ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Results
Discussion
Materials and Methods
Acknowledgements
Supplementary Material
References

Int J Biol Sci 2018; 14(9):1067-1080. doi:10.7150/ijbs.23845 This issue Cite
Research Paper
WDR1 Promotes Cell Growth and Migration and Contributes to Malignant Phenotypes of Non-small Cell Lung Cancer through ADF/cofilin-mediated Actin Dynamics
Baiyin Yuan1,*, ![]() , Ruirui Zhang1,*, Jisheng Hu1, Zhongying Liu1, Chao Yang1, Tongcun Zhang1,
, Ruirui Zhang1,*, Jisheng Hu1, Zhongying Liu1, Chao Yang1, Tongcun Zhang1, ![]() , Chenxi Zhang2,
, Chenxi Zhang2, ![]()
1. Biomedical Research Institute, College of Life Science and Health, Wuhan University of Science and Technology, Wuhan, Hubei Province 430081, China.
2. Central Laboratory, Nanjing Chest Hospital, Medical School of Southeast University, Nanjing, Jiangsu Province 210029, P.R. China.
* These authors contributed equally to this work.
Abstract

The characteristic of carcinoma is cell migration and invasion, which involve in strong actin dynamics. Regulations of actin dynamics have been implicated in cancer cell migration and tumor progression. WDR1 (WD-repeat domain 1) is a major cofactor of the actin depolymerizing factor (ADF)/cofilin, strongly accelerating ADF/cofilin-mediated actin disassembly. The role of WDR1 in non-small cell lung cancer (NSCLC) progression has been unknown. Here, we show that the expression levels of WDR1 are increased in human NSCLC tissues compared with adjacent non-tumor tissues, and high WDR1 level correlates with poor prognosis in NSCLC patients. Knockdown of WDR1 in NSCLC cells significantly inhibits cell migration, invasion, EMT process and tumor cell growth in vitro and in vivo. Otherwise, overexpression of WDR1 promotes NSCLC cell proliferation and migration. Mechanically, our data suggested WDR1 regulated tumor cells proliferation and migration might through actin cytoskeleton-mediated regulation of YAP, and we demonstrated that WDR1 contributes to NSCLC progression through ADF/cofilin-mediated actin disassembly. Our findings implicate that the ADF/cofilin-WDR1-actin axis as an activator of malignant phenotype that will be a promising therapeutic target in lung cancer.
Keywords: WDR1, Cancer, Non-small cell lung cancer, Actin dynamics, ADF/cofilin
Introduction
Lung cancer is the leading cause of cancer-related mortality, and is a serious public health problem worldwide [1]. Lung cancer includes two main histological types: small cell lung cancer (SCLC) and non-SCLC (NSCLC), NSCLC accounts for approximately 85% of all cases of lung cancer [2]. Despite advances in early diagnosis and clinical therapies, while lacking early symptoms characteristics, patients with NSCLC are often diagnosed at an advanced stage and have a poor prognosis, and the 5-year survival is only 16% independent of the tumor stage and size [3, 4]. Therefore, it is urgent to elucidate the underlying molecular pathways and mechanisms contributing to the NSCLC progression.
Actin filaments simultaneously undergo assembly at their barbed ends and disassembly at their pointed ends under physiological conditions, which was known as actin dynamics [5]. Actin dynamics are critical for cell shape regulation and motility, and are regulated by various actin-binding proteins [6, 7]. ADF (actin depolymerizing factor)/cofilin is the major factor enhancing severing of actin filaments (F-actin) and depolymerization of monomeric actin (G-actin) from the pointed ends [8]. WDR1 (WD-repeat domain 1), the mammalian homolog of AIP1 (actin interacting protein 1) in Drosophila, is a major cofactor of ADF/cofilin that can strongly accelerate the disassembly of ADF/cofilin-bound actin filaments by capping the barbed ends of ADF/cofilin-severed filaments and blocking their re-annealing and elongation [9, 10]. WDR1 is highly conserved across all eukaryotes, and has been shown to act together with ADF/cofilin to promote actin dynamics in various actin-based processes, including cell migration, cytokinesis [8, 9, 11]. In addition, our research group showed that WDR1 and ADF/cofilin are both required for Drosophila eye epithelial E-cadherin dynamics, and mammalian myocardial growth and maintenance [12, 13].
The characteristic of carcinoma is cell migration and invasion, which require strong actin dynamics: F-actin continuously undergoes rapid assembly and/or disassembly to form lamellipodia at the leading direction, and then pushes cell to migrate [14]. Actin dynamics have been related to cancer cell migration and tumor progression [15-17]. It has been shown that ADF/cofilin mediated actin dynamics is required for invasive cancer metastasis and migration in prostate cancer, breast cancer, astrocytoma and gastric cancer [18-21]. In addition, WDR1 was significantly upregulated in highly metastatic cell line compared to the low metastatic potential cell line in gallbladder carcinoma [22]. Consistently, WDR1 promoted breast cancer cells migration, and WDR1 overexpression was found in invasive ductal carcinoma and associated with poor survival in breast cancer patients [23, 24]. However, the role of WDR1 in NSCLC progression has not yet been comprehensively studied and involved molecular mechanisms are unknown.
Here, we showed that WDR1 was up-regulated in human NSCLC tissues and high WDR1 level correlated with reduced overall survival in NSCLC patients. For the first time we set out to comprehensively uncover the potential roles of WDR1 in NSCLC progression and the involved mechanism, and we showed that WDR1 contributed to malignant processes in NSCLC, such as tumor cell growth, migration, invasion and the epithelial-mesenchymal transition (EMT) process. Mechanically, our data suggested that WDR1 regulated tumor cells proliferation and migration might through actin cytoskeleton-mediated regulation of YAP, the key relay for the transduction of actin cytoskeleton reorganization to gene transcriptional program, and we demonstrated that WDR1 contributed to NSCLC progression through ADF/cofilin-mediated actin disassembly. Our findings suggest that the WDR1/cofilin-actin axis will be a promising therapeutic target in lung cancer.
Results
High WDR1 expression level correlates with reduced overall survival in NSCLC patients
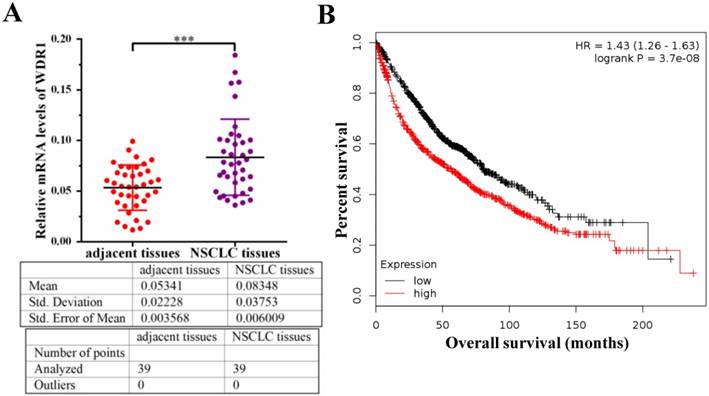
To investigate the potential role of WDR1 in NSCLC patients, we measured the mRNA level of WDR1 in human NSCLC tissues and its matched adjacent non-tumor tissues by quantitative real-time PCR (qPCR) assay. Our results showed that the mRNA level of WDR1 was significantly increased in NSCLC tissues compared to adjacent non-tumor tissues (Figure 1A). To evaluate the relationship between the expression level of WDR1 and patient prognosis, we performed Kaplan-Meier survival analysis (http://kmplot.com) [25]. Analysis of the cohort containing about 960 NSCLC patients revealed that high WDR1 expression level correlates with reduced overall survival (HR=1.43, log-rank P=3.7E-08) (Figure 1B). We also analyzed this relationship in another online tool (http://www.oncolnc.org), and found high WDR1 expression level correlates with reduced survival in lung adenocarcinoma (P=0.0428) and lung squamous carcinoma (P=0.193) (Figure S1). Thus, these results indicated that the expression of WDR1 was altered in NSCLC tissues relative to adjacent normal tissues, and patients with higher WDR1 expression levels exhibited shorter survival, suggesting that WDR1 might have an oncogenic role in the progression of NSCLC.
