ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2018; 14(14):2051-2064. doi:10.7150/ijbs.28576 This issue Cite
Research Paper
SRC-3 protects intestine from DSS-induced colitis by inhibiting inflammation and promoting goblet cell differentiation through enhancement of KLF4 expression
Wenbo Chen1,2*, Minghui Zhuo2*, Xuqiang Lu2*, Xiaochun Xia3, Yang Zhao2, Zhengrong Huang1, Jianming Xu4, Weihua Li1 ![]() , Chundong Yu2
, Chundong Yu2 ![]()
1. Department of Cardiology, The First Affiliated Hospital of Xiamen University, Xiamen, China;
2. State Key Laboratory of Cellular Stress Biology, Innovation Center for Cell Biology, School of Life Sciences, Xiamen University, Xiamen, China;
3. Xiamen Medical College, Xiamen Fujian 361023,China.
4. Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, Texas, USA.
* These authors contributed equally to this work
Abstract

Goblet cell loss, which leads to the reduction of mucin secretion, is a hallmark of ulcerative colitis (UC). We previously reported that steroid receptor coactivator 3 (SRC-3), a transcriptional coactivator, contributes to host defense against Citrobacter rodentium by recruiting neutrophils, suggesting a role of SRC-3 in intestine homeostasis. However, the biological role of SRC-3 in UC remains unclear. Here, we showed that SRC-3-/- mice were more susceptible to dextran sulfate sodium (DSS)-induced colitis compared with wild-type mice after oral administration of 2% DSS dissolved in drinking water. After oral administration of 2% DSS, SRC-3-/- mice displayed higher mortality rate, significant body weight loss, and higher clinical symptom scores compared to wild-type mice. SRC-3-/- mice suffered a severe loss of mature colonic goblet cells, leading to more severe histopathology and more proinflammatory cytokine production. Mechanistically, SRC-3-/- mice exhibited a decreased expression of transcription factor KLF4 in the colons, which is responsible for colonic goblet cell differentiation and maturation. At the molecular level, SRC-3 cooperated with c-Fos to promote KLF4 expression at the transcriptional level. These results demonstrate that SRC-3 can ameliorate DSS-induced colitis by inhibiting inflammation and promoting colonic goblet cell differentiation and maturation through enhancing the expression of transcriptional factor KLF4, which is responsible for colonic goblet cell differentiation and maturation.
Keywords: SRC-3, ulcerative colitis, goblet cell
Introduction
The inflammatory bowel diseases (IBD), comprising ulcerative colitis (UC) and Crohn's disease (CD), are inflammatory diseases of the intestine [1,2]. They mainly occur in developed countries, but their occurrence rate is increasing in developing countries [1,2]. Although UC and CD are characterized by injury of intestinal epithelial cells and chronic relapsing inflammation, they are different in clinical manifestations. CD patients exhibit discontinuous and transmural inflammation and CD etiology is associated with decreased antimicrobial peptides (e.g. defensin) [3,4,5,6], NOD2 expression in Paneth cells [7], autophagy and ER stress within Paneth cells [8]. Unlike CD, UC patients exhibit continuous and superficial inflammation, goblet cell depletion, and mucus layer attenuation, and UC etiology correlates with increased intestinal permeability [9,10] and decreased MUC2 production [11,12,13]. Although etiologies of IBD are not well known, the intestinal epithelial barrier dysfunction is crucial in the pathogenesis of IBD. Oral administering DSS can induce intestinal inflammation that is considered as an animal model of human UC [14]. It has been reported that the colonic mucus barrier is essential for protecting mice from DSS-induced colitis [15,16,17,18]. Goblet cell development regulated by the transcriptional program plays a critical role in protecting against DSS-induced colitis [19]. The intestinal epithelial cell proliferation is required for protecting against DSS-induced colitis [20,21]. Additionally, tight junctions are the major regulators of the epithelial barrier function and can protect against DSS-induced colitis through regulating intestinal epithelial homeostasis via Notch signaling [22]. Therefore, there is a great need to better understand intestinal epithelial barrier function, which can provide information for therapeutic restoration of barrier function.
Steroid receptor coactivator 3 (SRC-3), which belongs to a member of the p160 SRC family, can interact with multiple nuclear hormone receptors and transcription factors to regulate the expression of their target genes [23]. Our previous study has demonstrated that SRC-3-/- mice are more sensitive to LPS-induced endotoxic shock compared with wild-type mice, as SRC-3-/- macrophage can produce more proinflammatory cytokines (e.g. TNF-α, IL-6, and IL-1β) via regulating these cytokines mRNA translation [24]. Our recent study has shown that SRC-3-/- mice display delayed clearance of Citobacter rodentium and more severe tissue pathology after oral infection with C. rodentium, due to a decreased production of chemokines CXCL2 and CXCL5 and decreased recruitment of neutrophils [25], indicating that SRC-3 plays a critical protective role in bacteria-induced colitis. However, the biological role of SRC-3 in UC remains unclear.
In the current study, we accessed the biological role of SRC-3 in DSS-induced colitis by comparing the responses of SRC-3-/- mice and wild-type mice after oral administration of 2% DSS. We found that SRC-3-/- mice were dramatically sensitive to DSS-induced colitis compared with wild-type mice and SRC-3 could prevent DSS-induced colitis by promoting goblet cell differentiation and maturation via regulating the expression of transcriptional factors related to the goblet cell differentiation and maturation.
Materials and Methods
Mice
SRC-3-/- mice (on C57BL/6×129Sv background) were generated as described previously [26]. Wild-type mice serve as control mice. 6-8 weeks old female mice were used in all experiments. Animal experiments were approved by the Laboratory Animal Center of Xiamen University, Xiamen, Fujian, China.
DSS-induced colitis model
Colitis was induced by oral administration of 2% (w/v) of DSS (molecular mass 36,000-50,000; MP Biologicals) dissolved in autoclaved drinking water for 7 days, and then normal drinking water was replaced until the end of the experiment. The DSS solutions were changed every two days. For survival study, mice were monitored over the next 14 days. For other experiments, samples were collected at 0, 4, and 6 days after DSS administration.
Determination of clinical scores
Body weight, stool consistency and intestinal bleeding were determined up to 10 days after 2% DSS administration. Scoring for stool consistency and intestinal bleeding was accessed as described previously [27]. In a brief, stool consistency scores were determined as followed: well-formed stools were scored as 0, semi-formed stools which didn't adhere to the anus were scored as 1, semi-formed stools adhering to the anus were scored as 2, and diarrhea was scored as 3. Intestinal bleeding scores were determined as followed: no blood determined by using hemoccult kits (Nanjing Jiancheng bioengineering Institute, Nanjing, China) was scored as 0, positive hemoccult was scored as 1, visible blood traces in stool was scored as 2, and rectal bleeding was scored as 3. The averaged stool consistency and intestinal bleeding scores were calculated as the clinical score.
