ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2018; 14(5):508-517. doi:10.7150/ijbs.23318 This issue Cite
Research Paper
DNA damage checkpoint pathway modulates the regulation of skeletal growth and osteoblastic bone formation by parathyroid hormone-related peptide
Ying Zhang1,2, Guangpei Chen1, Zhen Gu1 ![]() , Haijian Sun1, Andrew Karaplis3, David Goltzman3, Dengshun Miao1,4
, Haijian Sun1, Andrew Karaplis3, David Goltzman3, Dengshun Miao1,4 ![]()
1. State Key Laboratory of Reproductive Medicine, The Research Center for Bone and Stem Cells, Department of Anatomy, Histology and Embryology, Nanjing Medical University, Nanjing, China;
2. Department of Anatomy, Histology and Embryology, Suzhou Vocational Health College, Suzhou, China;
3. Department of Medicine, McGill University, Canada;
4. The Research Center for Aging Research, Friendship Affiliated Hospital of Nanjing Medical University Plastic Surgery, Nanjing Medical University, Nanjing, China
Abstract

We previously demonstrated that parathyroid hormone-related peptide (PTHrP) 1-84 knockin (Pthrp KI) mice, which lacked a PTHrP nuclear localization sequence (NLS) and C-terminus, displayed early senescence, defective osteoblastic bone formation, and skeletal growth retardation. However, the mechanism of action of the PTHrP NLS and C-terminus in regulating development of skeleton is still unclear. In this study, we examined alterations of oxidative stress and DNA damage response-related molecules in Pthrp KI skeletal tissue. We found that ROS levels, protein expression levels of γ-H2AX, a DNA damage marker, and the DNA damage response markers p-Chk2 and p53 were up-regulated, whereas gene expression levels of anti-oxidative enzymes were down-regulated significantly. We therefore further disrupted the DNA damage response pathway by deleting the Chk2 in Pthrp KI (Chk2-/-KI) mice and did comparison with WT, Chk2-/- and Pthrp KI littermates. The Pthrp KI mice with Chk2 deletion exhibited a longer lifespan, improvement in osteoblastic bone formation and skeletal growth including width of growth plates and length of long bones, trabecular and epiphyseal bone volume, BMD, osteoblast numbers, type I collagen and ALP positive bone areas, the numbers of total colony-forming unit fibroblasts (CFU-f), ALP+ CFU-f and the expression levels of osteogenic genes. In addition, the genes associated with anti-oxidative enzymes were up-regulated significantly, whereas the tumor suppressor genes related to senescence were down-regulated in Chk2-/- KI mice compared to Pthrp KI mice. Our results suggest that Chk2 deletion in Pthrp KI mice can somewhat rescue defects in osteoblastic bone formation and skeletal growth by enhancing endochondral bone formation and osteogenesis. These studies therefore indicate that the DNA damage checkpoint pathway may be a target for the nuclear action of PTHrP to regulate skeletal development and growth.
Keywords: PTHrP, Chk2, osteoblastic bone formation, skeletal growth
Introduction
PTHrP was first discovered in the search for the hormone mediating hypercalcemia caused by tumors and was subsequently found to play a significant role in regulating cell proliferation and differentiation, morphogenesis, as well as transplacental transport of calcium [1-3]. Further study revealed that the different domains of the PTHrP protein play different functions. Thus, the N-terminal region of PTHrP, which demonstrates homology with parathyroid hormone (PTH), binds to the type I PTH/PTHrP receptor and exerts PTH-like hormonal actions; an amino acid sequence from 37 to 86 has a role in transplacental transport of calcium [4];and the region from amino acid 87 to 107 can function as a nuclear localization sequence (NLS), facilitating entry of PTHrP into the nucleus of target cells, and playing a significant role in cell cycle regulation causing enhanced proliferation and inhibition of apoptosis via an intracrine action [5]; finally the C-terminal domain of PTHrP has been shown to inhibit osteoclast activity and bone resorption [6].
To investigate the function of the NLS and C-terminus of PTHrP in vivo, a PTHrP “knock-in” (KI) mouse model was established by introducing a premature termination codon, TGA, into PTHrP gene to disrupt the mid-region and C-terminal region of PTHrP. Our previous studies demonstrated that Pthrp KI mice display a severe skeletal phenotype including the development of premature osteoporosis [7], however the underlying mechanisms remain unknown.
We demonstrated that ROS levels were increased, whereas the expression levels of antioxidant enzymes were down-regulated, the expression levels of p53 and caspase-3, γ-H2AX, and p16 were up-regulated significantly in bone tissue of Pthrp KI mice [8]. These results suggest that the growth arrest and senescence generated in Pthrp KI might be associated with oxidative stress and a DNA damage response.
Checkpoint kinase 2 (Chk2) is significant regulator and is rapidly phosphorylated in result to DNA damage [9]. When activated, the cycle of cell is blocked in G1, leading to alternative cellular responses such as DNA repair, programmed cell death and also arrest the cell cycle [9, 10]. Previous studies showed that deletion of the Chk2 gene (in Chk2-/- mice) could improve the multiple organ phenotypes caused by the deficiency of the polycomb ring finger oncogene Bmi1 in mice, and prolong their survival time [11]. Bmi1 is down-regulated in Pthrp KI mice and we therefore hypothesized that interruption of the DNA damage response pathway by Chk2 deletion might rescue the phenotypes of premature aging and osteoporosis in Pthrp KI mice.
