorigenicity of prostate cancer via caspase-1 activation. Oncotarget. 2017;8(24):39087-100
24. Hwang KE, Na KS, Park DS. et al. Apoptotic induction by simvastatin in human lung cancer A549 cells via Akt signaling dependent down-regulation of survivin. Invest New Drugs. 2011;29(5):945-52
25. Sendler M, Mayerle J, Lerch MM. Necrosis, apoptosis, necroptosis, pyroptosis: It matters how acinar cells die during pancreatitis. Cell Mol Gastroenterol Hepatol. 2016;2(4):407-8
Author contact
![]() Corresponding authors: Jianqun Ma, Department of Thoracic Surgery, Harbin Medical University Cancer Hospital, 150, Haping Road, Harbin, 150001, China. Tel: 0451-86298631; E-mail: jianqunmahydcom and Shidong Xu, Department of Thoracic Surgery, Harbin Medical University Cancer Hospital, 150, Haping Road, Harbin, 150001, China. Tel: 0451-85718182; E-mail: shidongxuhydcom
Corresponding authors: Jianqun Ma, Department of Thoracic Surgery, Harbin Medical University Cancer Hospital, 150, Haping Road, Harbin, 150001, China. Tel: 0451-86298631; E-mail: jianqunmahydcom and Shidong Xu, Department of Thoracic Surgery, Harbin Medical University Cancer Hospital, 150, Haping Road, Harbin, 150001, China. Tel: 0451-85718182; E-mail: shidongxuhydcom
Received 2017-10-29
Accepted 2018-2-26
Published 2018-3-10