Histopathology
Colon tissues were fixed in 10 % neutral formalin and embedded in paraffin. Five μm sections were prepared and stained with hematoxylin and eosin. Scoring for inflammation and tissue damage was determined as described previously [28]. Briefly, inflammation scores were determined as followed: casual inflammatory cells in the colonic lamina propria were assigned score 0, increased inflammatory cells in the colonic lamina propria were assigned score 1, confluent inflammatory cells extending into the colonic submucosa was assigned score 2, and transmural extension of inflammatory cells was assigned score 3. Crypt damage scores were accessed as followed: no crypt damage was scored as 0, basal 1/3 crypt damaged was scored as 1, basal 2/3 crypt damaged was scored as 2, only surface epithelium intact was scored as 3, and entire crypt and epithelium loss was scored as 4. Five random fields per colon of at least four mice per group were chosen to analyze histological scoring.
Colon culture
The colon segment was removed, cut open longitudinally, weighed and washed in PBS containing penicillin and streptomycin. The colon segment was incubated at 37 ℃ in 24 flat-bottom well culture plates containing 1 ml of fresh RPMI 1640 medium for 24 h. Culture supernatants were collected at 13,000 rpm for 5 min at 4 ℃ and stored at -80 ℃ until analysis.
Cytokines measure
The concentrations of IL-6, IL-1β, TNF-α, IFN-γ and CCL2 in the colon culture supernatants were determined by ELISA kits (eBioscience) according to manufacturer's instructions.
Goblet cell and mucus layer preservation ex vivo
The medium colon after excision were immediately placed in Ethanol-Carnoy's fixative (ethanol: acetic acid = 3:1) at 4 ℃ for 2 hours and then put in 100% ethanol. Fixed colon tissues were embedded in paraffin and 5 μm sections were prepared. Sections were stained with Alclan blue (AB)/Periodic Acid-Schiff (PAS). Images were acquired with microscope.
Cell PAS staining
ShCtrl and shSRC-3 stable knockdown LS174T cells were grown on glass chamber slides. After overnight culture, cells were fixed with ethanol-acetic acid (3:1) for 15 min at room temperature, and then stained with 0.5% periodic acid solution for 5 min at room temperature. Remove excess stain of the slides with tap water and then cells were stained with Schiffs reagent in dark for 15 min at room temperature. After washing with water for 5 min, cells were counterstained with hematoxylin.
Immunohistochemistry
4 μm paraffin sections were prepared, deparaffinized and rehydrated. Antigens were retrieved by citrate buffer (pH 6.0) under microwave heating for 15 min. To block nonspecific binding sites, sections were incubated with 10% goat serum for 10 min at room temperature and then were incubated with anti-SRC-3 (cell signaling, 5E11, #2116) and anti-MUC2 (Proteintech Group, 27675-1-AP) for overnight at 4℃. Sections were incubated with 3% H2O2 for 10 min at room temperature to eliminate endogenous peroxidase activity. Then sections were incubated with EliVision kits (Maixin) at room temperature. And sections were incubated with DAB reagent for 2-10 min at room temperature in dark to visualize stained proteins.
Quantitative real-time PCR
Total RNA from tissues and cells were extracted using Trizol reagent (Invitrogen). Total RNA (1 μg) was reversely transcribed using ReverTra Ace Qpcr RT Master Mix kit (TOYOBO). Real-time PCR reactions were performed using FastStart Universal SYBR Green Master (Rox) (Roche). Relative mRNA level was calculated by normalization to GAPDH. The sequences of primers for quantitative real-time PCR will be provided upon request.
Co-immunoprecipitation assays and immunoblot analysis
For Co-immunoprecipitation (Co-IP) assays, 293T cells or LS174T cells were lysed with lysis buffer (150 mM NaCl, 200 mM Tris, 25 mM sodium pyrophosphate, 5 mM EDTA, 10 mM sodium orythovanadate, 10 mM glycerolphosphate, 50 mM NaF, 1mM PMSF and protein inhibitors cocktail). Total cell lysates were immunoprecipitated with anti-SRC-3 antibodies (Santa Cruz, C-20, sc-7216), anti-c-Fos antibodies (Santa Cruz, H-125, sc-7202) or control rabbit immunoglobulin G (IgG) (Sigma-Alrich, I5006). After washing five times with cell lysis buffer, samples were analyzed by immunoblot. For immunoblot analysis, LS174T cells were lysed in RAPA buffer (150 mM NaCl, 50 mM Tris, 0.1% SDS, 1 mM EDTA, 1% Triton X-100, 50 mM NaF, 1 mM PMSF and protease inhibitors cocktail). Proteins were quantified with BCA assay. Equal amount of proteins were loaded onto 8% sodium dodecyl sulfate-polyacrylamide gels and transferred onto polyvinylidene difluoride membranes (Millipore), followed by immunoblotting with anti-SRC-3 (cell signaling, 5E11, #2116), anti-KLF4 (Proteintech Group, 11880-1-AP), MUC2 (Santa Cruz, B306.1, sc-59859)or anti-β-actin (Sigma-Alrich, A5441). Western blots were analyzed by using a Tonen Image.
Construction of plasmids and luciferase reporter assay
Human KLF4 full-length cDNA was amplified by PCR from LS174T cells, and then was inserted into pCR3.1-HA vector. The KLF4 promoter fragment -908-+405 bp was inserted into pGL3-basic vector to produce the KLF4 promoter reporter plasmid. Mutated c-Fos recognition site at the KLF4 promoter was generated by a one-step site-directed and site-saturation mutagenesis method [29]. The primers of site-directed mutation of c-Fos site were yielded as followed: mut-Fos, forward, 5'- TGGATGgacCACGCGGATAATCGCGCTCTT-3', reverse, 5'- CGCGTGgtcCATCCAGCCCTCCATCTCC-3'. Lowercase letters indicate mutated site. DNA sequencing was used to verify the nucleotide sequences of these constructs. Luciferase activity was performed by a Dual Luciferase Reporter Assay System (Promega, Madison, WI). Renilla luciferase activity was used to normalize transfection efficiency.
Chromatin immunoprecipitation assay
LS174T cells or SRC-3-knockdown LS174T cells were used for chromatin immunoprecipitation (ChIP) assay and were performed according to the method described by Abcam (Cambrige, MA). The primers were used as followed: c-Fos binding site at KLF4 promoter, forward, 5'-AGCGGACTCCTGCGAGCG-3' and reverse, 5'- GCGTCCGCACCCCTGCTA-3'. Anti-SRC-3 (C-20, sc-7216) and anti-c-Fos (H-125, sc-7202) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Statistical analysis
The log-rank methods were used to analyze mortality rate. Data were collected from at least two independent experiments. All data were expressed as mean + SD or mean + SEM. Statistical significance was examined by two-tailed Student t test.
Results
SRC-3-/- mice are more susceptible to DSS-induced colitis compared with wild-type mice
To study the role of SRC-3 in DSS-induced colitis, we first accessed the mortality rate of SRC-3-/- mice and wild-type mice after oral administration of 2% of DSS dissolved in sterile distill water for 7 days. Only 9.1% of wild-type mice died during study period, while a mortality rate of 54.8% was observed in SRC-3-/- mice (Fig. 1A). More susceptibility of SRC-3-/- mice noted in the survival assay was reflected in more body weight loss and a higher combined score of stool consistency and occult bleeding. DSS administration induced more body weight loss in SRC-3-/- mice at day 7 post-DSS administration compared with wild-type mice (Fig. 1B). SRC-3-/- mice exhibited more severe diarrhea (Fig. 1C) and fecal bleeding (Fig. 1D) compared with wild-type mice. To further investigate the severity of colitis, we measured the colon length of SRC-3-/- mice and wild-type mice at days 0, 4, 6, and 14 post-DSS administration. The colon length of SRC-3-/- mice and wild-type mice was comparable at day 0, whereas the colon length of SRC-3-/- mice was shorter than that of wild-type mice at days 4, 6, and 14 post-DSS administration (Fig.1 E and F). These results demonstrate that SRC-3 plays a critical protective role in DSS-induced colitis.
SRC-3-/- mice display more severe intestinal histopathology and produce more proinflammatory cytokines than do wild-type mice after DSS administration
It is well known that DSS administration could trigger histopathological changes in the colons of DSS-administrated wild-type mice characterized by crypt loss and inflammation [30]. Therefore, colon sections were used for histological examination by hematoxylin and eosin (H&E) staining. There were no signs of tissue damage and inflammation in the colons of wild-type mice and SRC-3-/- mice without DSS treatment (Fig. 2A). Only minimal evidence of crypt loss and tissue damage was observed in the colons of wild-type mice at days 4 and 6 post-DSS administration (Fig. 2A). In contrast, colonic sections from SRC-3-/- mice at days 4 and 6 post-DSS administration exhibited severe crypt loss and transmural inflammation in the lamina propria and submucosa (Fig. 2A). There were intact crypt and minimal inflammation in the colons of wild-type mice at day 14 post-DSS administration, while SRC-3-/- mice still exhibited serious inflammation and crypt damage (Fig. 2A). Histopathological scoring for crypt damage and inflammation revealed the markedly higher degree of epithelial injury and inflammation in SRC-3-/- mice at days 4 and 6 post-DSS administration compared with wild-type mice (Fig. 2B). During recovery, SRC-3-/- mice continued to exhibit higher scores for epithelial injury and inflammation, whereas wild-type mice recovered fast (Fig. 2B).
It has been reported that proinflammatory cytokines can modulate the severity of DSS-induced colitis [30], so we analyzed the levels of proinflammatory cytokines in the colonic culture supernatant of SRC-3-/- mice and wild-type mice. The concentrations of IL-1β, IL-6, and CCL2 were comparable between SRC-3-/- mice and wild-type mice without DSS administration, but the concentrations of IL-1β, IL-6, and CCL2 in the colonic culture supernatant of SRC-3-/- mice were significantly elevated at days 4 and 6 post-DSS administration (Fig. 2C). The concentrations of TNF-α and IFN-γ in the colonic culture supernatant of SRC-3-/- mice were significantly higher than those in the colonic cultured supernatant of wild-type mice at days 0, 4, and 6 post-DSS administration (Fig. 2C). Collectively, these results suggest that the dramatic susceptibility of SRC-3-/- mice to DSS could be partially due to a significant increase in production of proinflammatory cytokines after DSS administration.
SRC-3-/- mice suffer a severe loss of colonic goblet cells
Colonic goblet cells can secret mucus such as MUC2 to protect colons from different type of injuries [31]. Goblet cell loss is a hallmark of ulcerative colitis, which can lead to a reduced mucus secretion. Colonic goblet cell dysfunction can exacerbate DSS-induced colitis [19]. To determine whether SRC-3 deficiency could affect colonic goblet cell differentiation, we examined mature goblet cells by AB/PAS staining on the colonic sections from wild-type mice and SRC-3-/- mice at days 0, 4, and 6 post-DSS administration. AB/PAS staining showed a decreased abundance of mature PAS+ cells in the colons of SRC-3-/- mice compared with wild-type mice at day 0 post-DSS administration (Fig. 3A). The mature goblet cells were depleted in the colons of both SRC-3-/- mice and wild-type mice at days 4 and 6 post-DSS administration, but the depletion of mature goblet cells in the colons of SRC-3-/- mice was more severe when compared to wild-type mice (Fig. 3A). Consistently, MUC2 immunostaining showed that SRC-3-/- mice had reduced MUC2-positive colonic goblet cells at days 0, 4, and 6 post-DSS administration (Fig. 3B). These results indicate that loss of SRC-3 severely impairs the differentiation of goblet cells.
SRC-3-/- mice are more susceptible to DSS-induced colitis compared with wild-type mice. (A) Survival of SRC-3-/- mice and wild-type mice after oral administration of 2% DSS dissolved in sterile distill water for 7 days. Survival curve was calculated by the log-rank methods. Results were calculated from three independent experiments. Body weight change (B), combined scores of stool consistency (C) and bleeding scores (D) of SRC-3-/- mice (n = 13) and wild-type mice (n = 15) after oral administration of 2% DSS dissolved in sterile distill water for 7 days. Macroscopic pictures (E) and colonic length (F) of SRC-3-/- mice (n = 8) and wild-type mice (n = 8) after oral administration of 2% DSS dissolved in sterile distill water for 7 days. Pictures are representative of three independent experiments. *p<0.05, **p<0.01.
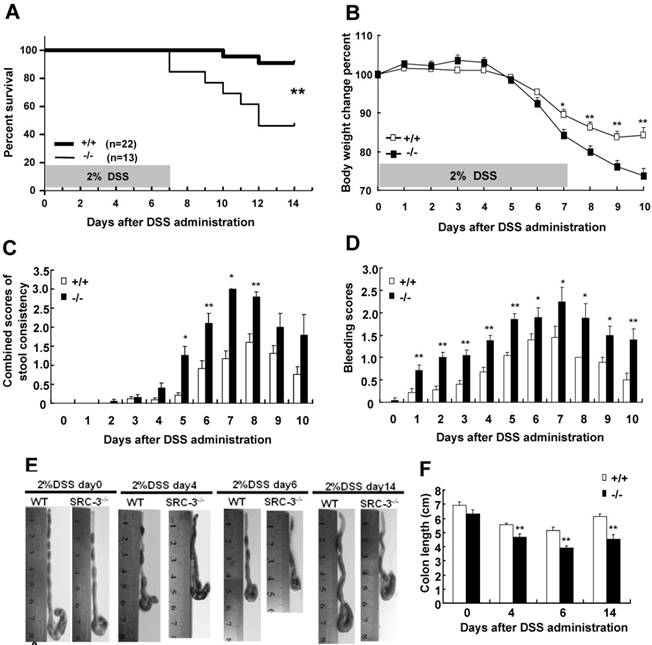
SRC-3-/- mice display more severe histopathology and produce more proinflammatory cytokines than do wild-type mice after DSS administration. (A) H&E staining of colon from SRC-3-/- mice and wild-type mice without or with DSS administration (n = 6-8). (B) Histopathological scoring of crypt damage and inflammation in the colon from SRC-3-/- mice and wild-type mice without or with DSS administration (n = 6-8). Arrow and arrowhead denote significant submucosal inflammatory cell infiltration and crypt damage. (C) The concentrations of IL-1β, IL-6, TNF-α, IFN-γ, and CCL2 in the colons of SRC-3-/- mice and wild-type mice without or with DSS administration (n = 6-8). Results are representative of three independent experiments. *p<0.05, **p<0.01.
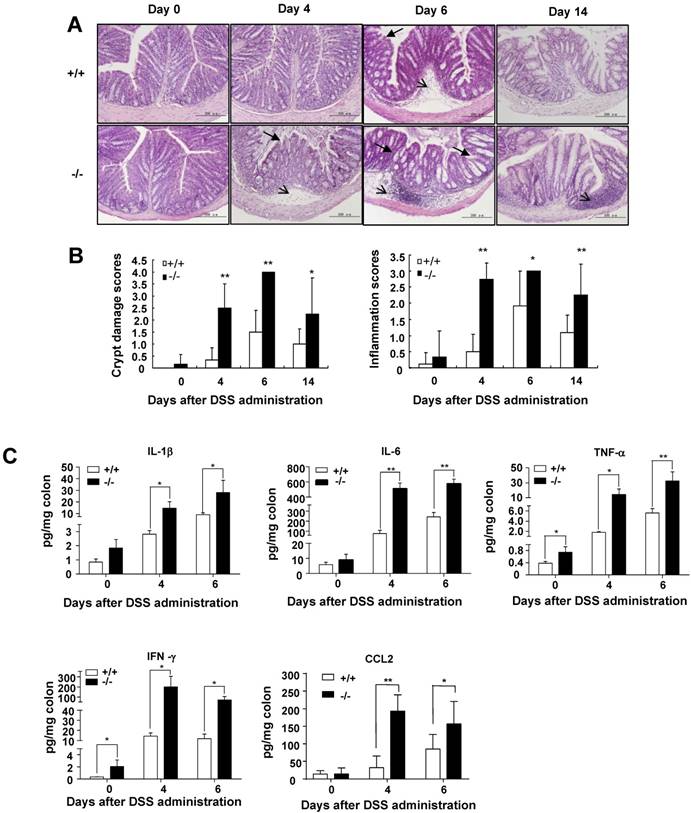
To determine whether reduced intestinal goblet cells in SRC-3-/- mice can influence the thickness of colonic inner mucus layer, which plays an essential role in protecting the intestine from diverse types of insults [31], we measured the thickness of inner mucus layer in the colons of SRC-3-/- mice and wild-type mice. We found that SRC-3-/- mice had reduced thickness of colonic inner mucus layer compared with wild-type mice (Fig. 3C). These results suggest that SRC-3 can protect against DSS-induced colitis by promoting the differentiation of mucus-secreting intestinal goblet cells.
Transcriptional factor KLF4 is decreased in the colons of SRC-3-/- mice compared to wild-type mice
To determine whether the defect in differentiation of intestinal goblet cells in SRC-3-/- mice is due to a transcriptional imbalance, we examined the expression of several transcription factors such as GFI1, SPDEF, HES5, and KLF4, which are essential for goblet cell differentiation and maturation [32]. The mRNA levels of GFI1, SPDEF, and HES5 in the colons of SRC-3-/- mice were similar to wild-type mice at day 0 post-DSS administration (Fig. 4A-C), but lower than wild-type mice at day 6 post-DSS administration (Fig. 4A-C). The mRNA levels of KLF4 in the colons of SRC-3-/- mice were significantly lower than wild-type mice at both days 0 and day 6 post-DSS administration (Fig. 4D). The expression pattern of colonic protein KLF4 was consistent with the expression pattern of its mRNA (Fig. 4E). Because the number of colonic goblet cells in SRC-3-/- mice was reduced at both days 0 and 6 post-DSS administration, and among four transcription factors examined, only KLF4 expression was reduced in the colons of SRC-3-/- mice at both days 0 and 6 post-DSS administration compared with wild-type mice, we speculated that reduced KLF4 most likely accounts for the reduced colonic goblet cell number.
SRC-3-/- mice suffer a severe loss of mature colonic goblet cells. (A) Representative AB/PAS staining of the colon sections from SRC-3-/- mice (n = 6) and wild-type mice (n = 10) at days 0, 4, and 6 post-DSS administration (left panels). Quantification of AB/PAS positive cells (right panels). Arrow and arrowhead denote AB-positive cells (blue) and PAS-positive cells (pink), respectively. (B) Representative MUC2 immunostaining of the colon sectins from SRC-3-/- mcie (n = 5) and wild-type mice (n = 5) at days 0, 4 and 6 post-DSS administration (left panels). Quantification of MUC2-positive cells (right panels). Arrow denotes MUC2-positive cells (brown). (C) Representative AB/PAS staining of the colon sections from SRC-3-/- mice and wild-type mice (left panels). Arrow represents inner mucus layer. Quantification of thickness of inner mucus layer (right panels). Data are mean + SEM. *p<0.05, **p<0.01.
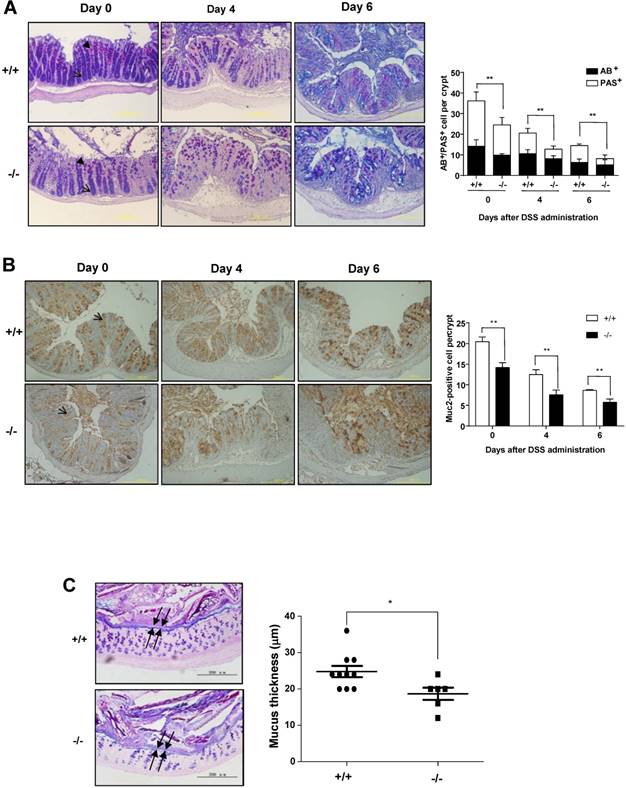
Transcription factor KLF4 is decreased in the colons of SRC-3-/- mice compared to wild-type mice. Quantitative RT-PCR of GFI1 (A), SPDEF (B), HES5(C), and KLF4 (D) in the colons of SRC-3-/- mice (n = 6) and wild-type mice (n = 10) at days 0, 4, and 6 post-DSS administration. (E) The protein levels of KLF4 were detected by western blot in the colonic epithelial cells of SRC-3-/- mice and wild-type mice at day 0 and 6 post-DSS administration. Data are mean + SEM. Results are representative of three independent experiments. *p<0.05, **p<0.01.
SRC-3 promotes KLF4 expression through cooperating with transcription factor c-Fos
Since KLF4 expression was reduced in the colons of SRC-3-/- mice and SRC-3 was mainly expressed in colonic enterocytes, including goblet cells, of wild-type mice without and with DSS administration (Supplementary Fig. 1), we hypothesized that KLF4 expression may be regulated by SRC-3 in colonic epithelial cells. To test this hypothesis, we determined whether SRC-3 could regulate the expression of KLF4 in colonic goblet cell-like LS174T cells. We established two SRC-3-knockdown stable LS174T cell lines using two different short hairpin RNA against SRC-3 (shSRC-3-1 and shSRC-3-2), and then examined the effects of SRC-3 knockdown on the expression of KLF4. The mRNA levels of KLF4 in two SRC-3-knockdown cell lines were significantly reduced compared with control LS174T cells (Fig. 5A). Consistently, the protein levels of KLF4 in SRC-3-knockdown LS174T cell lines were also markedly decreased (Fig. 5B). What's more, MUC2 protein levels in SRC-3-knockdown LS174T cells were significantly reduced (Fig. 5B). SRC-3 knockdown reduced the levels of PAS-staining (red color) of LS174T cells (Fig. 5C). We next determined whether KLF4 overexpression can rescue MUC2 expression and SRC-3-knockdown-induced losses of PAS staining. Western blot analysis showed that KLF4 overexpression could partly rescue MUC2 expression in SRC-3-knockdown LS174T cells (Fig. 5D). KLF4 overexpression could also rescue SRC-3-knockdown-induced losses of PAS staining (Fig. 5E). These results suggest that SRC-3 maintains goblet cell-like phenotype of LS174T cells at least in part via enhancing KLF4 expression.
Furthermore, we examined whether SRC-3 could promote KLF4 transcription by transfecting KLF4 promoter reporter into control and SRC-3-knockdown LS174T cells. KLF4 promoter activity was decreased in SRC-3-knockdown LS174T cells (Fig. 5F), suggesting that SRC-3 can promote KLF4 expression at the transcriptional levels.
To determine which transcription factor SRC-3 can cooperate to promote KLF4 expression, the sequence of human KLF4 promoter was screened in three databases (PROMO, http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3; Jaspar, http://jaspar.genereg.net/; signal scan, https://www-bimas.cit.nih.gov/molbio/ signal/ ) to look for the potential transcription factor binding sites. We found a transcription factor c-Fos binding site (-619TGAGTCA-613) at the KLF4 promoter, implicating that c-Fos may bind to this site to induce KLF4 expression. To assess whether SRC-3 could enhance c-Fos-induced KLF4 expression, LS174T cells were transfected with KLF4 promoter reporter and the expression vectors of SRC-3, c-Fos, and SRC-3 plus c-Fos, respectively. Although transfection of SRC-3 and c-Fos only induced KLF4 promoter activity 1.16-fold and 1.2-fold, respectively, cotransfection of SRC-3 and c-Fos markedly induced KLF4 promoter activity 2.58-fold (Fig.5G), indicating that SRC-3 can cooperate with c-Fos to induce KLF4 expression.
SRC-3 promotes KLF4 expression through cooperating with transcription factor c-Fos. (A) KLF4 mRNA expression was reduced in two SRC-3-knockdown stable LS174T cell lines (shSRC-3-1 and shSRC-3-2). Data are mean + SD (n = 3). (B) The expression of KLF4 and MUC2 proteins was reduced in two SRC-3-knockdown LS174T cell lines. (C) SRC-3 knockdown inhibited goblet cell differentiation of LS174T cells. Goblet cell differentiation was assessed by PAS staining. (D) KLF4 overexpression partly rescued MUC2 expression in SRC-3-knockdown LS174T cells. (E) KLF4 overexpression rescued SRC-3-knockdown-induced losses of PAS staining. (F) KLF4 promoter activity was decreased in shSRC-3-1 and shSRC-3-2 LS174T cells. Data are mean + SD (n = 3). (G) SRC-3 cooperated with c-Fos to enhance the activity of the KLF4 promoter. (H) Mutation analysis of the role of c-Fos binding site in c-Fos/SRC-3-mediated activation of the KLF4 promoter. LS174T cells were transfected with wild-type KLF4 promoter reporter (pKLF4-Fos-WT) and c-Fos binding site mutated KLF4 promoter reporter (pKLF4-Fos-Mut) together with SRC-3 expression plasmid and c-Fos expression plasmid, respectively. KLF4 promoter activity was assayed at 24 h post-transfection. Data are mean + SD (n = 3). (I) KLF4 protein was reduced in c-Fos-knockdown LS174T cells. (J) KLF4 mRNA expression was decreased in c-Fos-knockdown LS174T cells. Data are mean + SD (n = 3). (K) KLF4 promoter activity was decreased in c-Fos-knockdown LS174T cells. Data are mean + SD (n = 3). *p<0.05, **p<0.01.
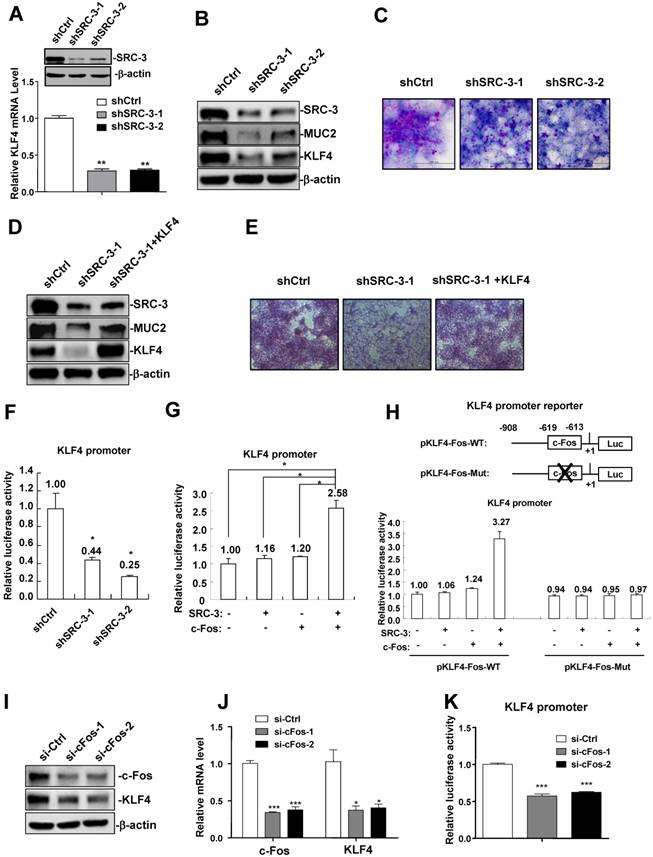
We next performed mutation analysis to determine the role of c-Fos binding site in c-Fos/SRC-3-meditated activation of KLF4 promoter. Cotransfection of SRC-3 and c-Fos expression plasmids could synergistically induce the activity of wild-type KLF4 promoter (pKLF4-Fos-WT) 3.27-fold (Fig. 5H), while it failed to induce the activity of KLF4 promoter carrying mutated c-Fos binding site (pKLF4-Fos-Mut) (Fig. 5H). What's more, si-c-Fos-mediated knockdown of c-Fos in LS174T cells reduced the expression of KLF4 protein and mRNA (Fig. 5I and J). KLF4 promoter activity was also decreased in c-Fos-knockdown LS174T cells (Fig. 5K). These results suggest that c-Fos binding site at the KLF4 promoter plays an important role in c-Fos/SRC-3-mediated activation of KLF4 promoter.
Both c-Fos and SRC-3 are recruited to c-Fos binding site at the KLF4 promoter
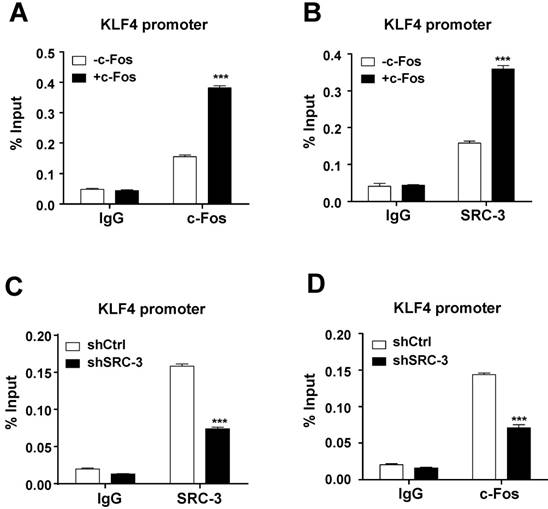
To determine whether c-Fos and SRC-3 can be recruited to c-Fos binding site at the KLF4 promoter in LS174T cells, ChIP assays were performed. Results showed that both c-Fos and SRC-3 could be recruited to KLF4 promoter, and the recruitment was increased after overexpressing c-Fos (Fig. 6A and B), indicating that SRC-3 is recruited to KLF4 promoter in a c-Fos-dependent manner. Interestingly, SRC-3 knockdown not only reduced SRC-3 recruitment to KLF4 promoter (Fig. 6C), but also reduced c-Fos recruitment to KLF4 promoter (Fig. 6D), suggesting that SRC-3 can also affect c-Fos recruitment to KLF4 promoter. These results implicate that both SRC-3 and c-Fos are recruited to KLF4 promoter to cooperatively enhance KLF4 promoter activity.
SRC-3 interacts with c-Fos through the S/T and HAT domains of SRC-3
To determine whether SRC-3 could interact with c-Fos to conduct its coactivating function, we transfected 293T cells with SRC-3 and c-Fos expression plasmids and then performed Co-IP assays. As shown in Fig. 7A, SRC-3 antibodies could immunoprecipitate c-Fos from cell lysates and c-Fos antibodies could immunoprecipitate SRC-3 from cell lysates. Additionally, the interaction of endogenous SRC-3 and c-Fos in LS174T cells could also be detected by Co-IP assays using SRC-3 antibodies and c-Fos antibodies, respectively (Fig. 7B).
Both c-Fos and SRC-3 are recruited to c-Fos binding site at the KLF4 promoter. (A) c-Fos could be recruited to KLF4 promoter, and the recruitment was increased after c-Fos overexpression. (B) SRC-3 could also be recruited to KLF4 promoter, and the recruitment was increased after c-Fos overexpression. (C) SRC-3 knockdown reduced SRC-3 recruitment to KLF4 promoter. (K) SRC-3 knockdown reduced c-Fos recruitment to KLF4 promoter. Data are mean +SD (n = 3). *p<0.05, ***p<0.001.
SRC-3 interacts with c-Fos through the S/T and HAT domains of SRC-3. (A) Co-IP analysis of the interaction of SRC-3 and c-Fos in 293T cells. (B) Co-IP analysis of the interaction of SRC-3 and c-Fos in LS174T cells. (C) SRC-3 interacted with c-Fos through its S/T and HAT domains.
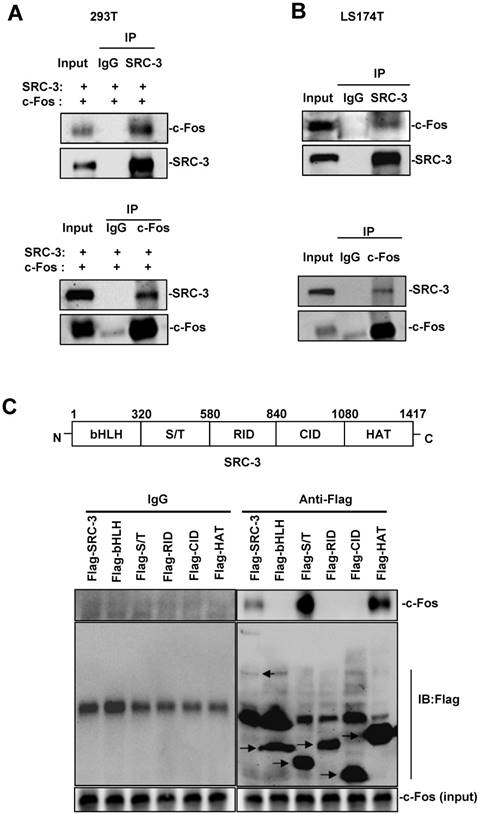
SRC-3 contains bHLH domain, serine/threonine-rich (S/T) domain, receptor interaction domain (RID), CBP interaction domain (CID), and histone acetyltransferas (HAT) domain (Fig. 7C, upper panel). To identify which domain within SRC-3 can interact with c-Fos, c-Fos were coexpressed with full-length SRC-3, bHLH domain, S/T domain, RID domain, CID domain, and HAT domain in 293T cells, and then Co-IP assays were performed. The results showed that SRC-3 interacted with c-Fos through its S/T and HAT domains (Fig. 7C, lower panel).
Discussion
The critical role of innate inflammatory cells in IBD is reflected by the underlying proinflammantory effects of their secreted cytokines, especially TNF-α and IL-6, which have implicated in the pathogenesis of IBD [31]. Anti-TNF-α antibody [33,34] or blockade of IL-6 signaling [35] can ameliorate colitis in mice and/or human IBD models, suggesting that these inflammatory cytokines play an important role in intestinal inflammation. In this study, we have showed that SRC-3-/- mice are more susceptible to the lethality caused by DSS-induced colitis and produce more proinflammatory cytokines compared with wild-type mice. Therefore, the increased sensitivity of SRC-3-/- mice to DSS-induced colitis is, at least in part, due to the elevated proinflammatory cytokines. Our previous study has shown that SRC-3 can regulate proinflammatory cytokines (e.g. TNF-α, IL-6, and IL-1β) translation via cooperating with translational repressors TIA-1 and TIAR [24].
UC is characterized by goblet cells loss, which leads to a reduced mucus secretion. As protective mucins, trefoil factors and other proteins secreted by goblet cells, are necessary for the integrity of barrier function and for inhibiting microflora-driven intestinal inflammation, therefore the mucus layer has a critical role in intestinal homeostasis [31]. The inner mucus with its barrier function is a crucial factor in limiting bacteria to get to the epithelium and its defect can initiate the inflammation [10]. Once barrier integrity is broken down, luminal bacteria and toxins can be recognized by innate immune cells residing in lamina propria, producing proinflammatory cytokines [36,37]. These proinflammantory cytokines trigger inflammatory cascades, leading to colonic tissue damage and chronic inflammation [38]. In this study, we found that SRC-3 was expressed in colonic goblet cell and SRC-3-/- mice displayed less mature goblet cells in the colons at days 0, 4, and 6 post-DSS administration compared with wild-type mice. Furthermore, SRC-3-/- mice has a thinner mucus layer than wild-type mice, lining with the results of lower number of goblet cells. These results suggest that colonic goblet cell dysfunction in SRC-3-/- mouse is one of the causes of severe DSS-induced colitis. Therefore, we focused on goblet cells but no other colonic cells for further study although we could not exclude the possible contributions of other colonic cells to the protection of intestine from DSS-induced colitis.
Several transcription factors essential for goblet cell differentiation and maturation have been identified, including HES5, GFI1, SPDEF, and KLF4 [32]. Notch signaling can drive progenitor cells to specifically differentiate into enterocytes [39], whereas inhibiting Notch signaling can induce progenitor cells to become secretory lineages in the intestine [40]. Notch mutant promotes HES5 expression in post-mitotic intestinal epithelium, resulting in an increase of differentiated goblet cells [41], suggesting HES5 can directly regulate goblet cell differentiation. GIF1 governs intestinal secretory cell differentiation and is necessary for the normal development of goblet cells [42,43]. SPDEF can promote goblet cell maturation [44,45]. The transcription factor KLF4 is required for goblet cell differentiation [46,47]. Moreover, deletion of GFI1, KLF4, or SPDEF, which are constitutively expressed in mature goblet cells, can lead to the loss of mature goblet cells [42,44,46]. In this study, our results showed that the expression of transcription factors SPDEF, HES5, GFI1, and KLF4 in the colons of SRC-3-/- mice at day 6 post-DSS administration was dramatically decreased compared with wild-type mice, implicating that SRC-3 may regulate goblet cell differentiation and maturation during intestinal inflammation by directly or indirectly regulating the expression of these transcription factors.
Before DSS administration, SRC-3-/- mice already exhibited colonic goblet cell dysfunction as demonstrated by a reduction of mature goblet cells and a thinner mucus layer as compared with wild-type mice, implying that goblet cell dysfunction is the cause of more severe colitis after DSS administration. Among four goblet cell differentiation-related transcription factors examined, only KLF4 expression in the colon of SRC-3-/- mice was markedly reduced at both days 0 and 6 post-DSS administration compared with wild-type mice, implicating that reduced KLF4 is the primary transcription factor responsible for goblet cell dysfunction in the colons of SRC-3-/- mice. In light of these results, we focused on studying how SRC-3 regulates KLF4 expression. Previous study has shown that SRC-3 can directly regulate KLF4 expression in embryonic stem cells [48], and SRC-3 can serve as a coactivator for AP-1 (including c-Fos and c-Jun) to enhance its transcriptional activity [49,50]. Consistently, our results showed that SRC-3 could interact with c-Fos, and both SRC-3 and c-Fos could be recruited to c-Fos binding site at the KLF4 promoter in a mutually dependent manner to enhance KLF4 transcription, suggesting that SRC-3 promotes goblet cell differentiation and maturation through cooperating with c-Fos to promote KLF4 expression.
It has been reported that IL-18 enhanced DSS-induced colitis and mucosal damage by downregulating the expression of transcriptional factors GFI1, SPDEF, and KLF4 to inhibit goblet cell maturation [19], demonstrating that goblet cell maturation plays a critical role in DSS-induced colitis. Our results are in agreement with this notion. Intriguingly, Ghaleb et al showed that deletion of KLF4 in mouse intestinal epithelium protected intestine from DSS-induced colitis by repressing NF-κB pathway inflammatory response [51]. We found that the expression of p-p65 and IκBα was comparable in the colonic epithelial cells of wild-type and SRC-3-/- mice without or with DSS administration (Supplementary Fig. 2), indicating that deletion of SRC-3 does not repress NF-κB pathway inflammatory response in mouse intestinal epithelium, although it reduces the expression of KLF4. This may explain the discrepancy between our study and Ghaleb's study.
We previously reported that SRC-3 protected against enteric bacteria-induced colitis by upregulaing CXCL2 expression to recruit neutrophils for enteric bacteria clearance [25]. In this study, we showed that SRC-3 protected against DSS-induce colitis by governing goblet cell differentiation via upregulating KLF4 expression. Therefore, our studies demonstrate that SRC-3 plays its protective role via different mechanisms in dealing with different types of colitis, highlighting the important protective function of SRC-3 against colitis.
Abbreviations
SRC-3: steroid receptor coactivator 3; UC: ulcerative colitis (UC); DSS: dextran sulfate sodium; KLF4: kruppel like factor 4; IBD: inflammatory bowel disease; CD: Crohn's disease; IP: immunoprecipitation; ChIP: chromatin immunoprecipitation.
Supplementary Material
Supplementary figures.
Acknowledgements
We thank Yuan Chen for breeding and genotyping the mice. This work was supported by: grants from National Basic Research Program of China (973 Program) [grant number 2015CB553800 to CY], the National Natural Science Foundation of China [grant number 81772942 to CY], the Natural Science Foundation of Fujian Province of China [grant number 2015J01563 to WL], the Science and Technology Benefit Project of Xiamen of China [grant number 3502Z20164008 to WL], the Doctoral Research Foundation of Xiamen Medical College [grant number K2016-12 to XX], and the National Institutes of Health [ grant number CA193455 to JX].
Competing Interests
The authors declare no financial conflict of interest and no conflict of interest with the contents of this article.
References
1. Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G. et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46-54
2. Bitton A, Vutcovici M, Sewitch M, Suissa S, Brassard P. Mortality trends in crohn's disease and ulcerative colitis: A population-based study in quebec, canada. Inflammatory Bowel Diseases. 2016;22:416-23
3. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014;14:329-42
4. Fellermann K, Wehkamp J, Herrlinger KR, Stange EF. Crohn's disease: A defensin deficiency syndrome?. Eur J Gastroenterol Hepatol. 2003;15:627-34
5. Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE. et al. Reduced paneth cell alpha-defensins in ileal crohn's disease. Proc Natl Acad Sci U S A. 2005;102:18129-34
6. Wehkamp J, Schmid M, Fellermann K, Stange EF. Defensin deficiency, intestinal microbes, and the clinical phenotypes of crohn's disease. J Leukoc Biol. 2005;77:460-5
7. Ogura Y, Lala S, Xin W, Smith E, Dowds TA, Chen FF. et al. Expression of nod2 in paneth cells: A possible link to crohn's ileitis. Gut. 2003;52:1591-7
8. Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Bock J, Martinez-Naves E. et al. Paneth cells as a site of origin for intestinal inflammation. Nature. 2013;503:272-6
9. Miki K, Moore DJ, Butler RN, Southcott E, Couper RT, Davidson GP. The sugar permeability test reflects disease activity in children and adolescents with inflammatory bowel disease. J Pediatr. 1998;133:750-4
10. Johansson ME, Gustafsson JK, Holmen-Larsson J, Jabbar KS, Xia L, Xu H. et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut. 2014;63:281-91
11. Tytgat KM, van der Wal JW, Einerhand AW, Buller HA, Dekker J. Quantitative analysis of muc2 synthesis in ulcerative colitis. Biochem Biophys Res Commun. 1996;224:397-405
12. Van Klinken BJ, Van der Wal JW, Einerhand AW, Buller HA, Dekker J. Sulphation and secretion of the predominant secretory human colonic mucin muc2 in ulcerative colitis. Gut. 1999;44:387-93
13. McGuckin MA, Eri R, Simms LA, Florin TH, Radford-Smith G. Intestinal barrier dysfunction in inflammatory bowel diseases. Inflamm Bowel Dis. 2009;15:100-13
14. Kitajima S, Takuma S, Morimoto M. Changes in colonic mucosal permeability in mouse colitis induced with dextran sulfate sodium. Exp Anim. 1999;48:137-43
15. Van der Sluis M, De Koning BAE, De Bruijn ACJM, Velcich A, Meijerink JPP, Van Goudoever JB. et al. Muc2-deficient mice spontaneously develop colitis, indicating that muc2 is critical for colonic protection. Gastroenterology. 2006;131:117-29
16. Cobo ER, Kissoon-Singh V, Moreau F, Holani R, Chadee K. Muc2 mucin and butyrate contribute to the synthesis of the antimicrobial peptide cathelicidin in response to entamoeba histolytica- and dextran sodium sulfate- induced colitis. Infect Immun. 2017;85:e00905-16
17. Klepsch V, Gerner RR, Klepsch S, Olson WJ, Tilg H, Moschen AR. et al. Nuclear orphan receptor nr2f6 as a safeguard against experimental murine colitis. Gut. 2017;67:1434-44
18. Podolsky DK, Gerken G, Eyking A, Cario E. Colitis-associated variant of tlr2 causes impaired mucosal repair because of tff3 deficiency. Gastroenterology. 2009;137:209-20
19. Nowarski R, Jackson R, Gagliani N, de Zoete MR, Palm NW, Bailis W. et al. Epithelial il-18 equilibrium controls barrier function in colitis. Cell. 2015;163:1444-56
20. Bussieres-Marmen S, Vinette V, Gungabeesoon J, Aubry I, Perez-Quintero LA, Tremblay ML. Loss of t-cell protein tyrosine phosphatase in the intestinal epithelium promotes local inflammation by increasing colonic stem cell proliferation. Cell Mol Immunol. 2017;15:367-76
21. Deng F, Peng L, Li Z, Tan G, Liang E, Chen S. et al. Yap triggers the wnt/beta-catenin signalling pathway and promotes enterocyte self-renewal, regeneration and tumorigenesis after dss-induced injury. Cell Death Dis. 2018;9:153
22. Pope JL, Bhat AA, Sharma A, Ahmad R, Krishnan M, Washington MK. et al. Claudin-1 regulates intestinal epithelial homeostasis through the modulation of notch-signalling. Gut. 2014;63:622-34
23. Xu J, Wu RC, O'Malley BW. Normal and cancer-related functions of the p160 steroid receptor co-activator (src) family. Nat Rev Cancer. 2009;9:615-30
24. Yu C, York B, Wang S, Feng Q, Xu J, O'Malley BW. An essential function of the src-3 coactivator in suppression of cytokine mrna translation and inflammatory response. Mol Cell. 2007;25:765-78
25. Chen W, Lu X, Chen Y, Li M, Mo P, Tong Z. et al. Steroid receptor coactivator 3 contributes to host defense against enteric bacteria by recruiting neutrophils via upregulation of cxcl2 expression. J Immunol. 2017;198:1606-15
26. Xu J, Liao L, