ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2018; 14(9):1054-1066. doi:10.7150/ijbs.25349 This issue Cite
Research Paper
CCL2-SQSTM1 positive feedback loop suppresses autophagy to promote chemoresistance in gastric cancer
Wenxia Xu1, Qi Wei1, Mengjiao Han2, Bingluo Zhou2, Hanying Wang2, Jianbing Zhang3, Qiang Wang4, Jie Sun1, Lifeng Feng1, Shouyu Wang4, Yang Ye5, Xian Wang2, Jianwei Zhou4, Hongchuan Jin1, ![]()
1. Labortaory of Cancer Biology, Key Laboratory of Biotherapy in Zhejiang, Sir Runrun Shaw hospital, Medical School of Zhejiang University, China
2. Department of Medical Oncology, Sir Runrun Shaw hospital, Medical School of Zhejiang University, China
3. Pathology Center, Shanghai General Hospital, Medical School of Shanghai Jiaotong University
4. Department of Molecular Cell Biology and Toxicology, School of Public Health, Nanjing Medical University, Nanjing, China
5. Department of Preventive Medicine and Public Health Laboratory Science, School of Medicine, Jiangsu University, Zhenjiang, Jiangsu, China
Abstract

Chemotherapy is one of the most important approaches for the treatment of various cancers. However, tumor cells often develop resistance to chemotherapeutic drugs. The tumor microenvironment reconstituted by various cytokines secreted from immune cells was recently found to play important roles in affecting therapeutic response of tumor cells. Herein, we reported that tumor cells can secrete autocrine cytokines to confer chemoresistance by inactivating proapoptotic autophagy. Through cytokine screening, we found that drug resistant cancer cells secreted more CCL2 than drug sensitive cells. Such secreted CCL2 could not only maintain chemoresistance in drug-resistant cancer cells but also confer drug resistance to drug-sensitive cancer cells. CCL2 attenuated drug-induced cytotoxicity by activating PI3K-Akt-mTOR signaling to inhibit proapoptotic autophagy and increase SQSTM1 expression. CCL2 expression in primary carcinoma tissues also correlated well with SQSTM1 expression. Either CCL2 knock-down or autophagy induction successfully reversed drug resistance of tumor cells. Moreover, increased expression of SQSTM1 in turn activated CCL2 transcription via NF-κB signal pathway, representing a positive feedback loop to maintain drug resistance. Therefore, our results provided a new insight to understand drug resistance, and indicated the potential value of CCL2 as a biomarker and intervention target for chemotherapy resistance.
Keywords: CCL2, drug resistance, apoptosis, autophagy, gastric cancer
Introduction
Chemotherapy is one of the most important approaches for the treatment of many cancers. However, tumor cells often develop resistance to chemotherapeutic drugs, leading to the failure of chemotherapy [1]. It has been well documented that tumor cells can resist various chemotherapeutic drugs. Drug resistance involves genetic and epigenetic changes to reprogram the expression of genes critical to drug response [2-3]. Evidences have indicated that the tumor microenvironment influences therapeutic response and drug resistance in various tumor cells [4]. In this regarding, the non-tumor host cells in the tumor microenvironment such as fibroblasts and immune cells secrete various cytokines to activate or inactivate distinct signaling pathways in tumor cells, resulting in the resistance of drug-induced cell death [5-6]. On the other hand, tumor cells can also recruit and educate non-tumor host cells to construct a beneficial microenvironment in support of their growth and expansion [7-8].
CCL2 (chemokine C-C motif ligand 2), also named MCP-1 (Monocyte Chemotactic Protein-1), is a well-known cytokine belonging to the CC chemokine family [9]. CCL2 is usually secreted by macrophages in response to infectious pathogens [10]. Interestingly, paracrine CCL2 can also be secreted by tumor-associated-macrophages (TAMs) in tumor microenvironment to activate PI3K/AKT pathway in the tumor cells [11]. CCL2 expression was associated with the development of various tumors, such as lung cancer, breast cancer, prostate cancer, and gastric cancer [12-15]. Recently, some tumor cells such as ovarian cancer cells were found to secrete autocrine CCL2 to confer the resistance to paclitaxel or carboplatin based chemotherapy [16]. Therefore, cytokines could affect drug sensitivity in both paracrine and autocrine manners. However, it is still unknown what the role of CCL2 plays in drug resistance of gastric cancer cells, nor the precise mechanism.
Autophagy is an evolutionarily conserved cellular degradation process in which damaged organelles and protein aggregates are sequestered in a double membrane vesicle followed by fusion with lysosomes for degradation [17]. Many chemotherapeutic drugs show significant effects on autophagy, whereby modulating autophagy can influence the anticancer effects [18-20]. Activation of autophagy can promote resistance to various treatments [21-24]. Therefore, autophagy inhibition was proposed to potentially increase the clinical efficacy of chemotherapies and the anti-cancer effect of autophagy inhibitors in combination with various chemotherapeutic regiments have been extensively evaluated in multiple clinical trials (www.clinicaltrials.gov).
Recently, we established two chemoresistant cancer cell lines BGC823/DDP cells and SGC7901/DDP [25-27]. By using these cell models, we reported two XRCC1 related proteins JWA and TXNL1 that contributed to drug resistance. These studies demonstrated the important role of DNA repair and apoptosis in gastric cancer chemoresistance. Here, in an effort to clarify the relevance of tumor cell-produced autocrine cytokines to drug resistance, we performed a cytokine screening and found that drug resistant cancer cells secreted more CCL2. Moreover, CCL2 was found to maintain or transduce drug resistance by activating PI3K-Akt-mTOR signaling pathway to suppress autophagy, leading to the increased expression of SQSTM1. In this regard, autophagy should be activated rather than inhibited to overcome chemoresistance. Moreover, increased expression of SQSTM1 activated CCL2 transcription via NF-κB signal pathway, demonstrating the CCL2-SQSTM1 positive feedback loop in chemoresistance. Therefore, CCL2 could be a potential biomarker and intervention target for chemotherapy resistance.
Materials and Methods
Chemicals and Cell lines
Rapamycin and LY294002 were bought from Sigma-Aldrich (Louis, MO, USA) and CCL2 recombinant protein was provided by Sino Biological Inc (Beijing, China). BAY11-7082 were bought from Selleck Chemicals (Shanghai, China). Human gastric cancer cell lines BGC823 and SGC7901 were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in RPMI 1640 medium supplemented with 10% of fetal bovine serum (FBS), 100 U/ml of penicillin, and 100 μg/ml of streptomycin (Life Technologies/Gibco).
Grand Island, NY, USA). The cells were grown at 37℃ in a humidified incubator with 5% CO2. Cisplatin was obtained from Sigma-Aldrich (St. Louis, MO, USA). The cisplatin-resistant cells were developed from the parental cells subjected to persistent gradient exposure to cisplatin for about 12 month, through increasing cisplatin concentration from 0.05 μg/ml until the cells acquired resistance to 1 μg/ml.
Co-culture experiments
Co-culture of BGC823 and BGC823/DDP, SGC7901 and SGC7901/DDP were performed with Corning Transwell polyester membrane cell culture system including 24 mm Transwell with 0.4 μm pore polyester membrane insert.