WDR1 promotes NSCLC cell growth in vitro
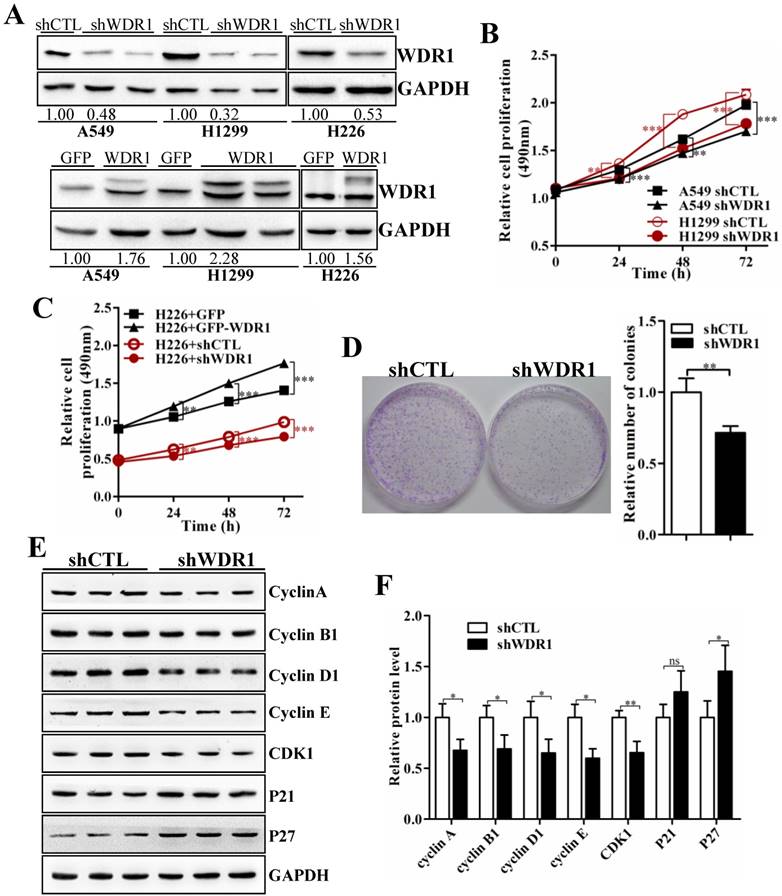
To further investigate the biological function of WDR1 in NSCLC in vitro, we chose A549, H1299 and H226 cell lines, in which the expression levels of WDR1 were shown in Figure S1. In A549, H1299 and H226 cells, we generated cells with stable knockdown and overexpression of WDR1 by infecting lentiviruses that stable expressing shRNA, and PCDH-WDR1 plasmid, respectively. The efficiencies of WDR1 knockdown and overexpression were determined by western blotting analysis. Compared with the responding control cells, the expression levels of WDR1 were profoundly reduced in shWDR1 cells, and were increased in WDR1 overexpressed cells (Figure 2A). To dissect the functions of WDR1 in NSCLC cell growth, cell proliferation assay and colony formation assay were carried out. As shown in Figure 2B-D, compared with shCTL, WDR1 stable knockdown cells showed significant declines in proliferative rate (A549, H1299 and H226 cells) and the potential of colony formation (A549 cells), whereas overexpression of WDR1 increased cell proliferation in H226 cells. TUNEL staining did not reveal any differences between shWDR1 and shCTL cells, indicating that knockdown of WDR1 did not induce cell apoptosis (Figure S2).
WDR1 is upregulated and correlates with poor prognosis in NSCLC patients. A: mRNA levels of WDR1 were determined by qPCR in NSCLC tissues and its matched adjacent non-tumor tissues. The expression levels of WDR1 were increased in NSCLC tissues, compared with adjacent non-tumor tissues. B: Kaplan-Meier plot showed the overall survival of NSCLC patients with all history stratified by high or low WDR1 expression. High WDR1 expression level correlates with reduced overall survival. Data are expressed as means ± SEM. ***P < 0.001.
To dissect the possible mechanism that WDR1 promotes NSCLC cell proliferation, we checked the effect of WDR1 on cell cycle progression. Many molecules involved in cell cycle were detected by western blotting analysis in A549 cells. As shown in Figure 2E and 2F, compared with shCTL cells, in shWDR1 cells, the protein level of cell cycle inhibitor p27 was elevated and that of P21 was not significantly increased, whereas, expression levels of cell cycle regulators cyclin A2, cyclin B1, cyclin D1, cyclin E and Cdk1 were substantially decreased. Further, qPCR results supported western blotting findings and revealed increased level of p27, decreased levels of cyclin A2, cyclin B1, cyclin D1, cyclin E and Cdk1 (Figure S3).
WDR1 positively regulates cell migration, invasion and induces EMT process in vitro
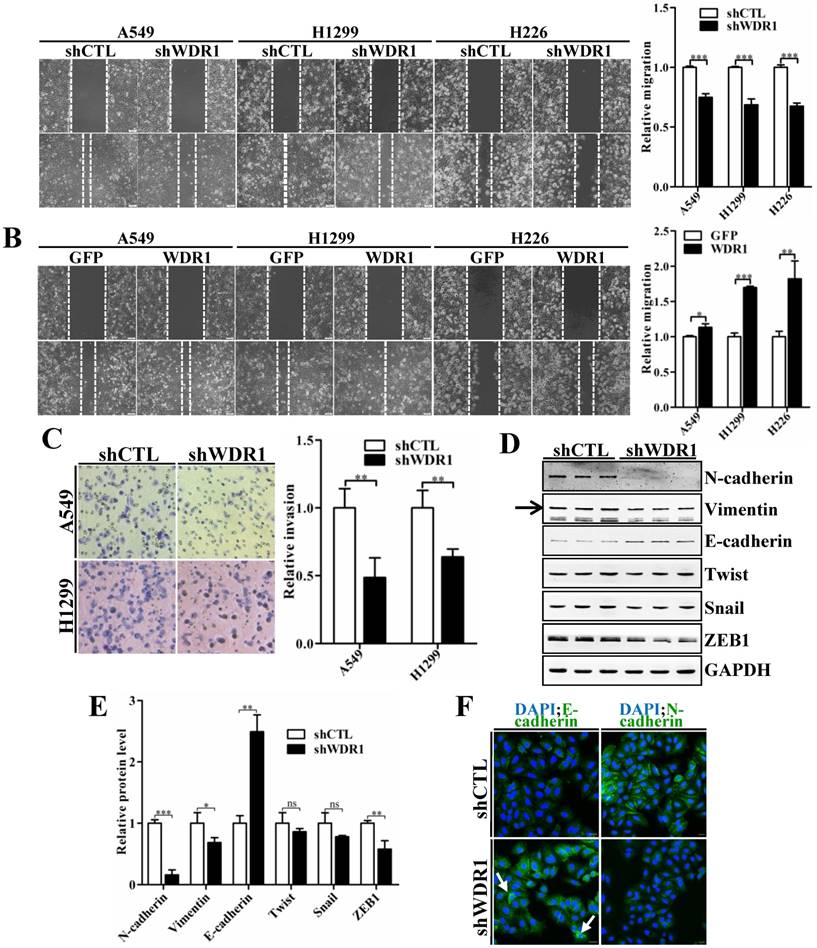
To characterize the effect of WDR1 on cell migration and invasion, wound healing assay and matrigel invasion assay were performed. Wound healing assay showed that, knockdown of WDR1 resulted in a significant inhibition of wound closure, compared with control cells (shCTL) (Figure 3A). In addition, overexpression of WDR1 promoted cell migration (Figure 3B). Consistent with the result of wound healing assay, Wdr1 depleted cells exhibited significantly decreased invading ability (Figure 3C). These data revealed that WDR1 promotes motility and invasion of NSCLC cells in vitro.