To examine this, we generated double Chk2 and Pthrp KI mutant mice (Chk2-/-KI) to compare the phenotypes with WT, Chk2-/-, and Pthrp KI littermates.
Materials and Methods
Animals
Mice used in experimental study were maintainned in the Animal Center of Nanjing Medical University (Nanjing, China). All experimental procedures were pre-approved by committee of the Institutional Animal Care and Use. Chk2+/- mice (C57BL/6J hybrid background) [12] were kindly provided by Professor Noboru Motoyama (Department of Geriatric Research, National Institute for Longevity Sciences, Japan). Chk2-/- mice were generated through breeding of heterozygous mice. The Pthrp KI mice were on a C57BL/6J hybrid background [7]. Chk2+/- and Pthrp KI+/- mice were mated to produce heterozygous offspring at both loci; these were then mated to generate Chk2-/-KI pups. Wild-type littermates were used as controls.
Radiography, micro-computed tomography, and histopathological analyses
Femurs or tibias were analyzed using radiography, histochemistry, micro-computed tomography (μ-CT), histology and immunohistochemistry as we described previously [13].
Western blot analysis
For the examination of protein expression levels, equivalent concentrations (20μg) of lysates extracted from mouse tibiae, from which the epiphyses and growth plates had been removed. They were separated on SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes. Incubation with 5% BSA at room temperature for 1 hr blocked the non-Specific sites and blocked membrane was blotted with primary antibodies against SOD1 (Abcam, MA), Chk2 (phosphor T68, Abcam, MA), Caspase-3 (Cell Signaling Technology, Danvers, MA) Phospho-Histone H2A.X (Cell Signaling Technology, Danvers, MA), p53 (Cell Signaling Technology, Danvers, MA), Bmi1 (Millpore, MA), and β-actin (Bioworld Technology, St. Louis Park, MN, USA) were used as loading control followed by appropriate secondary antibodies and chemiluminescent detection which was carried out as we described previously [14]. All the experiments were independently repeated three times.
Intracellular reactive oxygen species (ROS) Analysis
To investigate of the levels of intracellular ROS, 2-week-old mouse organs, collected from indicated groups of mice, were enzymatically dispersed into single cell suspensions. The 5 mM diacetyldichloro fluorescein (DCFDA, Invitrogen) were used for the incubation of isolated cells and placed it in a shaker for 30 min at 37°C and analyzed in a FACS calibur flow cytometer (Becton Dickinson, Heidelberg, Germany).
Real-time RT-PCR
To analyze gene expression, TRIzol reagent (Invitrogen, Carlsbad, CA) was used to isolate the total RNA from fresh tissue according to the standard protocol of the manufacturer. PrimeScript™ 1st Strand cDNA Synthesis Kit (Takara Bio, Shiga, Japan) was used for reverse transcription reactions. The primer sequences used for the real-time PCR are presented in Table 1 and real time RT-PCR was performed as described previously [15].
Sequences of primers employed for RT-PCR.
| Name | S/AS | Sequence | Tm (℃) | bp |
|---|---|---|---|---|
| Runx2 | Forward | GTGACACCGTGTCAGCAAAG | 55 | 356 |
| Reverse | GGAGCACAGGAAGTTGGGAC | |||
| ALP | Forward | CTTGCTGGTGGAAGGAGGCAGG | 55 | 393 |
| Reverse | GGAGCACAGGAAGTTGGGAC | |||
| COL-I | Forward | TCTCCACTCTTCTAGTTCCT | 55 | 269 |
| Reverse | TTGGGTCATTTCCACATGC | |||
| OCN | Forward | CAAGTCCCACACAGCAGCTT | 55 | 370 |
| Reverse | AAAGCCGAGCTGCCAGAGTT | |||
| OPG | Forward | TGGAGATCGAATTCTGCTTG | 57 | 719 |
| Reverse | TCAAGTGCTTGAGGGCATAC | |||
| RANK | Forward | CTGCCTCTGGGAACGTGACT | 60 | 108 |
| Reverse | GCGAGGTCTGGCTGACATAC | |||
| RANKL | Forward | GGTCGGGCAATTCTGAATT | 57 | 813 |
| Reverse | GGGGAATTACAAAGTGCACCAG | |||
| TRAP | Forward | ACACAGTGATGCTGTGTGGCAACTC | 57 | 466 |
| Reverse | CCAGAGGCTTCCACATATATGATGG | |||
| SOD1 | Forward | GGTGAACCAGTTGTGTTGTC | 56 | 203 |
| Reverse | CCGTCCTTTCCAGCAGTC | |||
| SOD2 | Forward | CAGACCTGCCTTACGACTATGG | 56 | 113 |
| Reverse | CTCGGTGGCGTTGAGATTGTT | |||
| GSR | Forward | GACACCTCTTCCTTCGACTACC | 56 | 116 |
| Reverse | CCCAGCTTGTGACTCTCCAC | |||
| GPX4 | Forward | CTCCATGCACGAATTCTCAG | 58 | 117 |
| Reverse | ACGTCAGTTTTGCCTCATTG | |||
| CAT | Forward | GCAGATACCTGTGAACTGTCCCT | 60 | 472 |
| Reverse | TTACAGGTTAGCTTTTCCCTTCG | |||
| P16 | Forward | GGGTTTTCTTGGTGAAGTTCG | 60 | 333 |
| Reverse | TTGCCCATCATCATCACCT | |||
| P19 | Forward | AACAGAAGAACAAAGGGAAGT | 55 | 568 |
| Reverse | GGCGACGAATCAACAATA | |||
| P21 | Forward | CAATCCTGGTGATGTCCGACCTGTT | 70 | 369 |
| Reverse | GAATCTTCAGGCCGCTCAGACACCA | |||
| P27 | Forward | GGAATTCATGTCAAACGTGAGAGTGTCT | 66 | 610 |
| Reverse | CCTCGAGTTACGTCTGGCGTCGAAGGCC | |||
| P53 | Forward | CTGTCTTCCAGATACTCGGGATAC | 55 | 400 |
| Reverse | CTGATCGTTACTCGGCTTGTCC | |||
| GAPDH | Forward | TGGATTTGGACGCATTGGTC | 55 | 211 |
| Reverse | TTTGCACTGGTACGTGTTGAT |
RT-PCR primers used with their name, orientation (S, sense; AS, antisense), sequence, annealing temperature (Tm), and length of amplicon (bp).