Cytokine screening and validation
1×106 BGC823, BGC823/DDP, SGC7901 and SGC7901/DDP cells were plated in 6-cm dish. After 24 h incubation, the mediums were replaced with serum free 1640 medium for 24 h. The conditional mediums were then collected to screen cytokines with cytokine antibody array according to protocols provided (RayBio® Human Cytokine Antibody Array G Series 1000). For the validation, the cytokines in the conditional medium were determined by ELISA according to the manufacturer's instructions (Boster, Wuhan, China).
siRNA and transfection
Small interfering RNA (siRNA) specific for CCL2 (1# 5'-CUCGCGAGCUAUAGAAGAATT-3', 2# 5'-CUCGCGAGCUAUAGAAGAATT-3'), SQSTM1 (1# 5'-GUGACGAGGAAUUGACAAUTT-3', 2# 5'-GGAGUCGGAUAACUGUUCATT-3') and control siRNA was synthesized in GenePharma (Shanghai, China). The siRNA was transfected into cells with Lipofectamine RNAiMAX Reagent (Life Technologies, CA, USA) according to manufacturer's instructions. In brief, cells were seeded overnight in 6-well plates (3-5×105/well). (a) Dilute 9μl Lipofectamine RNAiMAX Reagent in 150μl Opti-MEM Medium. (b) Dilute 30pmol siRNA in 150μl Opti-MEM Medium. Add (a) to (b) for 5min. Then Add siRNA-lipid complex to cells. Incubate cells for 1-3 days at 37°C. Then, analyze transfected cells.
Cytotoxicity assay
One day before treatment, BGC823, SGC7901, BGC823/DDP, SGC7901/DDP cells were plated at a density of 5000 cells per well in 96-well plates. The cells were treated with various concentrations of drugs. After 24 h treatment, the cell viability was determined using Cell Counting Kit-8 (CCK-8) according to the manufacturer's instructions (Dojindo, Kumamoto, Japan). The cell survival rates were expressed as mean±S.D. from at least three independent experiments.
TUNEL assay
Apoptosis was determined using the TUNEL apoptotic cell detection kit (Roche, Basel, Switzerland), according to the manufacturer's instructions. The percentages of apoptotic cells were counted from at least 1000 cells. The confocal images of cells were sequentially acquired with Zeiss AIM software on a Zeiss LSM 710 confocal microscope system (Carl Zeiss Jena, Oberkochen, Germany) and images were processed with ZEN LE software (Carl Zeiss).
Tandem mRFP-GFP fluorescence microscopy
A tandem monomeric RFP-GFP-tagged LC3 (tfLC3) was used to monitor autophagy flux as previous reported. For evaluating tandem fluorescent LC3 puncta, 48 h after tfLC3 transfected alone, or with siRNAs/plasmid cotransfected, cells were washed with 1 × PBS, and then sent out for confocal microscope analysis directly. Samples were examined under a Zeiss LSM 710 confocal microscope system (Carl Zeiss, Oberkochen, Germany) and images were processed with ZEN LE software (Carl Zeiss, Oberkochen, Germany)
Western blotting
Western blot analyses were performed as previously described [28]. In brief, cells were lysed in whole-cell lysate buffer containing 1% phosphatase inhibitor cocktail and NPC tumor tissues were lysed in RIPA buffer (Beyotime, Jiangsu, China) with 1% phosphatase inhibitor cocktail. Lysates containing 20-30 µg protein were loaded onto 8% or 12% sodium dodecyl sulfate-polyacrylamide gels for electrophoresis (SDS-PAGE) and the separated proteins transferred to poly vinylidene fluoride (PVDF) membranes (Pall, NY, USA). After blocking with 5% fat free milk for 1 h in Tris-buffered saline (TBS), the membranes were incubated with the primary antibody overnight at 4°C and then with the peroxidase labelled secondary antibody (agilent, CA, USA) for 1 h on the next day. WB bands were visualized with an enhanced chemiluminescence kit (EMD Millipore, MA, USA). The primary antibodies used were γH2AX (1:2000 dilution, Epitomics, Burlingame, CA, USA) and monoclonal anti-cleaved PARP-1, β-actin, p-AKT, p-mTOR, SQSTM1/p62, LC-3, P-P65 (1:2000 dilution, Cell Signaling Technology, Danvers, MA, USA).
Quantitative real-time RT-PCR
RT-PCR analyses were performed as previously described [29]. In brief, Total RNA was extracted using Trizol reagent (Life Technologies, CA, USA) according to the manufacturer's instructions. RNAs were quantified using a NanoDrop 2000c instrument (Thermo Scientific, MA, USA). Reverse transcription reaction was performed using 1μg of total RNA with Quantscript RT kit (Tiagen Biotechnology, Beijing, China). The mRNA expression level was determined by quantitative real-time PCR using SYBR Green Master Mix (Applied Biosystens, CA, USA) and ABI 7500 Real-Time PCR System (Applied Biosystems, CA, USA). Human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control of RNA integrity. The Primers (CCL2-F 5'-CAGCAGCAAGTGTCCCAAAG-3', CCL2-R 5'-TCCAGGTGGTCCATGGAATC-3'), (SQSTM1-F 5'-AGAACGTTGGGGAGAGTGTG, SQSTM1-R 5'-GCGATCTTCCTCATCTGCTC-3'), (GAPDH-F 5'-GGAGTCAACGGATTTGGT-3', GAPDH-R 5'-GTGATGGGATTTCCATTGAT-3').
Immunohistochemistry
All tissue samples were obtained from the tissue bank of Sir Runrun Shaw Hospital. This study was approved by the hospital ethics committee. Both anti-CCL2 (1:300 dilution) and anti-SQSTM1 (1:400 dilution) antibodies were optimized for immunohistochemistry. Subsequently, an identical formalin-fixed and paraffin-embedded human gastric cancer tissue array was immunostained using microwave antigen retrieval in 0.01 M citrate buffer, pH 6.0. After washing, signal was detected using a suitable HRP-labeled second antibody with DAB as the chromagen. The staining results were judged by 2 independent pathologists in the department of pathology of our hospital. The positive staining was defined as uniform staining of more than 5% of 1000 tumor cells and the staining of less than 5% of 1000 tumor cells was defined as negative. Slides were assessed using Chi-Square analysis to determine the correlation of CCL2 and SQSTM1 expression.