Tumor cells invasion and metastasis through a series of critical steps that involve morphological changes known as EMT, which requires strong cytoskeletal reorganization [26]. To dissect whether WDR1 induced EMT, various key molecules involved in EMT process were examined by western blotting analysis in A549 cells. Protein levels of two mesenchymal markers N-cadherin and Vimentin were remarkably decreased, and that of the epithelial marker E-cadherin was dramatically increased, indicating an inactivated EMT process in shWDR1 cells (Figure 3D and E). Further, we analyzed the expression levels of EMT-inducing transcription factors and found that, in shWDR1 cells, the level of ZEB1 was significantly decreased, snail and twist levels were reduced, but not significantly (Figure 3D and E). Immunofluorescence staining for E-cadherin and N-cadherin further confirmed western blotting results: compared with shCTL cells, E-cadherin staining was strongly increased in shWDR1 cells, while that of N-cadherin was dramatically reduced; interestingly, E-cadherin staining appeared strong and sometimes punctate in shWDR1 cells (Figure 3F). These results indicated that WDR1 induced the EMT process and regulated E-cadherin distribution in NSCLC cells.
WDR1 promotes NSCLC growth in vitro. A: In A549, H1299 and H226 cell lines, we generated stable WDR1 knockdown (mediated by shRNA), and overexpressed cells, respectively. The efficiencies of WDR1 knockdown and overexpression were detected by western blotting analysis. The results showed that shWDR1 markedly down-regulated the expression levels of WDR1; and WDR1 was increased in cells transfected with PCDH-WDR1 plasmid (WDR1), relative to cells transfected with PCDH-GFP plasmid (control). The relative protein level was quantified, and the results were showed below the corresponding bands. B: CCK-8 assay showed that WDR1 knockdown significantly inhibited the proliferative rates in A549 and H1299 cells. C: CCK-8 assay in H226 cells. WDR1 knockdown significantly inhibited the proliferative rates, whereas WDR1 overexpression promoted the proliferative rates, compared to their corresponding controls. D: The cloning ability was determined by colony formation assay in A549 cells. Compared with shCTL cells, the colony formation was dramatically inhibited in shWDR1 cells. E-F: Western blotting analysis (E) and quantification (F) of protein levels of cell cycle regulators in A549 cells. Compared with shCTL, Cyclin A2, cyclin B1, cyclin D1, cyclin E, Cdk1 were decreased in shWDR1 cells, and p27 was significantly increased. Data are expressed as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001.
WDR1 positively regulates cell migration, invasion and induces the EMT process in vitro. A: Wound healing assay in A549, H1299 and H226 cell lines. Knockdown of WDR1 (shWDR1) significantly inhibited cell migration (48h) in A549, H1299 and H226 cells, compared with control cells (shCTL). B: Wound healing assay and quantification analysis showed that overexpression of WDR1 (WDR1) increased cell migration (36h) in A549, H1299 and H226 cell lines, relative to the control cells (GFP). C: Matrigel invasion assay and quantification analysis showed that WDR1 depletion greatly inhibited invasive abilities of H1299 and A549 cells. D-E: Western blotting analysis (D) and quantification (E) revealed decreased expression levels of N-cadherin, Vimentin and ZEB1 in shWDR1 cells, and increased level of E-cadherin. Snail and twist were reduced but not significantly in shWDR1 cells. The target protein band is indicated by an arrow. F: Immunofluorescence staining analysis of E-cadherin and N-cadherin in A549 cells. N-cadherin staining was dramatically reduced in shWDR1 cells, while that of E-cadherin was increased and sometimes punctate (arrow). Data are expressed as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001. Scale bars = 20 μm.
WDR1 promotes tumor growth and induces EMT process in vivo
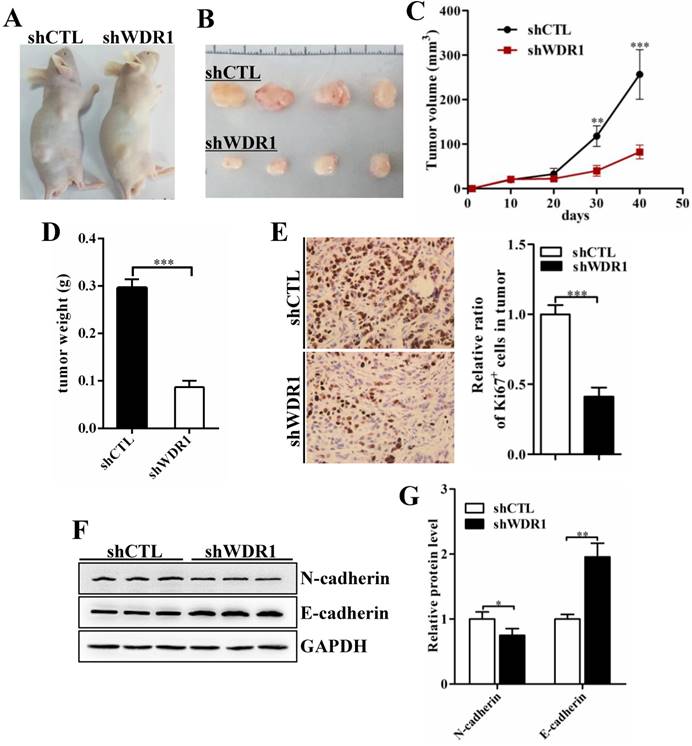
To assess the effects of WDR1 on tumor growth in vivo, we established xenograft tumor models using A549 cells transfected with scrambled shRNA (shCTL) or shRNA against WDR1 (shWDR1). All nude mice developed xenograft tumors at the injection site, and xenograft tumors were harvested 40 days after injection (Figure 4A and B). Consistent with in vitro results, in vivo experiments showed that WDR1 deficient A549 cells exhibited significantly reduced growth rate in mice, as the average tumor volume and tumor weight in the shWDR1 group were dramatically lower than those of shCTL group (Figure 4C and D). The immunohistochemical staining of Ki67 further revealed that knockdown of WDR1 inhibited NSCLC cell proliferation in vivo (Figure 4E). We also detected the EMT process in tumors derived from shWDR1 cells and shCTL cells, and found that N-cadherin was decreased but E-cadherin was increased in the shWDR1 group, relative to shCTL group (Figure 4F and G). Collectively, our data indicated that WDR1 promoted tumor growth and induced the EMT process in vitro and in vivo.
WDR1 promotes tumor growth and induces EMT process in vivo. A: Xenograft model in nude mice. A549 cells were transfected with scrambled shRNA (shCTL) or shRNA against WDR1 (shWDR1) and 2*106 cells were injected into male nude mice. B: Nodules harvested from shCTL group and shWDR1 group. Tumor nodules derived from shWDR1 cells were remarkably smaller than those in shCTL group. C: The tumor growth curve. The length and width of tumor were measured every 10 days. D: Tumor weight from shCTL and shWDR1 groups was shown. E: Immunohistochemical staining of Ki67 and quantification of the number of Ki67-positive cells in tumors from shCTL and shWDR1 groups. F-G: Detection of EMT-related proteins by western blotting analysis in protein extracted from tumors derived from shCTL and shWDR1 cells. Western blotting analysis and quantification revealed decreased expression level of N-cadherin and increased level of E-cadherin in tumors derived from shWDR1 cells, relative to those from shCTL cells. Data are expressed as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001.