Bone marrow mesenchymal stem cell cultures
Bone marrow cells were extracted from tibias and femurs of 2-week-old mice and cultured it in 36 cm2 petri dishes at 2×106 cells/dish with a modified essential medium (α-MEM) containing 10% fetal calf serum (FBS, Hyclone Laboratories, Logan, UT, USA), 10 mM β-glycerophosphate and 50 mg/mL ascorbic acid for 18 days. The cultured cells were stained with methyl blue or cytochemically for ALP. The positive areas of ALP+ CFU-f (CFU-fap) and CFU-f were measured as we described previously [16].
Statistical analysis
All the described data represent at least three separate experiments (n>5/group) and are expressed as the Mean ± SEM. Two-way ANOVA statistic with Bonferroni post-hoc test was used for statistical comparison. Significant level was taken as P<0.05.
Results
Effect of Pthrp KI on oxidative stress and DNA damage response pathways
To determine whether oxidative stress and DNA damage response pathways were involved in the regulation, by PTHrP, of skeletal growth and development in vivo, we examined the alterations of oxidative stress and DNA damage response related molecules in Pthrp KI skeletal tissue. Our results demonstrated that ROS levels in multiple organs (Fig. 1A), and protein expression levels of DNA damage response proteins and related molecules γ-H2AX, p-Chk2 and p53 were up-regulated significantly in skeletal tissue (Fig. 1C and 1D), whereas mRNA expression levels of anti-oxidative enzymes including catalase, superoxide dismutase (SOD) 1 and 2, glutathione reductase (GSR) and glutathione peroxidase 4 (GPX4) were down-regulated in Pthrp KI skeletal tissues (Fig. 1B). Therefore, we not only confirmed that Pthrp KI increased oxidative stress and DNA damage response [8], but also found that Pthrp KI activated Chk2.
Effect of deletion of Chk2 on growth and lifespan in Pthrp KI mice
To assess if deletion of Chk2 can improve the premature aging and growth retardation occurring in Pthrp KI mice, the effect of deletion of Chk2 on lifespan, body weight and skeletal growth parameters was examined in Pthrp KI mice. We found that the mean survival age of Pthrp KI mice was only 2 weeks (5-18 days), whereas it was extended to 3 weeks (12-24 days) in Chk2-/-KI mice (Fig. 2A). Body weight, the length of tibiae, the width of the cartilaginous growth plate and proliferative area were significantly reduced in both Pthrp KI and Chk2-/-KI mice compared with WT littermates but were significantly augmented in Chk2-/- mice; furthermore, body weight, the width of the cartilaginous growth plate and proliferative area were increased 46.6%, 31.1% and 32.8%, respectively, in Chk2-/-KI mice compared to Pthrp KI mice (Fig 2B-G). These results suggest that the prolonged lifespan and improved skeletal growth in Pthrp KI mice by deletion of Chk2 are only partial.
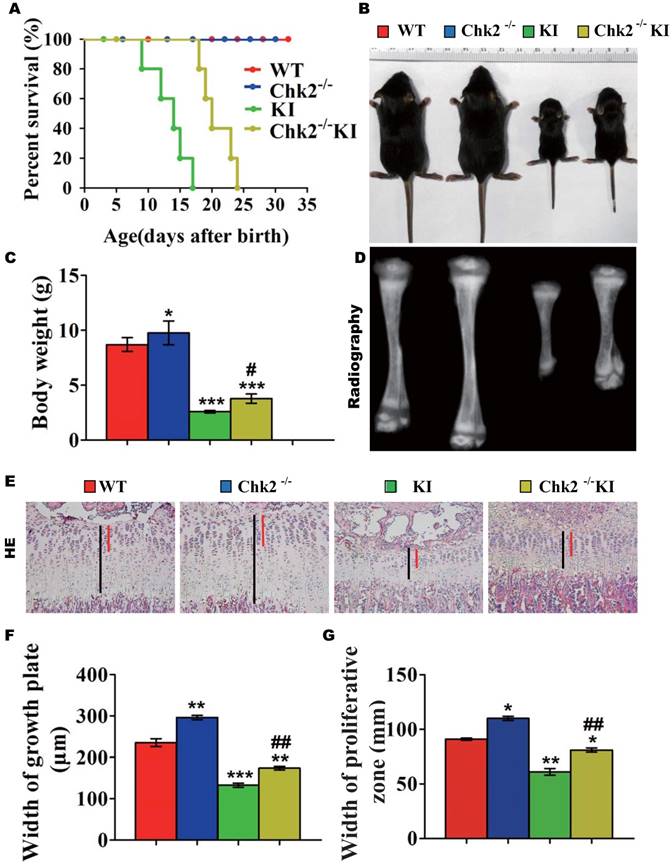
Effect of deletion of Chk2 on osteoblastic bone formation in Pthrp KI mice
We next examined the effect of deletion of Chk2 on osteoblastic bone formation parameters. The BMD, epiphyseal and trabecular bone volume, alkaline phosphatase (ALP) and type I collagen (Col I) positive bone areas, and osteoblast numbers were all significantly increased in Chk2-/- mice but were significantly decreased in both Pthrp KI and Chk2-/-KI mice compared to WT mice. However, the BMD, epiphyseal and trabecular bone volume, ALP and Col I positive bone areas, and osteoblast numbers were increased 81%, 84%, 34%, 145%, 35% and 53%, respectively, in Chk2-/-KI mice compared to Pthrp KI mice, therefore, the improved osteoblastic bone formation in Pthrp KI mice by deletion of Chk2 are only partial. (Figs. 3A-K).