Identification of CCL2 as a critical autocrine cytokine to transmit drug resistance. (a) The viability of cells treated with cisplatin as indicated was determined CCK-8 assay. (b) The graphic illustration of the co-culture system. (c) BGC823 co-cultured with BGC823 or BGC823/DDP cells and SGC7901 co-cultured with SGC7901 or SGC7901/DDP cells were treated with cisplatin for additional 24h, and collected for Western blotting as indicated. (d) ELISA was used to validate the CCL2 levels in the conditional medium of BGC823, BGC823/DDP, SGC7901 and SGC7901/DDP cells. (e) BGC823 and SGC7901 co-cultured with various cells as indicated were treated with cisplatin and collected to determine the cleaved PARP-1 by Western blotting. (*P<0.05, ***P<0.001)
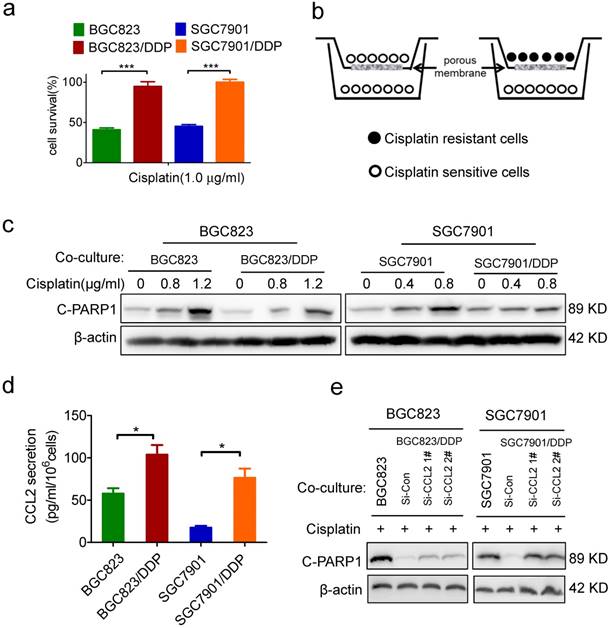
Statistical analysis
Data are expressed as the mean ± SD. The statistical significance of the differences between the cell lines was determined by the parametric unpaired Student's t test. Differences were considered significant when P < 0.05.
Results
Identification of CCL2 as a critical autocrine cytokine to transmit drug resistance
Recently, we have established two cisplatin-resistant GC cell lines BGC823/DDP cells and SGC7901/DDP. These cells can keep vigorous growth in the medium containing various chemotherapeutic drugs such as cisplatin or arsenic trioxide.[26-27] Here, we confirmed the drug resistance character of BGC823/DDP and SGC7901/DDP cells. Compared with BGC823/DDP and SGC7901/DDP cells, their parental sensitive cells BGC823 and SGC7901 were significantly killed in the presence of 1.0 μg/ml cisplatin (Figure 1a). In order to identify autocrine cytokines relevant to drug resistance in gastric cancer cells, we constructed an in vitro model to co-culture drug resistant cells with sensitive cells. In this co-culture transwell system, drug resistant BGC823/DDP or SGC7901/DDP cells were cultured in the top insert and the drug sensitive parental cells were cultured in the bottom well (Figure 1b), allowing the incorporation of cytokines potentially produced from drug resistant cells into the culture medium for drug sensitive cells. PARP1 plays a role in various pathological processes such as DNA damage repair and cell death. When PARP1 is cleaved by caspase-3, a truncated fragment of 89 KD is produced and mediates apoptosis [30]. As a result, the cleaved form of PARP1 (C-PARP1) was significantly elevated in cisplatin-treated BGC823 and SGC7901 cells co-cultured with drug sensitive cells (Figure 1c). However, such ciaplatin-induced cleavage of PARP1 was significantly impaired after the co-culture of drug resistant cells (Figure 1c). In line with this result, the TUNEL assay also showed that cisplatin-induced apoptosis was significantly decreased in BGC823 cells and SGC7901 cells co-cultured with drug-resistant BGC823/DDP and SGC7901/DDP cells, respectively (Supplementary Figure 1a and b). These data demonstrated that drug resistant cancer cells could transmit the resistance to drug sensitive cells, most likely by secreting autocrine cytokines.
To identify which cytokines were responsible for the transmission of drug resistance, we employed a cytokine antibody array to screen potential cytokines in the conditional medium of drug-resistant cells (Supplementary Figure 1c, supplementary table). As a result, CCL2 was 5.6 fold up-regulated in the conditional medium of SGC7901/DDP cells when compared with its level in the conditional medium of SGC7901 cells (Supplementary Figure 1d). Similarly, CCL2 was also increased in the culture medium of another drug-resistant cell line, BGC823/DDP (Supplementary Figure 1d). As confirmed by ELISA assay, CCL2 secretion was indeed significantly increased in BGC823/DDP and SGC7901/DDP cells compared with parental BGC823 and SGC7901 cells, respectively (Figure 1d). Importantly, when CCL2 secretion was effectively inhibited by siRNA-mediated gene knock-down (Supplementary Figure 1e), drug resistant cells partly lost their ability to transmit drug resistance to sensitive cells (Figure 1e). These findings indicated that autocrine CCL2 might be responsible for the transmission of drug resistance from resistant cancer cells to sensitive cancer cells.
Relevance of CCL2 to acquired drug resistance
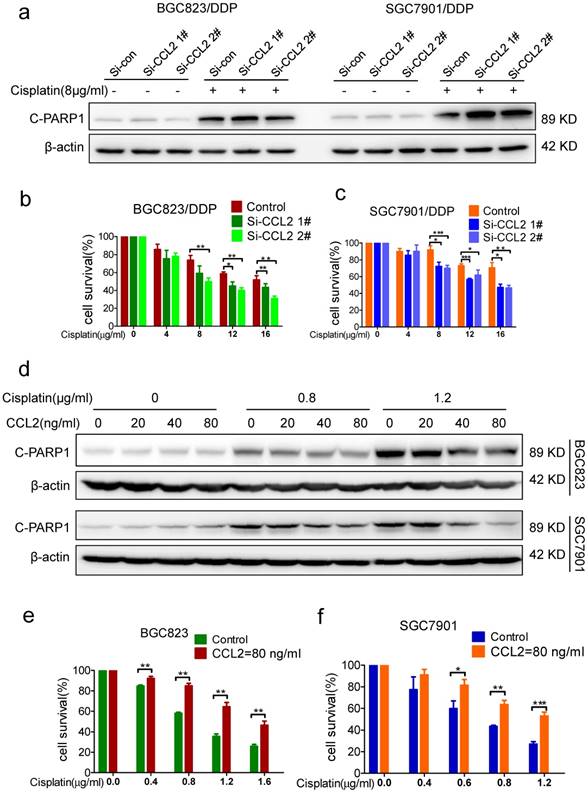
Next, we wonder whether CCL2 is relevant to maintain drug resistance in drug-resistant cells. Indeed, cisplatin-induced PARP1 cleavage in BGC823/DDP and SGC7901/DDP cells was significantly elevated after CCL2 knockdown by transfection of CCL2 siRNA (Figure 2a). Consistently, knockdown of CCL2 in BGC823/DDP and SGC7901/DDP cells resulted in increased cell death induced by cisplatin (Figure 2b and c). The TUNEL assay further confirmed that the apoptosis of BGC823/DDP and SGC7901/DDP cells were significantly increased upon CCL2 knockdown (Supplementary Figure 2a and b).
To validate the importance of CCL2 to acquired drug resistance in drug sensitive cells, we compared the effect of cisplatin on drug-sensitive BGC823 and SGC7901 cells with or without the addition of recombinant CCL2 protein. Interestingly, cisplatin-induced PARP1 cleavage was significantly decreased with the addition of recombinant CCL2 protein (Figure 2d). In support of this, the addition of recombinant CCL2 protein significantly attenuated cisplatin-induced viability inhibition of BGC823 and SGC7901 cells (Figure 2e and f). The TUNEL assay further confirmed that the apoptosis of BGC823 and SGC7901 cells were attenuated significantly in the presence of recombinant CCL2 protein. (Supplementary Figure 2c and d) Therefore, exogenous CCL2 protein could confer drug resistance to drug sensitive cells.