WDR1 promotes tumor cell proliferation and migration might through actin cytoskeleton-regulated YAP activity
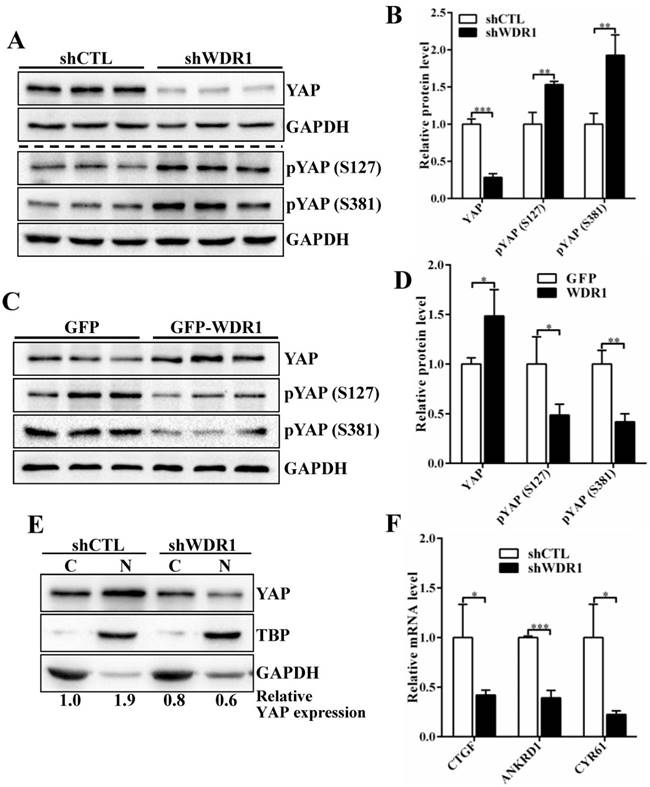
To investigate the possible mechanisms by which WDR1-mediated actin dynamics regulated tumor cell proliferation and migration, we analyze the signaling pathways that involved in the proliferation and migration. Yes-associated protein (YAP) is an oncogene capable of promoting cell growth and EMT in cultured cells, acting as a pivotal relay for the transmission of mechanical cues into gene expression program[27-29]. Actin cytoskeletal structure and tension have been shown to be dominant upstream regulators of YAP activity [29-31]. To test whether YAP provide a link between WDR1-mediated actin dynamics and tumor cell proliferation and migration, we first examined the expression levels of YAP, pYAP (S127) and pYAP (S381) in A549 cell line. The data showed that Wdr1 depletion resulted in a significantly decreased level of total YAP protein, increased levels of pYAP (S127) and pYAP (S381), whereas Wdr1 overexpression increased total YAP and decreased pYAP (S127), pYAP (S381) (Figure 5A-D). To clarify YAP localization, we prepared nuclear and cytoplasmic fractional lysates and found that, shWDR1 cells exhibited dramatically decreased nuclear YAP and comparable level of cytoplasmic YAP, relative to shCTL cells (Figure 5E). It has been reported that nuclear YAP associates with TEAD to accelerate the transcription of pro-proliferative and pro-migratory genes such as CTGF and Cyr61 [27]. We examined transcriptional activation of YAP target genes CTGF, ANKRD1, and CYR61 in shWDR1 and shCTL cells. As shown in Figure 5F, expression levels of the three target genes were significantly decreased in shWDR1 A549 cells. Our data suggested that WDR1 regulated tumor cells proliferation and migration might through actin cytoskeleton-mediated regulation of YAP.
WDR1 knockdown cells exhibit severe disrupted cellular morphology, F-actin accumulations, and increased levels of actin and cofilin
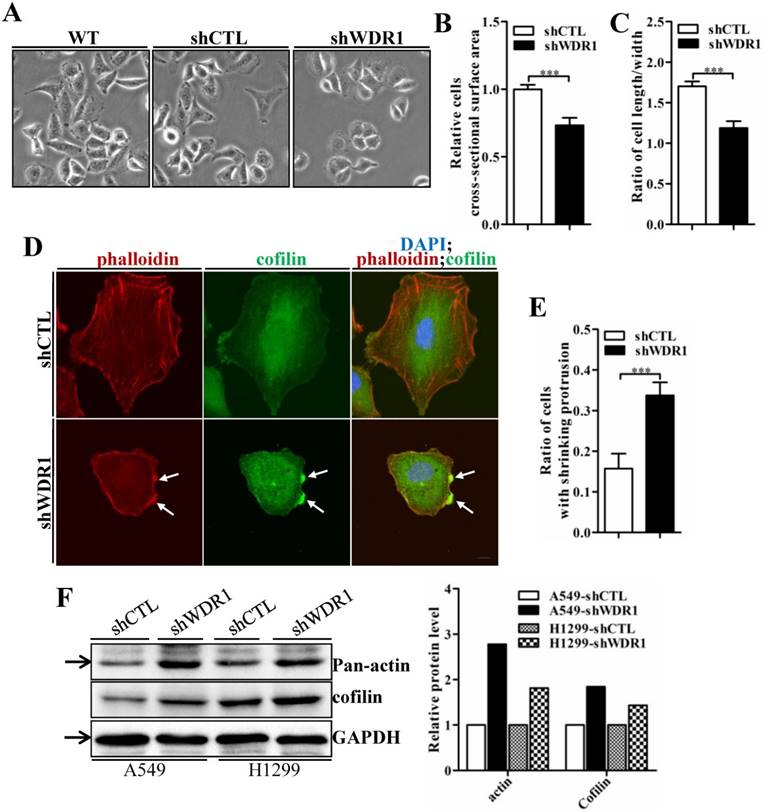
Actin cytoskeleton is required for cell motility, cell division, cytokinesis and cell shape regulation. Microscopic analysis showed that, in A549 cells, compared with shCTL cells, the morphology of most shWDR1 cells became smaller and rounder, as revealed by measuring cell cross-sectional surface area and the ratio of cell length/width (Figure 6A-C). To determine whether WDR1 is required for actin dynamics and turnover in NSCLC cells, we used phalloidin staining to specifically label F-actin. Compared with shCTL, shWDR1 cells exhibited shrinking cellular protrusions, and strong F-actin accumulations in the shortened protrusions; what's more, through an antibody detecting both isoforms of cofilin (ie, both cofilin-1 and cofilin-2), we found the localization of cofilin was also altered: in shCTL cells and in regions of shWDR1 cells without actin accumulations, cofilin displayed a diffuse and cellular center's stronger staining distribution pattern; by contrast, in shWDR1 cells, robust cofilin staining strongly colocalized with F-actin accumulations in the shortened protrusions (Figure 6D and E). Western blotting analysis also revealed increased levels of actin and total cofilin in shWDR1 cells (Figure 6F). These data revealed that WDR1 regulates cellular morphology, the formation of protrusions and actin dynamics in NSCLC cells.
Enforced overexpression of ADF/cofilin in WDR1 knockdown cells partly rescues F-actin accumulations, and the impaired migratory and invasive capacities
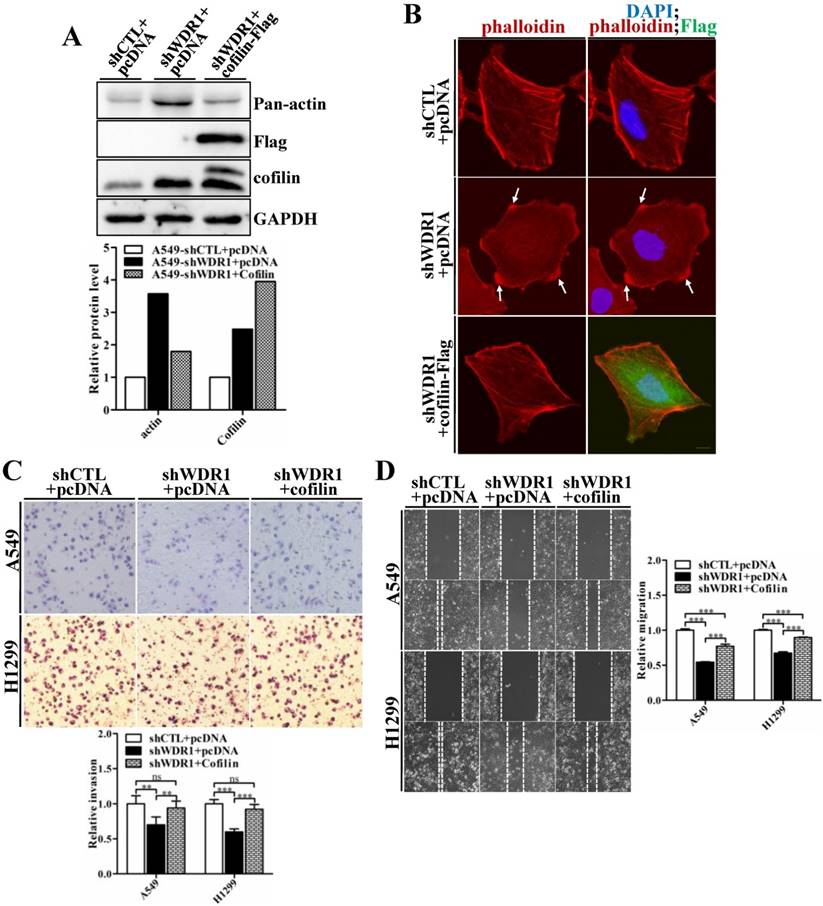
WDR1 acts as a major cofactor of ADF/cofilin, and WDR1 knockdown results in significantly increased protein level of cofilin. To examine whether WDR1 functions in NSCLC cells via ADF/cofilin mediated actin dynamics, shWDR1-treated A549 cells were transfected with pCDNA-Flag-cofilin plasmid to enforced overexpress cofilin. Flag-cofilin overexpression was confirmed by western blotting (Figure 7A). The increased pan-actin level was significantly downregulated after overexpression of cofilin in shWDR1 cells (Figure 7A). To examine F-actin and cellular morphology, we performed double immunostaining for phalloidin and Flag-cofilin, using the antibody of Flag. F-actin accumulations and the disrupted cellular morphology in shWDR1 cells were almost recovered after overexpression of cofilin (Figure 7B). As shown in Figure 7C and D, in A549 and H1299 cells, enforced overexpression of cofilin significantly rescued impaired cell migratory and invasive abilities in WDR1 knockdown cells. Taken together, the recoveries of disrupted cellular morphology, F-actin accumulations, and impaired migration and invasion indicate that WDR1 plays roles in NSCLC cells through ADF/cofilin-mediated actin dynamics.
Discussion
Our study is the first to investigate the role of WDR1 in NSCLC. The goal of this study was to comprehensively dissect the functions of WDR1 in NSCLC and the involved mechanisms. We found that the expression level of WDR1 was increased in NSCLC tissues, and high WDR1 expression levels correlated with reduced overall survival in NSCLC patients. With the present study, we have demonstrated that WDR1 silencing-induced disruptions of actin disassembly in NSCLC cells can directly affect cell shape, proliferation, migration and invasion. These findings are consistent with recent studies in breast cancer, in which WDR1 overexpression promoted cell migration and was associated with a shorter distant metastasis-free survival [24]. Another study in gallbladder carcinoma also showed WDR1 was significantly upregulated in highly metastatic cell lines [22]. In the light of pilot data indicating a positive correlation between WDR1 level and the progression of NSCLC, and showing an oncogenic role of WDR1 in the progression of NSCLC, further studies will be performed to evaluate the use of WDR1 as a biomarker to predict the progression and prognosis of NSCLC in the future.
WDR1 promotes tumor cell proliferation and migration might through actin cytoskeleton-regulated YAP activity. A-D: The protein levels of total YAP and p-YAP were examined by western blotting analysis. Western blotting and quantification data showed that, Wdr1 depletion resulted in a significantly decreased level of total YAP, increased levels of pYAP (S127) and pYAP (S381) (A,B), whereas Wdr1 overexpression increased total YAP and decreased pYAP (S127), pYAP (S381) (C,D). E: Immunoblotting for the indicated proteins. Cell lysates were fractionated into nuclear and cytoplasmic fractions. TBP and GAPDH were shown as loading controls for each fractionation. shWDR1 cells exhibited dramatically decreased nuclear YAP and comparable level of cytoplasmic YAP. Quantitation of the relative protein levels was also shown. F: qPCR analyses of relative normalized expression of YAP target genes CTGF, ANKRD1 and CYR61. The expression levels of the three target genes were significantly decreased in shWDR1 A549 cells. Data are expressed as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001.
The phenotype of inhibited migration and invasion in WDR1 knockdown cells was not surprising. Firstly, both cell motility and cell shape changes require the remodeling of actin cytoskeleton [6, 14]. Silencing of WDR1 blocks actin deploymerization and inhibits the repeated disassembly and assembly of the actin filament, results in F-actin accumulations, and then impairs actin dynamics mediated migration and invasion. Interestingly, in WDR1 knockdown cells, F-actin accumulations colocalized with enriched total-cofilin in the shortened protrusions, indicating the functional importance of the actin-based protrusions in tumor cell migration. Our study provided evidence that protrusions probably require strong actin turnover to drive tumor cells' migration. In addition, enforced overexpression of cofilin in WDR1 knockdown cells rescued the phenotype of F-actin accumulations, shortened protrusions, disrupted cellular morphology, and impaired cell migration and invasion further demonstrated that WDR1 functions in NSCLC through ADF/cofilin-mediated actin disassembly.
WDR1 depletion results in cell morphological disruptions, F-actin accumulations, increased levels of actin and total-cofilin. A: Microscopic analysis of A549 cells. Compared with shCTL cells, cells became smaller and the cellular shape turned rounder in most of shWDR1 cells. B: A549 cell cross-sectional area calculated by ImageJ, which was significantly smaller in shWDR1 cells, compared with CTL. C: The ratio of cell length/width measured by ImageJ was significantly reduced in shWDR1 cells. D: Immunofluorescence staining for phalloidin and total-cofilin in A549 cells. In shWDR1 cells, F-actin accumulations were observed in the shrinking protrusions, and total-cofilin was rich where F-actin accumulations were present (arrows). E: The number of A549 cells with shrinking protrusion(s) was quantified. Compared with shCTL, the ratio of cells with shrinking protrusion(s) was dramatically increased in shWDR1 cells. F: In A549 and H1299 cells, western blotting analysis revealed increased levels of pan-actin and total-cofilin in shWDR1 cells. The target protein band is indicated by an arrow. Data are expressed as means ± SEM. ***P < 0.001. Scale bar = 10 μm.
Overexpression of cofilin rescues phenotypes of F-actin accumulations, shrinking protrusions, and impaired cell migration and invasion in WDR1 knockdown cells. A: shWDR1-treated A549 cells were transfected with pCDNA-Flag-cofilin plasmid, and transfection efficiency of Flag-cofilin was determined by western blotting. Enforced overexpression of cofilin declined the increased level of actin induced by WDR1 knockdown. Quantitation of the relative proteins to the corresponding control was also shown. B: Double immunostaining for F-actin and Flag-cofilin, using the antibody of Flag. Overexpression of Flag-cofilin in shWDR1 cells rescued phenotypes of F-actin accumulations and cell morphological disruptions. C-D: Matrigel invasion assay (C) and wound healing assay (D) in A549 and H1299 cells. Cell migratory rate and invasive ability were partly recovered after enforced overexpression of cofilin. Data are expressed as means ± SEM. **P < 0.01, and ***P < 0.001. Scale bar = 10μm.
Secondly, EMT process plays important roles in tumor cells metastasis and chemoresistance [26, 32]. WDR1 promotes tumor cell migration and invasion might through the EMT, as knockdown of WDR1 resulted in decreased levels of N-cadherin, Vimentin and EMT-inducing transcription factor ZEB1, and an increase of E-cadherin level, indicating the EMT process was significantly inhibited. It should be noted that the distribution pattern of E-cadherin was also disrupted, and E-cadherin staining sometimes was punctate in shWDR1 cells. The impaired distribution pattern of E-cadherin was consistent with observations in mouse germ cells and sertoli cells and Drosophila eye epithelial cells, in which loss of WDR1 results in E-cadherin distribution defects [11, 12]. Our present results further support that WDR1 mediated actin dynamics is required for membrane-associated adherens junction remodeling in tumor cells. Based on the above results, disruptions of actin dynamics and cytoskeletal reorganization by silencing WDR1 might be an intervention target for tumor therapy.
Furthermore, knockdown of WDR1 inhibited cell growth in vitro and in vivo. Cell proliferation requires the precise regulation of cell cycle progression mediated by various regulators; mutation or deregulation of cell cycle regulators are frequently observed in most of the human malignancies, thus, regulation of cell cycle might be an effective strategy to inhibit tumor cell growth [33, 34]. Here, we showed that knockdown of WDR1 significantly reduced the expression levels of cell cycle positive regulators cyclin A2, cyclin B1, cyclin D1, cyclin E and Cdk1, while increased the expression of cell cycle inhibitor P27, suggesting that silence of WDR1 resulted in a uncontrolled cell cycle progression, and that may be a mechanism by which WDR1 promoted the proliferation or activated the cell viability. YAP, as a potent oncoprotein, associates with TEAD to promote the transcription of pro-proliferative and pro-migratory genes, acting as a pivotal relay for the transmission of mechanical cues into gene expression program [27-29, 35]. We found Wdr1 depletion significantly decreased the protein level of total YAP, increased the levels of pYAP (S127) and pYAP (S381), whereas WDR1 overexpression resulted in an increased level of total YAP and decreased levels of pYAP (S127), pYAP (S381). In addition, in shWDR1 A549 cells, the nucleus-localized YAP, the ratio of nucleus-localized YAP to cytoplasm-localized YAP, and the expression levels of YAP target genes CTGF, ANKRD1 and CYR61 were significantly decreased. Our data suggested that WDR1 regulates the cell cycle regulators and tumor cell migration might through YAP, the key relay for the transduction of actin cytoskeleton reorganization to gene transcriptional program.
It has been reported that the cytoskeleton tension and stress fibers, but not F-actin polymerization per se, are required for YAP regulation [29]. Actin cytoskeletal tension regulates YAP nuclear translocation and prevents degradation in both Hippo-dependent an independent ways [29-31, 36]. Interestingly, another study found that Wdr1 and cofilin/destrin-mediated actomyosin remodeling are essential for generating or maintaining epidermal cortical tension, and Wdr1-depleted epidermis exhibited reduced cortical tension [37]. Our results suggest that WDR1 might regulate YAP nuclear localization and/or activity through WDR1-mediated actin cytoskeleton tension in both Hippo-dependent and independent ways. However, the detailed molecular mechanism between WDR1, actin cytoskeleton and YAP localization/activity remains unclear, and needs to be further explored.
In conclusion, this study demonstrates the functional roles of WDR1 in NSCLC progression. The expression levels of WDR1 in human NSCLC specimens may be a valuable biomarker for aggressive behavior and poor prognosis, and disrupttions of actin dynamics might be a potential target for the therapeutic strategy in NSCLC.
Materials and Methods
Human NSCLC samples
39 cases of NSCLC samples were provided by Nanjing Chest Hospital of Southeast University. The study was approved by the ethical committee of Nanjing Chest Hospital of Southeast University. Informed consent from the patients was obtained in all cases. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee. NSCLC tissues and matched non-tumor tissue samples were collected and immediately frozen in liquid nitrogen until use. Quantitative real-time PCR (qPCR) was performed to determine the levels of WDR1 in tissue samples.
Cell lines, cell culture, protein overexpression and short hairpin-mediated knockdown
All cell lines were obtained from American Type Culture Collection (ATCC, USA). All cells were grown in RPMI1640 media (Hyclone, USA) supplemented with 10% fetal bovine serum (FBS) and penicillin/ streptomycin, and cultured at 37°C in a humidified incubator containing 5% CO2. Lentivirus production and transduction of cells was performed as described previously, lentiviral vector for shRNA expression was pLKO.1-TRC cloning vector [38]. The sequence of shRNA against WDR1 (shWDR1) was 5′-GCTGGGAAGATCAAAGACATT-3′. shRNA directed against GFP (Target sequence: 5'GCAAGCTGACCCTGAAGTTCAT3') was used as control scrambled shRNA (shCTL). To construct stable WDR1 overexpression cells, WDR1 cDNA was cloned into PCDH-Flag lentiviral vector, and PCDH-Flag-GFP lentiviral vector was used as a negative control. Transduced cells were selected for puromycin resistance. For overexpressing of cofilin, cofilin cDNA were cloned into pCDNA-Flag vector to construct overexpressed plasmid, and an empty vector was used as a negative control.
Quantitative real-time PCR (qPCR)
Total RNA was extracted from lung tissues and cells using TRIzol reagent (Life Technologies, Carlsbad, CA) according to the manufacturer's protocol. One microgram of total RNA from each specimen was reverse-transcribed to cDNA using a Promega (Madison, WI) Reverse Transcription System kit. qPCR was performed on an ABI StepOnePlus instrument (Life Technologies) using AceQ qPCR SYBR Green Master Mix (High ROX Premixed) (Vazyme, Nanjing, China). All of the primers were designed with Primer Premier 5.0 (PREMIER Biosoft, Palo Alto, CA). The primers were as follows: Wdr1 forward 5'-TTGTCAACTGTGTGCGATTCTC-3' and reverse 5'-GCTGTCGGGACTCCAACTAA-3'; E-cadherin forward 5'-CTTGGTCTACGCCTGGGACT-3' and reverse 5'-GAGGAGTTGGGAAATGTGAGC-3'; N-cadherin forward 5'-AGAATCGTGTCTCAGGCTCCA-3' and reverse 5'- GATTGCCTTCCATGTCTGTAGC-3'; Vimentin forward 5'-ACCAGCTAACCAACGACAAAG-3' and reverse 5'-CAAGGTCAAGACGTGCCAGA-3'; Cyclin A2 forward 5'-CATTGGTCCCTCTTGATT-3' and reverse 5'-TAACCTCCATTTCCCTAAG -3'; Cyclin B1 forward 5'-AAGATGGAGCTGATCCAAAC-3' and reverse 5'-TACATGGTCTCCTGCAACAA-3'; Cyclin D1 forward 5'-ATTTCCAATCCGCCCTCC-3' and reverse 5'-GGCTTCGATCTGCTCCTGG-3'; Cyclin E forward 5'-CAGGGAGACCTTTTACTTG-3' and reverse 5'-CCATCTGTCACATACGCA-3'; Cdk1 forward 5'-TGCTGGGGTCAGCTCGTTACTCA-3' and reverse 5'-TGGGATGCTAGGCTTCCTGGTT-3'; P21 forward 5'-CCTGGCACCTCACCTGCTCT-3' and reverse 5'-CGGCGTTTGGAGTGGTAGAA-3'; P27 forward 5'-CGGCTAACTCTGAGGACAC-3' and reverse 5'-CTGTTCTGTTGGCTCTTTTGT-3'; Twist forward 5'-AAGAAGTCTGCGGGCTGTGG-3' and reverse 5'-GAGGGCAGCGTGGGGATGA-3'; Snail forward 5'-CCCTCAAGATGCACATCCGAAGC-3' and reverse 5'-TGGAGCGGTCAGCGAAGGCAC-3'; ZEB1 forward 5'-AAGTGGCGGTAGATGGTA -3' and reverse 5'-TGTTGTATGGGTGAAGCA-3'; GAPDH forward 5'-GACCTGACCTGCCGTCTAG-3' and reverse 5'-AGGAGTGGGTGTCGCTGT-3'; ANKRD1 forward 5'- AAGAAGAATGGCAATGGG -3'and reverse 5'- AGGGTCACAGGGTGGGCTA -3'; CTGF forward 5'- ACGAGCCCAAGGACCAAACC -3'and reverse 5'- TGGAACAGGCGCTCCACTCT -3'; CYR61 forward 5'- TGCGAGGAGTGGGTCTGT -3'and reverse 5'- GCGAGGCTCCATTCCAAA -3'.
The relative mRNA levels were normalized to house-keeping gene actin in human NSCLC tissues, and house-keeping gene GAPDH in cells, respecttively, and determined by calculating the ΔΔCt value as per manufacturer's guidelines. The relative amount of target gene expression in shWDR1 cells was calculated as fold change relative to shCTL cells.
Western blot
Western blot was performed as described previously [13]. Nuclear/cytoplasmic fractional lysates were separated using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific) according to the manufacturer's instructions. The Proteins were probed with antibodies against WDR1 (13676-1-AP; Proteintech Group, Rosemont, IL), pan-actin (AAN01; Cytoskeleton, Denver, USA), β-actin (sc-47778; Santa Cruz Biotechnology, Dallas, Texas), Flag (20543-1-AP; Proteintech Group), total-cofilin (BS2183; Bioworld Technology), N-cadherin (13116; Cell Signaling technology, Danvers, MA), Vimentin (ab92547; Abcam, Cambridge, MA), E-cadherin (BS1098; Bioworld Technology), Cyclin A2 (4656; Cell Signaling technology), Cyclin B1 (BS1392; Bioworld Technology), Cyclin D1 (2922; Cell Signaling technology), Cyclin E (sc-481; Santa Cruz Biotechnology, Dallas, Texas), Cdk1 (BS1820; Bioworld Technology), P21 (AP0713; Bioworld Technology), P27 (554069; BD Biosciences, San Jose, CA), Snail (3879; Cell Signaling technology), Twist (ab50887; Abcam), ZEB1 (BS7670; Bioworld Technology), YAP (Ab52771; Abcam), pYAP (S127) (13008S; Cell Signaling technology), pYAP (S381) (13619; Cell Signaling technology), GAPDH (AP0063; Bioworld Technology), TBP (Ab818; Abcam), and then were visualized using enhanced chemiluminescence. Quantification of protein levels normalized to the internal control was performed using ImageJ software version 1.44 (NIH, Bethesda, MD). Experiments for quantitative data were performed in triplicate.
Immunohistochemistry
Cells were seeded on 1% gelatin coated coverslips in 6-well plates, and then fixed in 4% paraformaldehyde for 15 minutes at room temperature (RT), permeabilized in 0.2% Triton X-100 for 3 minutes at RT, blocked in blocking buffer (PBST with 5% BSA) for 1 hour at RT. Following removing blocking buffer, cells were incubated with anti-cofilin antibody (BS2183; Bioworld Technology), or anti-Flag antibody (20543-1-AP; Proteintech Group) or E-cadherin (562869, BD Biosciences, San Jose, CA), or N-cadherin (13116, Cell Signaling technology) in blocking buffer for over-night at 4℃. Secondary antibodies were Alexa Fluor 488-conjugated mouse IgG (115-545-166; Jackson ImmunoResearch Laboratories, West Grove, PA) and Alexa Fluor 488-conjugated anti-rabbit IgG (111-545-144; Jackson ImmunoResearch Laboratories). For phalloidin staining, cells were incubated with TRITC-conjugated phalloidin (p1951; Sigma-Aldrich, St. Louis, MO) and DAPI (1 mg/mL working concentration) (D6584; Sangon Biotech, Shanghai, China) for 2 hours at RT. For Ki67 immunohistochemical staining, tumor tissues were embedded in paraffin, sectioned in 6-μm thickness, and stained with ki67 antibody (9129; Cell Signaling Technology) using UltraSensitiveTM SP (Rabbit) IHC Kit (KIT-9707; Fuzhou Maixin Biotechnology, Fuzhou, China).
Cell proliferation assay
Cell Counting Kit-8 (Vazyme) was used to monitor cell proliferation. Each experiment was repeated independently in quadruplicate.
Clonal formation assay
For colony formation assay, cells were plated into 6-cm cell culture dishes (1000 cells), cultured for 2 weeks in RPMI1640 medium containing 10% FBS. Then plates were washed with PBS and stained with Giemsa. Cell colonies were photographed using a digital camera (Olympus, Tokyo, Japan).
Matrigel invasion assay
Matrigel invasion assay was performed using a 24-well Transwell chamber from Costar (8-μm pore size; Corning, USA) coated with 70μl diluted Matrigel (1:2 dilution with serum-free medium) (BD Bioscience) in the upper chamber. Twenty-thousand cells were resuspended in 100 μl serum-free medium and gently transferred to the upper chamber coated with matrigel. Lower chamber was filled with medium supplemented with 10% FBS. 24 hours after cell seeding, The Matrigel and the cells remaining in the upper chamber were removed by cotton swabs. Invaded cells in the lower surface of the membrane were stained with Crystal violet after fixed with 4% formaldehyde.
Wound healing assay
For wound healing assays, cells were seeded into 6-well plates. When a confluency of 90-100% was reached, a sterilized micro-pipette tip was used to generate a scratch across the cell layer. Cells at multiple points (36h, 48h) along the scratch were imaged using microscope (Olympus, Tokyo, Japan). Images were captured at 100× magnification. Each experiment was repeated independently in triplicate.
Xenograft experiments
All the animal protocols used in this study were approved by Institutional Animal Care and Use Committee (IACUC) of the Model Animal Research Institute of Nanjing University. The experiments and mouse maintenance were performed in the Model Animal Research Center of Nanjing University. Athymic male nude mice (nu/nu, 5 weeks old) were obtained from the Model Animal Research Center of Nanjing University. Cells stably expressing scrambled shRNA (shCTL) or shRNA against WDR1 (shWDR1) were generated as described above. 2*106 cells resuspended in PBS were mixed with matrigel (BD Bioscience, Bedford, MA) at a 1:1 volume ratio to 100 μl total volume and injected subcutaneously into the flanks of mice. After 40 days, mice were sacrificed and tumors were resected for further analysis.
Statistical analyses
Statistical calculations were performed using unpaired two tailed Student's t-tests. Data are expressed as means ± SEM. P < 0.05 was considered statistically significant, P < 0.01 (**) and P < 0.001 (***) statistically very significant.
Acknowledgements
This work was supported by National Natural Science Foundation of China [31701266] and the Natural Science Foundation of Hubei Province [2016CFB172], and Nanjing Medical Science and Technology Development Foundation [YKK16206]. This work was also partially supported by the Youth Science and Technology Backbone Training Program, Wuhan University of Science and Technology.
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA: a cancer journal for clinicians. 2017;67:7-30
2. Langer CJ, Besse B, Gualberto A, Brambilla E, Soria JC. The evolving role of histology in the management of advanced non-small-cell lung cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2010;28:5311-20
3. Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clinic proceedings. 2008;83:584-94
4. Marwitz S, Depner S, Dvornikov D, Merkle R, Szczygiel M, Muller-Decker K. et al. Downregulation of the TGFbeta Pseudoreceptor BAMBI in Non-Small Cell Lung Cancer Enhances TGFbeta Signaling and Invasion. Cancer research. 2016;76:3785-801
5. Lee SH, Dominguez R. Regulation of actin cytoskeleton dynamics in cells. Molecules and cells. 2010;29:311-25
6. Pollard TD, Cooper JA. Actin, a central player in cell shape and movement. Science. 2009;326:1208-12
7. Cooper JA, Schafer DA. Control of actin assembly and disassembly at filament ends. Current opinion in cell biology. 2000;12:97-103
8. Bamburg JR. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annual review of cell and developmental biology. 1999;15:185-230
9. Ono S. Regulation of actin filament dynamics by actin depolymerizing factor/cofilin and actin-interacting protein 1: new blades for twisted filaments. Biochemistry. 2003;42:13363-70
10. Okada K, Blanchoin L, Abe H, Chen H, Pollard TD, Bamburg JR. Xenopus actin-interacting protein 1 (XAip1) enhances cofilin fragmentation of filaments by capping filament ends. The Journal of biological chemistry. 2002;277:43011-6
11. Xu J, Wan P, Wang M, Zhang J, Gao X, Hu B. et al. AIP1-mediated actin disassembly is required for postnatal germ cell migration and spermatogonial stem cell niche establishment. Cell death & disease. 2015;6:e1818
12. Chu D, Pan H, Wan P, Wu J, Luo J, Zhu H. et al. AIP1 acts with cofilin to control actin dynamics during epithelial morphogenesis. Development. 2012;139:3561-71
13. Yuan B, Wan P, Chu D, Nie J, Cao Y, Luo W. et al. A cardiomyocyte-specific Wdr1 knockout demonstrates essential functional roles for actin disassembly during myocardial growth and maintenance in mice. The American journal of pathology. 2014;184:1967-80
14. Blanchoin L, Boujemaa-Paterski R, Sykes C, Plastino J. Actin dynamics, architecture, and mechanics in cell motility. Physiological reviews. 2014;94:235-63
15. Hall A. The cytoskeleton and cancer. Cancer metastasis reviews. 2009;28:5-14
16. Nurnberg A, Kitzing T, Grosse R. Nucleating actin for invasion. Nature reviews Cancer. 2011;11:177-87
17. Hemstreet GP 3rd, Bonner RB, Hurst RE, Bell D, Bane BL. Abnormal G-actin content in single prostate cells as a biomarker of prostate cancer. Cancer detection and prevention. 2000;24:464-72
18. Collazo J, Zhu B, Larkin S, Martin SK, Pu H, Horbinski C. et al. Cofilin drives cell-invasive and metastatic responses to TGF-beta in prostate cancer. Cancer research. 2014;74:2362-73
19. Madak-Erdogan Z, Ventrella R, Petry L, Katzenellenbogen BS. Novel roles for ERK5 and cofilin as critical mediators linking ERalpha-driven transcription, actin reorganization, and invasiveness in breast cancer. Molecular cancer research: MCR. 2014;12:714-27
20. Nagai S, Moreno O, Smith CA, Ivanchuk S, Romagnuolo R, Golbourn B. et al. Role of the cofilin activity cycle in astrocytoma migration and invasion. Genes & cancer. 2011;2:859-69
21. Wang H, Tao L, Jin F, Gu H, Dai X, Ni T. et al. Cofilin 1 induces the epithelial-mesenchymal transition of gastric cancer cells by promoting cytoskeletal rearrangement. Oncotarget. 2017;8:39131-42
22. Wang JW, Peng SY, Li JT, Wang Y, Zhang ZP, Cheng Y. et al. Identification of metastasis-associated proteins involved in gallbladder carcinoma metastasis by proteomic analysis and functional exploration of chloride intracellular channel 1. Cancer letters. 2009;281:71-81
23. Kang S, Kim MJ, An H, Kim BG, Choi YP, Kang KS. et al. Proteomic molecular portrait of interface zone in breast cancer. Journal of proteome research. 2010;9:5638-45
24. Lee JH, Kim JE, Kim BG, Han HH, Kang S, Cho NH. STAT3-induced WDR1 overexpression promotes breast cancer cell migration. Cellular signalling. 2016;28:1753-60
25. Gyorffy B, Surowiak P, Budczies J, Lanczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PloS one. 2013;8:e82241
26. Morris HT, Machesky LM. Actin cytoskeletal control during epithelial to mesenchymal transition: focus on the pancreas and intestinal tract. British journal of cancer. 2015;112:613-20
27. Zhao B, Ye X, Yu J, Li L, Li W, Li S. et al. TEAD mediates YAP-dependent gene induction and growth control. Genes & development. 2008;22:1962-71
28. Halder G, Dupont S, Piccolo S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nature reviews Molecular cell biology. 2012;13:591-600
29. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M. et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179-83
30. Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes & development. 2012;26:54-68
31. Kim MH, Kim J, Hong H, Lee SH, Lee JK, Jung E. et al. Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation. The EMBO journal. 2016;35:462-78
32. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST. et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472-6
33. Molinari M. Cell cycle checkpoints and their inactivation in human cancer. Cell proliferation. 2000;33:261-74
34. Singh T, Prasad R, Katiyar SK. Inhibition of class I histone deacetylases in non-small cell lung cancer by honokiol leads to suppression of cancer cell growth and induction of cell death in vitro and in vivo. Epigenetics. 2013;8:54-65
35. Qiao Y, Chen J, Lim YB, Finch-Edmondson ML, Seshachalam VP, Qin L. et al. YAP Regulates Actin Dynamics through ARHGAP29 and Promotes Metastasis. Cell reports. 2017;19:1495-502
36. Wada K, Itoga K, Okano T, Yonemura S, Sasaki H. Hippo pathway regulation by cell morphology and stress fibers. Development. 2011;138:3907-14
37. Luxenburg C, Heller E, Pasolli HA, Chai S, Nikolova M, Stokes N. et al. Wdr1-mediated cell shape dynamics and cortical tension are essential for epidermal planar cell polarity. Nature cell biology. 2015;17:592-604
38. Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G. et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283-98
Author contact
![]() Corresponding authors: Baiyin Yuan, Ph.D., Biomedical Research Institute, College of Life Science and Health, Wuhan University of Science and Technology, 947 Heping Avenue, Qingshan District, Wuhan 430081, P.R. China; Tel: +86 27 68893368; Email: baiyinyuannjcom. Chenxi Zhang, Ph.D., Central Laboratory, Nanjing Chest Hospital, Medical School of Southeast University, 215 Guangzhou Road, Gulou, Nanjing 210029, P.R. China; Tel: +86 25 58619883; Email: chenxi4262com. Tongcun Zhang, Ph.D., Biomedical Research Institute, College of Life Science and Health, Wuhan University of Science and Technology, 947 Heping Avenue, Qingshan District, Wuhan 430081, P.R. China; Tel: +86 27 68893368; Email: zhangtongcunedu.cn.
Corresponding authors: Baiyin Yuan, Ph.D., Biomedical Research Institute, College of Life Science and Health, Wuhan University of Science and Technology, 947 Heping Avenue, Qingshan District, Wuhan 430081, P.R. China; Tel: +86 27 68893368; Email: baiyinyuannjcom. Chenxi Zhang, Ph.D., Central Laboratory, Nanjing Chest Hospital, Medical School of Southeast University, 215 Guangzhou Road, Gulou, Nanjing 210029, P.R. China; Tel: +86 25 58619883; Email: chenxi4262com. Tongcun Zhang, Ph.D., Biomedical Research Institute, College of Life Science and Health, Wuhan University of Science and Technology, 947 Heping Avenue, Qingshan District, Wuhan 430081, P.R. China; Tel: +86 27 68893368; Email: zhangtongcunedu.cn.
Received 2017-11-14
Accepted 2018-5-5
Published 2018-6-8