Effect of deletion of Chk2 on the proliferation and differentiation of BM-MSCs in Pthrp KI mice
To investigate whether the improvement of osteoblastic bone formation in Pthrp KI mice caused by the deletion of Chk2 was associated with alterations of the osteogenesis of bone marrow mesenchymal stem cells, CFU-f assays were performed and osteoblastic differentiation related gene expression levels were examined using RT-PCR. CFU-f, CFU-fap area and mRNA levels of Runx2, ALP, osteocalcin and type I collagen were all significantly increased in Chk2-/- mice but were decreased significantly in Pthrp KI and Chk2-/-KI mice relative to WT mice. However, CFU-f and CFU-fap area were increased 60% and 48%, respectively, and and mRNA levels of Runx2, ALP, osteocalcin and type I collagen were up-regulated comparable levels with increased CFU-fap area in Chk2-/-KI mice relative to Pthrp KI mice (Figs. 4A-E).
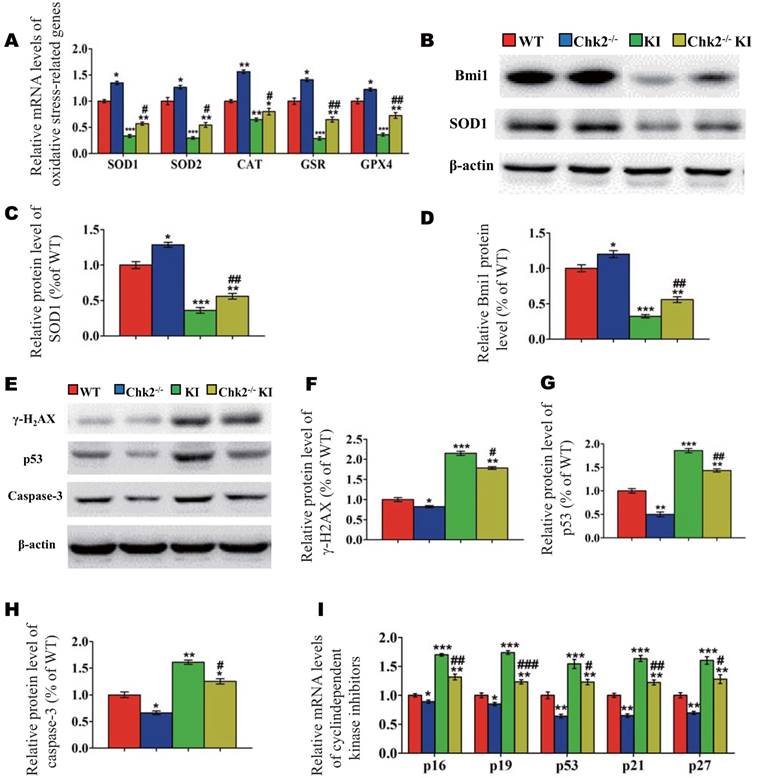
Effect of deletion of Chk2 on oxidative stress and DNA damage in Pthrp KI mice
To further investigate whether the improvement of premature osteoporosis in the Pthrp KI mice caused by the deletion of Chk2 is related with inhibition of oxidative stress, genes encoding antioxidant enzymes and their protein expression levels were assessed. Expression levels of SOD1/2, CAT, GSR and GPX4 mRNA and of SOD1 and Bmi1 protein were increased significantly in Chk2-/- mice but were decreased expressively in Pthrp KI and Chk2-/-KI mice relative to WT mice; these parameters were however significantly increased in Chk2-/-KI mice relative to Pthrp KI mice (Fig. 5A-D). Presented data indicate that deletion of Chk2 can inhibit the oxidative stress induced by PTHrP KI by up-regulating expression of Bmi1 and anti-oxidative enzymes.
Effect of Pthrp KI on oxidative stress and DNA damage response pathways. (A) Flow cytometry analysis of ROS in different tissues from 2-week-old WT and Pthrp KI littermates. (B) Western blot analyses of γ-H2AX, p-Chk2 and p53 expression in the bone tissue of WT and Pthrp KI mice. β-actin was used as a loading control. (C) Gene expression levels of anti-oxidative enzymes. (D) Protein expression levels of γ-H2AX, p-Chk2 and p53. Each value is the mean±SEM of determinations in 5 mice of each group. *: p< 0.05,**: p< 0.01,***: p<0.001,compared to WT mice.
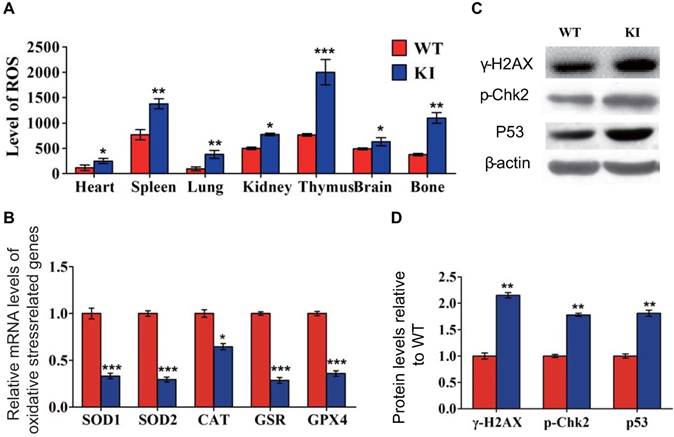
Effect of deletion of Chk2 on lifespan and growth in Pthrp KI mice. (A) Survival curves of wild-type (WT), Chk2-/-, Pthrp KI and Chk2-/- KI mice. (B) Whole-body view of 2-week- littermates. (C) Body weight of 2-week-old mice. (D) Representative radiographs of the femurs from 2-week-old WT, Chk2-/-, Pthrp KI and Chk2-/-KI mice. (E) Representative micrographs from paraffin-embedded sections of tibias stained with H&E. (F) The width of the cartilaginous growth plate. (G) The width of the proliferative zone of the cartilaginous growth plate. Each value is the mean±SEM of determinations in 5 mice of each group. *p < 0.05, **p < 0.01, **p <0.001compared with WT mice; ##p < 0.01compared with Pthrp KI mice.
We then further assessed whether omission of Chk2 in the Pthrp KI mice also suppressed DNA damage response related parameters. The results showed that the protein expression levels of γ-H2AX, Caspase-3, p53 and the mRNA expression levels of cyclin dependent kinase inhibitors including p16, p19, p53, p21 and p27 were significantly decreased in Chk2-/- mice but were expressively increased equally in Pthrp KI and Chk2-/-KI mice related to WT mice; nevertheless, these parameters were significantly lower in Chk2-/-KI mice compared to Pthrp KI mice (Fig 5E-I). These outcomes indicate that deletion of Chk2 can inhibit the DNA damage response pathway, which is up-regulated in Pthrp KI mice.
Effect of deletion of Chk2 on osteoblastic bone formation in Pthrp KI mice. (A) Representative longitudinal and cross sectional images of three-dimensional reconstructed distal ends of femurs and mid shaft diaphyses utilizing micro-CT. (B) Representative micrographs of paraffin-embedded sections of tibias stained histochemically for total collagen. Magnification is ×50. (C) BMD measurements, (D) Bone volume/total volume (BV/TV), and (E) Epiphyseal volume, as determined in 2-week-old mice. Representative micrographs of paraffin sections of tibial metaphyseal regions stained (F) with hematoxylin and eosin (H&E) (400x), (G) histochemically for alkaline phosphatase (ALP) (400x), and (H) immunohistochemically for type I collagen (Col I) (400x). (I) Osteoblast number relative to tissue area (N.Ob/T.Ar, #/mm2), (J) ALP positive areas and (K) Col I immune positive areas were measured by computer-assisted image analysis. Each value is the mean±SEM of determinations in 5 mice of each group. *p < 0.05, **p < 0.01, ***p < 0.001 compared with WT mice; #p < 0.05, ##p < 0.01 compared with Pthrp KI mice.
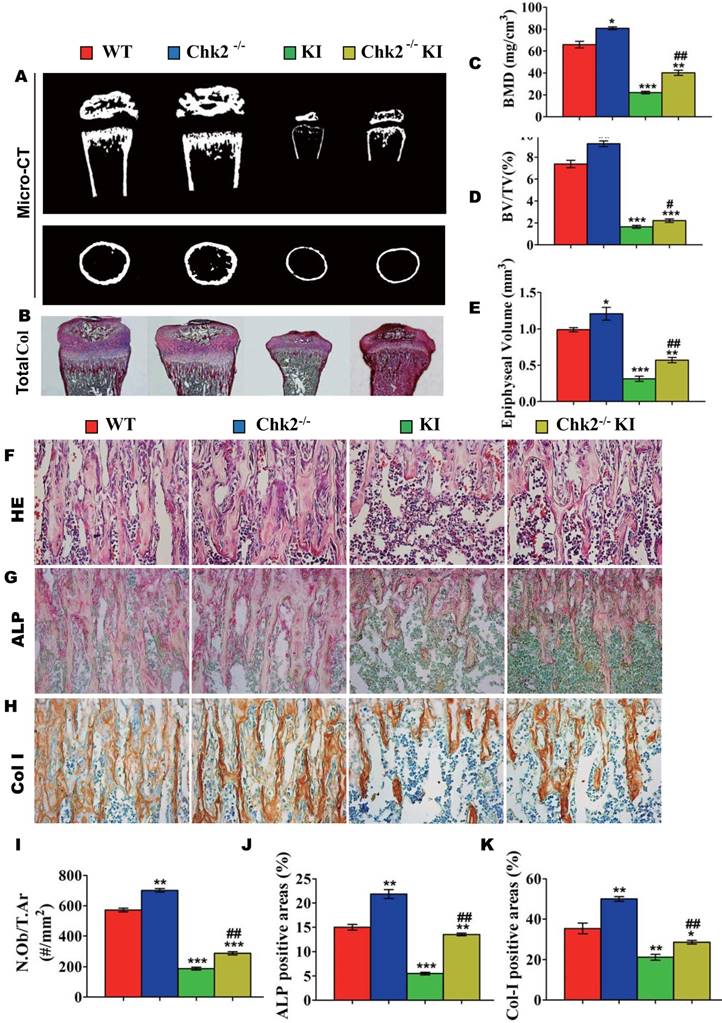
Effect of deletion of Chk2 on the proliferation and differentiation of BM-MSCs in Pthrp KI mice. Primary bone marrow cells from 2-week-old WT, Chk2-/-, Pthrp KI, and Chk2-/- KI mice were cultured ex vivo in osteogenic differentiation medium for 18 days and resulting cultures were stained with (A) methylene blue for total number of colonies (CFU-F) and (B) cytochemically for ALP to show CFU-Fap. (C) Total CFU-f-positive areas and (D) ALP-positive areas relative to culture dish area. (E) Relative levels of osteoblast- related genes. Each value is the mean±SEM of determinations in 5 mice of each group. * p < 0.05, ** p < 0.01, *** p < 0.001 compared with WT mice; ## p < 0.01 compared with Pthrp KI mice.
Discussion
To better understand the mechanisms underlying osteoporosis, we studied Pthrp KI mice which manifest both accelerated aging and osteoporosis. We assessed the effects of deletion of CHK2, by examining double Chk2 and Pthrp mutant mice (Chk2-/-KI), and then comparing the resultant phenotype to those of Chk2-/-, Pthrp KI, and WT littermates. At the age of 2 weeks, the body weight, skeletal growth parameters, osteoblastic bone formation and the numbers of bone marrow mesenchymal stem cells markers were reduced in Pthrp KI mice. In addition, ROS levels, protein expression levels of the DNA damage pathway and related molecules γ-H2AX, p-Chk2 and p53 were up-regulated significantly in skeletal tissues, whereas the gene expression levels of anti-oxidant enzymes were down-regulated. Therefore, intact PTHrP appears to exert intracrine action to inhibit oxidative stress and DNA damage, in order to facilitate osteoblastic bone formation and skeletal growth.
Effect of deletion of Chk2 on oxidative stress and DNA damage in Pthrp KI mice. (A) Gene expression levels of anti-oxidative enzymes. (B) Western blots of bone tissue extracts from 2-week-old WT, Chk2-/-, Pthrp KI, and Chk2-/-KI mice for expression of Bmi1 and SOD1. β-actin was used as loading control for western blots. (C) SOD1 and (D) Bmi1 protein levels relative to β-actin protein level were assessed by densitometric analysis and expressed relative to levels of 2-week-old WT mice. (E) Western blots of bone tissue extracts from 2-week-old WT, Chk2-/-, Pthrp KI, and Chk2-/-KI mice for expression of γ-H2AX,p53 and Caspase-3. β-actin was used as loading control for Western blots. (F) γ-H2AX, (G) p53 and (H) Caspase-3 protein levels relative to β-actin protein level were assessed by densitometric analysis and expressed relative to levels of 2-week-old WT mice. (I) Gene expression levels of cyclin dependent kinase inhibitors. Each value is the mean±SEM of determinations in 5 mice of each group. *p < 0.05, **p < 0.01, ***p < 0.001 compared with WT mice; #p < 0.05, ##p <0.01, ###p <0.001compared with Pthrp KI mice.
We previously found that protein levels of Bmi1 were meaningfully down-regulated in Pthrp KI skeletal tissues and mouse embryonic fibroblasts (MEFs), and although expressed in the nuclei of MEFs from wild-type mice, Bmi1 was not expressed in nuclei of MEFs derived from Pthrp KI mice [7]. Bmi1 was initially identified as an oncogene that has synergistic action with the C-Myc proto oncogene [17, 18] . Bmi1 also plays an important part in maintaining mitochondrial function and redox homeostasis [11]. Mice with low expression of Bmi1 caused by the deletion of the PTHrP NLS showed high ROS levels and decreasing mRNA expression levels of anti-oxidant enzymes in Pthrp KI mice. The elevation of oxidative stress can activate the DNA damage checkpoint pathway, up-regulating the proteins which are associated with DNA damage, such as γ-H2AX, p-Chk2 and p53. Therefore, Bmi1 may function as a downstream target of PTHrP action via the NLS by regulating mitochondrial function and redox homeostasis.
Bmi1 deficiency can decrease cell proliferation and increase cell apoptosis of BM-MSCs, and up-regulate p16, p19 and Chk2 [19]. Conversely Bmi1 can maintain the self-renewal ability of BM-MSCs, promote their osteogenesis and reduce their adipogenesis by inhibiting p16, P19 and Chk2. We previously showed that the expression of Bmi1 and its nuclear translocation were decreased significantly in the bone tissues of Pthrp KI mice [20]. Our current results demonstrated that the reduced expression of Bmi1 was partially corrected by deletion of Chk2 in Pthrp KI mice. Consequently Chk2 may reciprocally modulate the expression of Bmi1 in the absence of the intracrine action of PTHrP.
In our studies, we cultured BM-MSC and found that CFU-f number and cell proliferation ability were decreased significantly in Pthrp KI mice. Deletion of Chk2 from Pthrp KI mice partially reversed these changes, Therefore intact PTHrP may, via its intracrine action promote the osteogensis of BM-MSCs, at least in part by decreasing Chk2.
Chk2 is a multifunctional protein kinase and can performed as a tumor suppressor gene. Chk2 may be mutated in a variety of tumors, including ovarian cancer, bladder cancer, liver cancer, breast cancer, prostate cancer and colon cancer [21, 22]. Recent studies have shown that Chk2 also plays a vital role in maintaining normal mitosis and chromosome stability [23], and deletion of Chk2 can partially rescue phenotypes of mice with the deficiency of Bmi1 [11]. The tumor suppressor gene p53 is a key target of Chk2 in DNA damage response and Chk2 can phosphorylate p53 on Ser-20 and dissociate p53 from Mdm2 to maintain its stabilization and accumulation after DNA damage [24, 25]. Thus, Chk2 can regulate p53 directly and mediate the p53 dependent cell cycle arrest and apoptosis.
Although gene expression levels of Chk2 were significantly up-regulated in the bone tissue of Pthrp KI mice, the deletion of Chk2 could not completely correct the abnormal skeletal phenotype caused by deletion of the N and C-terminus of PTHrP. It was previously reported that the expression of p27 was also significantly increased in the bone tissue of Pthrp KI mice, however p27 deletion could also only partially correct the abnormal skeletal development occurring in Pthrp KI mice [15]. Therefore, Chk2 is likely not the only downstream target of the intracrine action of PTHrP, which may explain why Chk2 deletion cannot completely correct the skeletal abnormalities occurring in Pthrp KI mice.
To summarize,Pthrp KI mice with deletion of the PTHrP nuclear localization arrangement and the C terminal domain, displayed a short lifespan, skeletal growth retardation and premature osteoporosis with increased osteoclastic bone resorption and decreased osteoblastic bone formation associated with high oxidative stress and up-regulation of the DNA damage response pathway. We showed that the deletion of Chk2 in Pthrp KI mice could prolonged lifespan, increase body weight, facilitate skeletal growth, increase osteogenesis and osteoblastic bone formation and decreased osteoclastic bone resorption in association with enhanced antioxidant capacity and a decreased DNA damage response. These data thus indicate that the DNA damage checkpoint pathway may play role as a downstream target in the action of PTHrP to regulate skeletal development and growth.
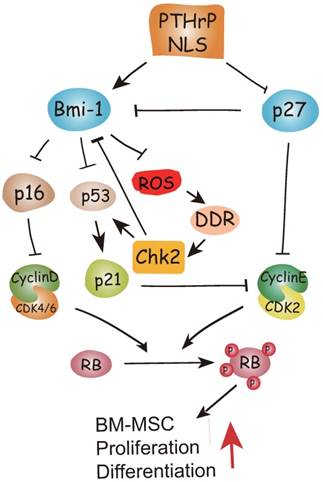
To conclude, therefore, the results of this study and previous study provide a model that suggests that in addition to inhibiting p27 [15], the PTHrP NLS and C-terminus activate Bmi1 signaling [7, 8], thus enhancing the osteogenesis of BM-MSCs and increasing skeletal development and growth. Bmi1 not only inhibits p16/Rb and P19-p53-p21 signaling but also modulates mitochondrial function to lower ROS levels and suppress activation of the DNA damage response pathway. The activation of the DNA damage response pathway not only activates p53 signaling, but also inhibits Bmi1 signaling in a feedback manner mediated by Chk2 (Fig. 6). Enhanced understanding of the intracrine mechanism of action of PTHrP may facilitate new approaches to improving skeletal growth and function.
Model of the action of of PTHrP via its nuclear localization signal (NLS).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81370465 to G.Z. and 81230009 and 81730066 to D.M.), from Natural Science Foundation of Jiangsu Province in China (BK20131384 to G.Z.), from the National Basic Research Program of China (2014CB942900 to D.M.) and from the Canadian Institutes of Health Research (CIHR) to D.G..
Competing Interests
The authors have declared that no competing interest exists.
References
1. McCauley LK, Martin TJ. Twenty-five years of PTHrP progress: from cancer hormone to multifunctional cytokine. J Bone Miner Res. 2012;27:1231-9
2. Wysolmerski JJ, Stewart AF. The physiology of parathyroid hormone-related protein: an emerging role as a developmental factor. Annu Rev Physiol. 1998;60:431-60
3. Burtis WJ, Wu T, Bunch C, Wysolmerski JJ, Insogna KL, Weir EC. et al. Identification of a novel 17,000-dalton parathyroid hormone-like adenylate cyclase-stimulating protein from a tumor associated with humoral hypercalcemia of malignancy. J Biol Chem. 1987;262:7151-6
4. Wu TL, Vasavada RC, Yang K, Massfelder T, Ganz M, Abbas SK. et al. Structural and physiologic characterization of the mid-region secretory species of parathyroid hormone-related protein. J Biol Chem. 1996;271:24371-81
5. Henderson JE, Amizuka N, Warshawsky H, Biasotto D, Lanske BM, Goltzman D. et al. Nucleolar localization of parathyroid hormone-related peptide enhances survival of chondrocytes under conditions that promote apoptotic cell death. Molecular and cellular biology. 1995;15:4064-75
6. Cornish J, Callon KE, Nicholson GC, Reid IR. Parathyroid hormone-related protein-(107-139) inhibits bone resorption in vivo. Endocrinology. 1997;138:1299-304
7. Miao D, Su H, He B, Gao J, Xia Q, Zhu M. et al. Severe growth retardation and early lethality in mice lacking the nuclear localization sequence and C-terminus of PTH-related protein. Proc Natl Acad Sci U S A. 2008;105:20309-14
8. Zhou X, Dai X, Wu X, Ji J, Karaplis A, Goltzman D. et al. Overexpression of Bmi1 in Lymphocytes Stimulates Skeletogenesis by Improving the Osteogenic Microenvironment. Scientific reports. 2016;6:29171
9. Scafoglio C, Smolka M, Zhou H, Perissi V, Rosenfeld MG. The co-repressor SMRT delays DNA damage-induced caspase activation by repressing pro-apoptotic genes and modulating the dynamics of checkpoint kinase 2 activation. PLoS One. 2013;8:e59986
10. Zannini L, Delia D, Buscemi G. CHK2 kinase in the DNA damage response and beyond. J Mol Cell Biol. 2014;6:442-57
11. Liu J, Cao L, Chen J, Song S, Lee IH, Quijano C. et al. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature. 2009;459:387-92
12. Kobayashi Y, Watanabe M, Okada Y, Sawa H, Takai H, Nakanishi M. et al. Hydrocephalus, situs inversus, chronic sinusitis, and male infertility in DNA polymerase lambda-deficient mice: possible implication for the pathogenesis of immotile cilia syndrome. Molecular and cellular biology. 2002;22:2769-76
13. Xue Y, Zhang Z, Karaplis AC, Hendy GN, Goltzman D, Miao D. Exogenous PTH-related protein and PTH improve mineral and skeletal status in 25-hydroxyvitamin D-1alpha-hydroxylase and PTH double knockout mice. J Bone Miner Res. 2005;20:1766-77
14. Liu J, Lv F, Sun W, Tao C, Ding G, Karaplis A. et al. The abnormal phenotypes of cartilage and bone in calcium-sensing receptor deficient mice are dependent on the actions of calcium, phosphorus, and PTH. PLoS Genet. 2011;7:e1002294
15. Zhu M, Zhang J, Dong Z, Zhang Y, Wang R, Karaplis A. et al. The p27 Pathway Modulates the Regulation of Skeletal Growth and Osteoblastic Bone Formation by Parathyroid Hormone-Related Peptide. J Bone Miner Res. 2015;30:1969-79
16. Miao D, Bai X, Panda D, McKee M, Karaplis A, Goltzman D. Osteomalacia in hyp mice is associated with abnormal phex expression and with altered bone matrix protein expression and deposition. Endocrinology. 2001;142:926-39
17. Haupt Y, Alexander WS, Barri G, Klinken SP, Adams JM. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell. 1991;65:753-63
18. van Lohuizen M, Frasch M, Wientjens E, Berns A. Sequence similarity between the mammalian bmi-1 proto-oncogene and the Drosophila regulatory genes Psc and Su(z)2. Nature. 1991;353:353-5
19. Enomoto-Iwamoto M, Enomoto H, Komori T, Iwamoto M. Participation of Cbfa1 in regulation of chondrocyte maturation. Osteoarthritis Cartilage. 2001;9(Suppl AS):76-84
20. Lin T, Chao C, Saito S, Mazur SJ, Murphy ME, Appella E. et al. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol. 2005;7:165-71
21. Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421-9
22. Antoni L, Sodha N, Collins I, Garrett MD. CHK2 kinase: cancer susceptibility and cancer therapy - two sides of the same coin?. Nat Rev Cancer. 2007;7:925-36
23. Stolz A, Ertych N, Kienitz A, Vogel C, Schneider V, Fritz B. et al. The CHK2-BRCA1 tumour suppressor pathway ensures chromosomal stability in human somatic cells. Nat Cell Biol. 2010;12:492-9
24. Chehab NH, Malikzay A, Appel M, Halazonetis TD. Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes & Development. 2000;14:278-88
25. Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites (vol 14, pg 289, 2000). Genes & Development. 2000;14(3):289-300
Author contact
![]() Corresponding authors: Dengshun Miao, The Research Center for Aging Research, Friendship Affiliated Hospital of Nanjing Medical University Plastic Surgery, Nanjing Medical University, Nanjing, China. E-mail: dsmiaoedu.cn Or Zhen Gu, State Key Laboratory of Reproductive Medicine, Research Center for Bone and Stem Cells, Nanjing Medical University, Nanjing, China. E-mail: 183651532com
Corresponding authors: Dengshun Miao, The Research Center for Aging Research, Friendship Affiliated Hospital of Nanjing Medical University Plastic Surgery, Nanjing Medical University, Nanjing, China. E-mail: dsmiaoedu.cn Or Zhen Gu, State Key Laboratory of Reproductive Medicine, Research Center for Bone and Stem Cells, Nanjing Medical University, Nanjing, China. E-mail: 183651532com
Received 2017-10-13
Accepted 2018-2-21
Published 2018-4-5