Activation of PI3K/AKT-mTOR signaling and inhibition of autophagy in CCL2-promoted drug resistance
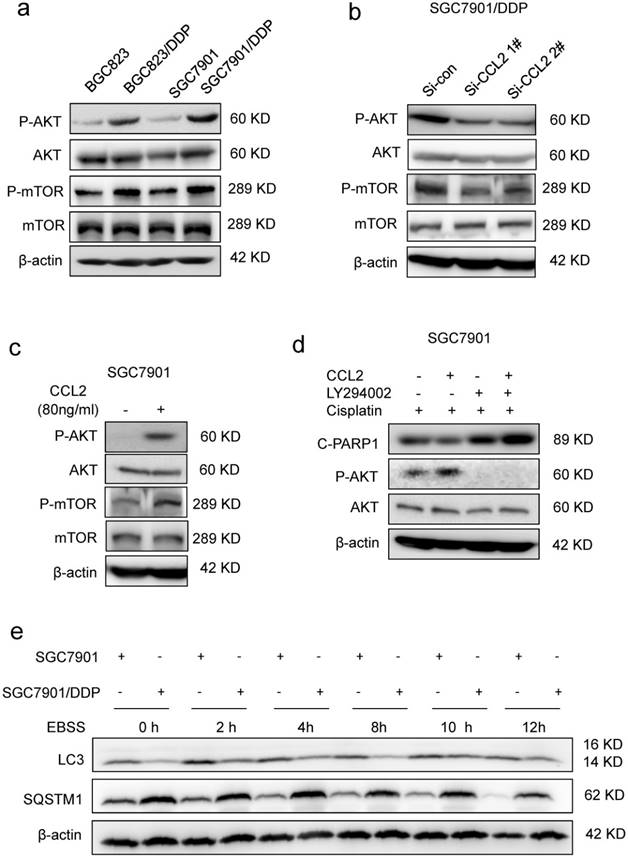
Then, we explored how CCL2 suppressed cisplatin-induced apoptosis. Cisplatin was well-known to induce DNA double strand breaks (DSBs) in the form of platinum-DNA adducts. The level of phosphorylated histone H2AX (γH2AX), a sensitive surrogate marker of DSBs, was increased in BGC823 and SGC7901 cells after cisplatin treatment (Supplementary Figure 3a). However, addition of recombinant CCL2 failed to reverse cisplatin-induced increase of γH2AX level in both cell lines (Supplementary Figure 3a), indicating that CCL2-transduced drug resistance was most likely due to the interference of downstream events in DNA damage response. It has been reported that CCL2 could activate intracellular signaling pathways such as PI3K/Akt signaling in myeloid-derived cells [11]. Therefore, we explored the activity of PI3K/Akt-mTOR signaling in response to changes in CCL2 expression. As a result, we found that the phosphorylation of Akt (P-AKT) and mTOR (P-mTOR) was increased in drug resistant cells when compared with drug sensitive cells (Figure 3a). In contrast, the P-Akt level was significantly decreased if cells were transfected with CCL2 siRNA (Figure 3b). Moreover, the P-AKT level was significantly elevated after the treatment of CCL2 (Figure 3c). LY294002, the PI3K/Akt pathway inhibitor, blocked CCL2-induced phosphorylation of Akt and restored cisplatin-activated cleavage of PARP1 even in the presence of CCL2 (Figure 3d).
PI3K/AKT-mTOR signaling pathway regulates numerous physiologic and pathologic processes [31]. Among them, autophagy was recently found to be related with apoptosis and drug resistance [32-33]. Indeed, the level of microtubule-associated protein 1 light chain-3 II (LC-3 II) was reduced in drug resistant cells when compared with drug sensitive cells (Figure 3e). Meanwhile, the level of SQSTM1, one of the specific substrates that are degraded by autophagy, was increased in drug resistant cells (Figure 3e). In addition, after the activation of autophagy by starvation, the elevation of LC-3 II and degradation of SQSTM1 were significantly reduced in drug resistant cells (Figure 3e). We further introduced the mRFP-GFP-LC3 reporter to determine the autophagy flux. When located in autolysosomes, this form of LC3 displays only red fluorescence since the GFP signal is sensitive to the acidic condition in the lysosome lumen, whereas the RFP signal is more stable. Increased red spots appeared in the merged section of EBSS-treated BGC823 and SGC7901 cells, indicating the maturation of autolysosome in these cells. In contrast, less red spots were observed in drug resistant cells (Figure 4a, Supplementary Figure 3b). These results suggested that autophagy induction was significantly suppressed in drug resistant cells compared with drug sensitive cells.
Importance of CCL2 to acquired drug resistance (a) The level of cleaved PARP-1 in CCL2 knockdown BGC823/DDP and SGC7901/DDP cells treated with 8μg/ml cisplatin was determined by Western blotting. (b-c) The viability of cisplatin treated cells (BGC823/DDP in B and SGC7901/DDP in C) before and after CCL2 knockdown was determined by CCK-8 assay. (d) The level of cleaved PARP-1 in BGC823 and SGC7901 cells treated with recombinant CCL2 protein and cisplatin was determined by Western blotting.(e and f) The viability of cisplatin-treated cells (BGC823 in E and SGC7901 in F) in the presence or absence of recombinant CCL2 protein was determined by CCK-8 assay. (*P<0.05, **P<0.01, ***P<0.001)
To further investigate the role of autophagy in drug resistance, we compared the drug response of drug resistant cells before and after activation of autophagy. When autophagy was activated by EBSS incubation (data was not shown) or rapamycin treatment (Figure 4b, Supplementary Figure 4a), more viability inhibition was observed in drug resistant cells (Figure 4c, Supplementary Figure 4b). Consistently, in drug resistant cells with autophagy activation, more cleavage of PARP1 was induced by cisplatin (Figure 4d, Supplementary Figure 4c). Therefore, autophagy was indeed suppressed in drug resistant cells and activation of autophagy could restore drug sensitivity.
CCL2 suppressed apoptosis via attenuating PI3K/AKT-mTOR pathway. (a) The level of phosphorylated AKT (P-AKT), phosphorylated mTOR (P-mTOR) in different cells as indicated was determined by Western blotting. (b) The level of phosphorylated P-AKT and P-mTOR in SGC7901/DDP cells before and after CCL2 knockdown was determined by Western blotting. (c) The level of P-AKT and P-mTOR in SGC7901 cells incubated with or without recombinant CCL2 was determined by Western blotting. (d) The level of cleaved PARP1 induced by 1.2 μg/ml cisplatin in SGC7901 cells treated as indicated was determined by Western blotting. (e) The LC3 and SQSTM1 protein levels in SGC7901 cells and SGC7901/DDP cells before and after EBSS treatment were determined by Western blotting.
Autophagy was suppressed in drug resistant cells. (a) Quantification of LC-3 puncta in SGC7901 and SGC7901/DDP cells treated with EBSS 2h. (b) The LC-3 and SQSTM1 levels in SGC7901/DDP cells before and after rapamycin treatment were determined by Western blotting. (c) The viability of cisplatin-treated SGC7901 cells in the presence or absence of rapamycin or EBSS were determined by CCK-assay. (d) 12 μg/ml Cispatin-induced apoptosis in SGC7901/DDP cells before and after 20 nM rapamycin treatment were determined by Western blotting. (e) The LC-3 and SQSTM1 levels in CCL2 knockdown SGC7901/DDP cells after EBSS treatment were determined by Western blotting. (f) Quantification of LC-3 puncta in CCL2 knockdown SGC7901/DDP cells treated with EBSS 2h or cisplatin 24h. (*P<0.05, **P<0.01).
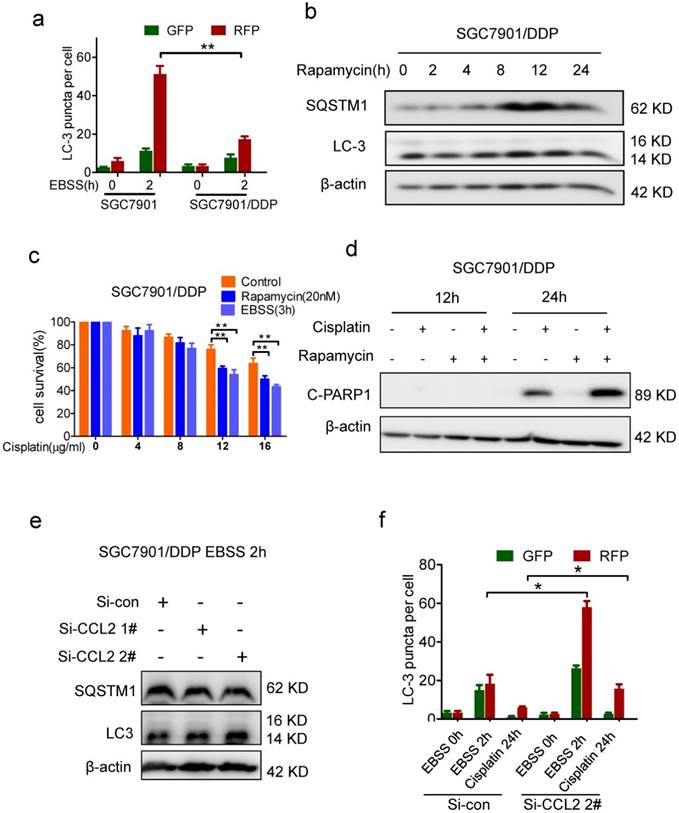
Based on above findings, we assumed that autocrine CCL2 promotes drug resistance by PI3K/Akt-mTOR activation and subsequent autophagy inhibition. Indeed, there was much more starvation-induced increase of LC-3 II and SQSTM1 degradation in CCL2-depleted resistant cells (Figure 4e). Moreover, after CCL2 depletion, increased red spots appeared in starved or cisplatin treated resistant cells with GFP-RFP-LC3 expression (Figure 4f, Supplementary Figure 3c), indicating that autocrine CCL2 was important to inhibit autophagy.
SQSTM1 promotes CCL2 transcription and secretion
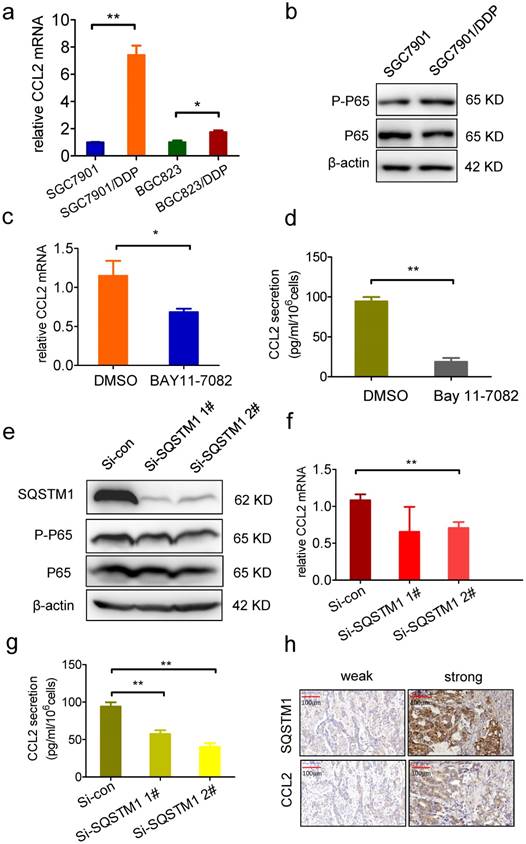
Next, we explored the mechanism of CCL2 overexpression in drug resistant cells. We found that the mRNA level of CCL2 was much higher in drug resistant cells (Figure 5a), indicating that CCL2 gene transcription was enhanced. It has been reported that c-Jun controls histone modifications, NF-κB recruitment, and RNA Polymerase II function to activate the transcription of CCL2 gene [34]. We found MAPK signal pathways were actually inhibited in drug resistant cells (Supplementary Figure 4d). However, P-P65 level was elevated, indicating the activation of NF-κB signal pathway in drug resistant cells (Figure 5b). Moreover, both the mRNA level and secretion of CCL2 were significantly decreased by treatment with NF-κB inhibitor BAY-11-7082 (Figure 5c and d). Since SQSTM1, which is important for the activation NF-κB signal pathway [35-36], was specifically upregulated in drug resistant cancer cells, we investigated whether SQSTM1 regulated the expression of CCL2. As a result, P-P65 was decreased by knockdown expression of SQSTM1 (Figure 5e). Similarly with NF-КB inhibition, both the mRNA level and secretion of CCL2 were significantly decreased after SQSTM1 knockdown (Figure 5f and g). These results indicated that SQSTM1 promoted CCL2 transcription and secretion, forming a positive feedback between CCL2 and SQSTM1.
SQSTM1 promote CCL2 transcription and secretion. (a) The CCL2 mRNA level in BGC823, BGC823/DDP, SGC7901, SGC7901/DDP cells was detected by RT-PCR. (b) The P-P65 level in SGC7901 and SGC7901/DDP cells was detected by Western blotting. (c) The CCL2 mRNA level in SGC7901/DDP cells treated with 5μM BAY11-7082 24h was detected by RT-PCR. (d) The CCL2 secretion level in 5μM BAY11-7082 treated SGC7901/DDP medium was detected by ELSIA. (e) The SQSTM1 and P-P65 levels in SQSTM1 knockdown SGC7901/DDP cells were detected by Western blotting. (f) The CCL2 mRNA level in SQSTM1 knockdown SGC7901/DDP cells were detected by RT-PCR. (g) The CCL2 secretion level in SQSTM1 knockdown SGC7901/DDP cells were detected by ELISA. (h) The representative photographs of CCL2 and SQSTM1 in gastric cancer tissues. (*P<0.05, **P<0.01)
CCL2 and SQSTM1 expression in gastric cancer tissues
| SQSTM1 | weak | strong | ||
|---|---|---|---|---|
| CCL2 | weak | 7 | 5 | |
| strong | 6 | 25 | P=0.018 |
As BGC823 and SGC7901 cells derived from stomach adenocarcinoma, a total of 43 gastric adenocarcinoma tissues were employed to analyze the relationship of CCL2 expression and SQSTM1 in vivo. SQSTM1 positivity was statistically more frequent in CCL2 positive tissues compared with CCL2 negative tissues (Fisher's exact test, P= 0.018) (Figure 5h, table 1), indicating that CCL2 indeed inhibited autophagy in vivo.
Clinical relevance of CCL2 in gastric cancer
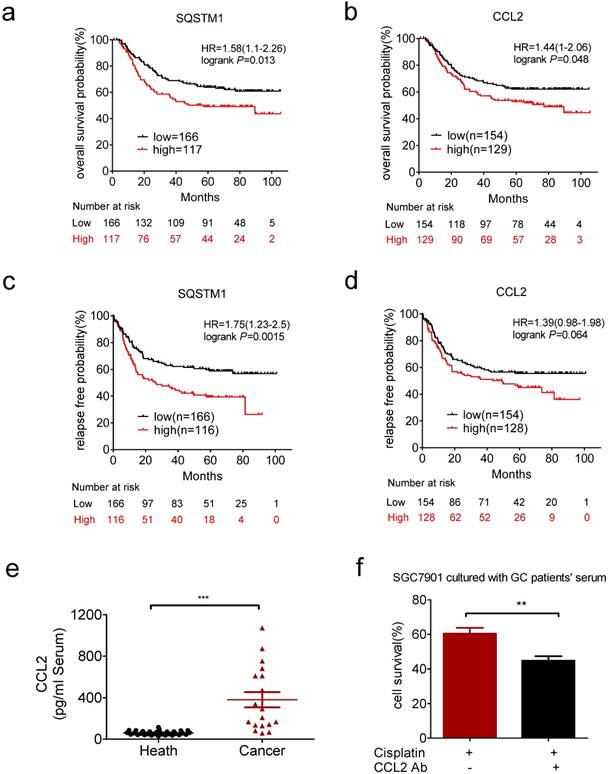
We next explored the clinical relevance of CCL2 and SQSTM1 mRNA level in gastric cancer patients by Kaplan-Meier plotter (http://kmplot.com/analysis/). We analyzed the GSE62254 dataset, which includes nearly 300 patients [37]. High expression of SQSTM1 and CCL2 mRNAs were significantly associated with shorter overall survival (Figure 6a and b). High expression of SQSTM1 and CCL2 mRNAs were also significantly associated with shorter relapse free survival (Figure 6c and d).
Clinical relevance of CCL2 in gastric cancer. (a) Overexpression of SQSTM1 mRNA predicted shorter overall survival of gastric cancer patients. (b) Overexpression of CCL2 mRNA predicted shorter overall survival of gastric cancer patients. (c) Overexpression of SQSTM1 mRNA predicted shorter relapse free survival of gastric cancer patients. (d) Overexpression of CCL2 mRNA predicted shorter relapse free survival of gastric cancer patients. (e) The levels of CCL2 in health people and gastric cancer patients' serums were detected by ELSIA(health n=31, patients n=18). (f) The viability of conditional medium cultured SGC7901 cells in the presence or absence of CCL2 neutralize antibody and cisplatin were determined by CCK-assay. (**P<0.01, ***P<0.001)
The positive feedback of CCL2 and SQSTM1 contributes to chemoresistance. Autocrine CCL2 suppresses proapoptotic autophagy to promote chemoresistance. Drug resistant cancer cells secreted CCL2 to maintain or transduce drug resistance by activating PI3K-Akt-mTOR signaling pathway to suppress proapoptotic autophagy. Up-regulated SQSTM1 activated CCL2 transcription and secretion.
We also found the CCL2 level was much higher in serum of gastric cancer patients than health people (Figure 6e). CCL2 level was significantly decreased after surgery in patients with high level of CCL2 (>249.5pg/ml, Supplementary Figure 4e). Then we used the conditional medium containg serum of patients with high serum level of CCL2 to culture gastric cancer cells. As a result, patient-derived CCL2 conferred drug resistance to gastric cancer cells which was impaired in the presence of CCL2 neutralizing antibody (Figure 6f). These results indicated the potential value of CCL2 as a biomarker and intervention target for chemotherapy resistance.
Discussion
Despite many advances that have been achieved, drug resistance is still the major obstacle for cancer treatment. Tumor cells acquired chemoresistance by various mechanisms including increased drug efflux, altered drug metabolism, enhanced capability of DNA repair, and activation of prosurvival signaling pathways [1]. However, how the communication between heterogeneous tumor cells and tumor microenvironment contributed to drug resistance was still obscure.
Tumor microenvironment created a complicated ecological niche for tumor growth and survival [38]. Co-culturing melanoma cells with fibroblasts led to a diminished therapeutic response at least partly due to the production of fibronectin by fibroblasts [38]. Interestingly, a subgroup of intrinsically resistant melanoma cells was capable of secreting fibronectin in response to a number of cellular stresses including BRAF inhibitor therapy and chemotherapy [39]. In this study, we found that the drug resistant cancer cells secreted chemokine CCL2 to activate PI3K/AKT-mTOR pathway in response to chemotherapy. Moreover, co-culture with drug resistant cells led to a diminished therapeutic response in drug sensitive cells. Therefore, CCL2 may function as a key molecule to transmit drug resistance among heterogeneous tumor cells. CCL2 was usually secreted by host cells to promote tumor progression such as tumor metastasis by recruiting macrophages [40-42]. In this study, we proved that CCL2 could be secreted by tumor cells and promote chemoresistance in an autocrine manner. Similarly, drug-resistant ovarian cancer cells can also secrete more CCL2 and blocking CCL2 increased sensitivity of ovarian cancer cells to chemotherapeutic drugs [16]. Enhanced the expression and secretion of CCL2 also lead to neovascularization and increased resistance to chemotherapy in glioblastoma [43]. Indeed, we succeeded in detecting increased level of CCL2 in cancer patients before surgical tumor removal. Blockage of CCL2 in conditional media by CCL2 neutralizing antibody restored the sensitivity of gastric cancer cells to cisplatin. Taken together, CCL2 could be a potential biomarker and intervention target for chemotherapy resistance. Moreover, CCL2 protein has been recognized as a potential independent adverse prognosticator to improve the predictive accuracy of overall survival in gastric cancer patients [44]. Consistently, we found that higher CCL2 mRNA level related to shorter overall survival in gastric cancer patients by using Kaplan-Meier plotter. Whether targeting CCL2 could improve the clinical efficacy of conventional chemotherapy in vivo remains further investigations.
Autophagy is an evolutionarily-ancient mechanism by which cellular materials is delivered to lysosomes for degradation [29, 45]. In tumor cells, autophagy can be induced by various stresses such as nutrient starvation, hypoxia, chemotherapy or radiotherapy. The pro-survival role of autophagy in chemotherapy resistance has been widely reported, leading to the extensive investigation of autophagy inhibitors for anti-cancer treatments in clinical trials [46-48]. However, autophagy acts as a double-edged sword in cancer development [49]. Activation of autophagy has also been reported to enhance chemosensitivity [50]. For example, autophagy can lead to the autophagic cell death which was also known as type 2 programmed cell death and difficult to differentiate from type 1 (apoptotic) cell death [51]. In addition, the crosstalk between apoptosis and autophagy is complex. Autophagy and apoptosis can be simultaneously detected in response to their common stimuli [32]. Our previous study reported that apoptosis was suppressed in drug resistant gastric cell lines [25]. Here, we found autophagy was also suppressed in drug resistant cells and activation of autophagy by rapamycin reversed chemoresistance through the induction of apoptosis. Interestingly, autophagy occurred earlier than apoptosis, indicating that activation of autophagy triggered apoptosis. Therefore, CCL2 promoted chemoresistance by inhibiting autophagy and autophagy inducer rather than inhibitor should be employed to overcome chemoresistance.
SQSTM1, a member of the autophagic receptors that promote autophagy activation, was degraded together with its cargo like misfolded proteins in autolysosomes [52-53]. Here, due to the inhibition of autophagy by CCL2, SQSTM1 was up-regulated in drug resistant cells. SQSTM1 expression correlated well with CCL2 expression in primary stomach carcinoma tissues. In addition, higher SQSTM1 and CCL2 expression level were related to shorter overall survival and relapse free survival in gastric cancer patients. Interestingly, SQSTM1 also directly contributes to tumor development through the regulation of autophagy and oncogenic signaling pathways like NF-κB [54]. NF-κB in turn can activate CCL2 transcription [55]. These data revealed a positive feedback of CCL2 and SQSTM1 that contributed to drug resistance in gastric cancer.
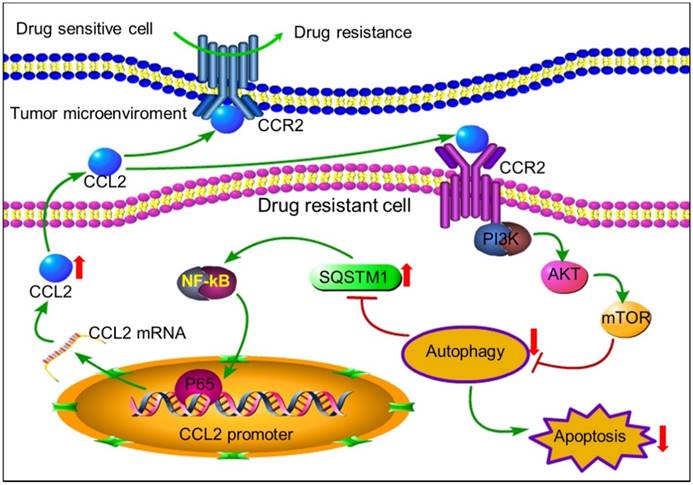
In conclusion, tumor cells can secrete autocrine CCL2 to maintain or transmit chemoresistance by activating PI3K-Akt-mTOR signaling to inhibit proapoptotic autophagy (Figure 7). CCL2 could be a potential biomarker and intervention target for chemotherapy resistance. In tumors with high level of CCL2 expression, autophagy inducers rather than inhibitors should be used to overcome chemoresistance.
Abbreviations
CCL2, chemokine C-C motif ligand 2; DDP, cis-diammine dichloridoplatinum(II); EBSS, Earle's Balanced Salt Solution; GFP, green fluorescent protein; LC3, microtubule-associated protein 1 light chain-3; PARP1, poly(ADP-ribose) polymerase 1; RFP, red fluorescent protein; SQSTM1, sequestosome 1; TAMs, tumor-associated-macrophages; TUNEL, TdT-mediated dUTP Nick-End Labeling; γH2AX, phosphorylated histone H2AX.
Supplementary Material
Supplementary figures and table.
Acknowledgements
This work was supported by Natural Science foundation of China (81502609, 8171101166; 91740106), High Level InnovativeTalents Program and 151 Talents program in Zhejiang, Natural Science Foundation of Zhejiang province (LQ15H160004), and Department of Health in Zhejiang Province (WKJ-ZJ-1520; 2016143571) and the Fundamental Research Funds for the Central Universities (2016FZA700 1). We also acknowledged Chaying Guan, who helped us collect clinical specimens.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O. et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869-83
2. Woods D, Turchi J J. Chemotherapy induced DNA damage response: convergence of drugs and pathways. Cancer Biol Ther. 2013;14:379-89
3. Roos W P, Kaina B. DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013;332:237-48
4. Junttila M R, de Sauvage F J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346-54
5. Gilbert L A, Hemann M T. DNA damage-mediated induction of a chemoresistant niche. Cell. 2010;143:355-66
6. Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian Z R, Du J. et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500-4
7. Tripathi C, Tewari B N, Kanchan R K, Baghel K S, Nautiyal N, Shrivastava R. et al. Macrophages are recruited to hypoxic tumor areas and acquire a pro-angiogenic M2-polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget. 2014;5:5350-68
8. Lee H J, Zhuang G, Cao Y, Du P, Kim H J, Settleman J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell. 2014;26:207-21
9. O'Boyle G, Brain J G, Kirby J A, Ali S. Chemokine-mediated inflammation: Identification of a possible regulatory role for CCR2. Mol Immunol. 2007;44:1944-53
10. Kolattukudy P E, Niu J. Inflammation, endoplasmic reticulum stress, autophagy, and the monocyte chemoattractant protein-1/CCR2 pathway. Circ Res. 2012;110:174-89
11. Li X, Xu Q, Wu Y, Li J, Tang D, Han L. et al. A CCL2/ROS autoregulation loop is critical for cancer-associated fibroblasts-enhanced tumor growth of oral squamous cell carcinoma. Carcinogenesis. 2014;35:1362-70
12. Giopanou I, Lilis I, Papaleonidopoulos V, Agalioti T, Kanellakis N I, Spiropoulou N. et al. Tumor-derived osteopontin isoforms cooperate with TRP53 and CCL2 to promote lung metastasis. Oncoimmunology. 2017;6:e1256528
13. Fang W B, Yao M, Brummer G, Acevedo D, Alhakamy N, Berkland C. et al. Targeted gene silencing of CCL2 inhibits triple negative breast cancer progression by blocking cancer stem cell renewal and M2 macrophage recruitment. Oncotarget. 2016;7:49349-49367
14. Zhang J, Yan Y, Cui X, Zhang J, Yang Y, Li H. et al. CCL2 expression correlates with Snail expression and affects the prognosis of patients with gastric cancer. Pathol Res Pract. 2017;213:217-221
15. Wu C Y, Yang Y H, Lin Y Y, Kuan F C, Lin Y S, Lin W Y. et al. Anti-cancer effect of danshen and dihydroisotanshinone I on prostate cancer: targeting the crosstalk between macrophages and cancer cells via inhibition of the STAT3/CCL2 signaling pathway. Oncotarget. 2017
16. Moisan F, Francisco E B, Brozovic A, Duran G E, Wang Y C, Chaturvedi S. et al. Enhancement of paclitaxel and carboplatin therapies by CCL2 blockade in ovarian cancers. Mol Oncol. 2014;8:1231-9
17. Green D R, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65-75
18. Tian L, Ma L, Guo E, Deng X, Ma S, Xia Q. et al. 20-Hydroxyecdysone upregulates Atg genes to induce autophagy in the Bombyx fat body. Autophagy. 2013;9:1172-87
19. Tan Q, Joshua A M, Saggar J K, Yu M, Wang M, Kanga N. et al. Effect of pantoprazole to enhance activity of docetaxel against human tumour xenografts by inhibiting autophagy. Br J Cancer. 2015;112:832-40
20. Qin L, Xu T, Xia L, Wang X, Zhang X, Zhang X. et al. Chloroquine enhances the efficacy of cisplatin by suppressing autophagy in human adrenocortical carcinoma treatment. Drug Des Devel Ther. 2016;10:1035-45
21. Zhao J, Nie Y, Wang H, Lin Y. MiR-181a suppresses autophagy and sensitizes gastric cancer cells to cisplatin. Gene. 2016;576:828-33
22. Han W, Pan H, Chen Y, Sun J, Wang Y, Li J. et al. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS One. 2011;6:e18691
23. Han W, Sun J, Feng L, Wang K, Li D, Pan Q. et al. Autophagy inhibition enhances daunorubicin-induced apoptosis in K562 cells. PLoS One. 2011;6:e28491
24. Wang Y, Ding L, Wang X, Zhang J, Han W, Feng L. et al. Pterostilbene simultaneously induces apoptosis, cell cycle arrest and cyto-protective autophagy in breast cancer cells. Am J Transl Res. 2012;4:44-51
25. Ni P, Xu W, Zhang Y, Chen Q, Li A, Wang S. et al. TXNL1 induces apoptosis in cisplatin resistant human gastric cancer cell lines. Curr Cancer Drug Targets. 2015;14:850-9
26. Xu W, Wang S, Chen Q, Zhang Y, Ni P, Wu X. et al. TXNL1-XRCC1 pathway regulates cisplatin-induced cell death and contributes to resistance in human gastric cancer. Cell Death Dis. 2014;5:e1055
27. Xu W, Chen Q, Wang Q, Sun Y, Wang S, Li A. et al. JWA reverses cisplatin resistance via the CK2-XRCC1 pathway in human gastric cancer cells. Cell Death Dis. 2014;5:e1551
28. Zhou J, Ye J, Zhao X, Li A, Zhou J. JWA is required for arsenic trioxide induced apoptosis in HeLa and MCF-7 cells via reactive oxygen species and mitochondria linked signal pathway. Toxicol Appl Pharmacol. 2008;230:33-40
29. Feng L, Ma Y, Sun J, Shen Q, Liu L, Lu H. et al. YY1-MIR372-SQSTM1 regulatory axis in autophagy. Autophagy. 2014;10:1442-53
30. Luo X, Kraus W L. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012;26:417-32
31. Kim Y C, Guan K L. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125:25-32
32. Maiuri M C, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741-52
33. Wang X, Zhang S, Zhang J, Lam E, Liu X, Sun J. et al. Annexin A6 is down-regulated through promoter methylation in gastric cancer. Am J Transl Res. 2013;5:555-62
34. Wolter S, Doerrie A, Weber A, Schneider H, Hoffmann E, von der Ohe J. et al. c-Jun controls histone modifications, NF-kappaB recruitment, and RNA polymerase II function to activate the ccl2 gene. Mol Cell Biol. 2008;28:4407-23
35. Duran A, Linares J F, Galvez A S, Wikenheiser K, Flores J M, Diaz-Meco M T. et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343-54
36. Moscat J, Diaz-Meco M T. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137:1001-4
37. Szasz A M, Lanczky A, Nagy A, Forster S, Hark K, Green J E. et al. Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget. 2016;7:49322-49333
38. Fedorenko I V, Smalley K S. The complexity of microenvironment-mediated drug resistance. Genes Cancer. 2015;6:367-8
39. Fedorenko I V, Abel E V, Koomen J M, Fang B, Wood E R, Chen Y A. et al. Fibronectin induction abrogates the BRAF inhibitor response of BRAF V600E/PTEN-null melanoma cells. Oncogene. 2015
40. Li X, Yao W, Yuan Y, Chen P, Li B, Li J. et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. 2017;66:157-167
41. Rao Q, Chen Y, Yeh C R, Jie D, Li L, Chang C. et al. Recruited mast cells in the tumor microenvironment enhance bladder cancer metastasis via modulation of ERbeta/CCL2/CCR2 EMT/MMP9 signals. Oncotarget. 2015
42. Svensson S, Abrahamsson A, Rodriguez G V, Olsson A K, Jensen L, Cao Y. et al. CCL2 and CCL5 Are Novel Therapeutic Targets for Estrogen-Dependent Breast Cancer. Clin Cancer Res. 2015;21:3794-805
43. Fianco G, Mongiardi M P, Levi A, De Luca T, Desideri M, Trisciuoglio D. et al. Caspase-8 contributes to angiogenesis and chemotherapy resistance in glioblastoma. Elife. 2017:6
44. Liu H, Shen Z, Wang X, Zhang H, Qin J, Qin X. et al. Increased expression of C-C motif ligand 2 associates with poor prognosis in patients with gastric cancer after gastrectomy. Tumour Biol. 2015
45. Dalby K N, Tekedereli I, Lopez-Berestein G, Ozpolat B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy. 2010;6:322-9
46. Kaminskyy V O, Piskunova T, Zborovskaya I B, Tchevkina E M, Zhivotovsky B. Suppression of basal autophagy reduces lung cancer cell proliferation and enhances caspase-dependent and -independent apoptosis by stimulating ROS formation. Autophagy. 2012;8:1032-44
47. Yang M C, Wang H C, Hou Y C, Tung H L, Chiu T J, Shan Y S. Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Mol Cancer. 2015;14:179
48. Shteingauz A, Boyango I, Naroditsky I, Hammond E, Gruber M, Doweck I. et al. Heparanase Enhances Tumor Growth and Chemoresistance by Promoting Autophagy. Cancer Res. 2015;75:3946-57
49. Qiu Y, Li P, Ji C. Cell Death Conversion under Hypoxic Condition in Tumor Development and Therapy. Int J Mol Sci. 2015;16:25536-51
50. Khurana A, Roy D, Kalogera E, Mondal S, Wen X, He X. et al. Quinacrine promotes autophagic cell death and chemosensitivity in ovarian cancer and attenuates tumor growth. Oncotarget. 2015;6:36354-69
51. Edinger A L, Thompson C B. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663-9
52. Jiang T, Harder B, Rojo D L V M, Wong P K, Chapman E, Zhang D D. p62 links autophagy and Nrf2 signaling. Free Radic Biol Med. 2015;88:199-204
53. Bitto A, Lerner C A, Nacarelli T, Crowe E, Torres C, Sell C. P62/SQSTM1 at the interface of aging, autophagy, and disease. Age (Dordr). 2014;36:9626
54. Moscat J, Karin M, Diaz-Meco M T. p62 in Cancer: Signaling Adaptor Beyond Autophagy. Cell. 2016;167:606-609
55. Wei M, Li Z, Xiao L, Yang Z. Effects of ROS-relative NF-kappaB signaling on high glucose-induced TLR4 and MCP-1 expression in podocyte injury. Mol Immunol. 2015;68:261-71
Author contact
![]() Corresponding author: Dr. Hongchuan Jin, email: jinhcedu.cn
Corresponding author: Dr. Hongchuan Jin, email: jinhcedu.cn
Received 2018-2-4
Accepted 2018-5-10
Published 2018-6-3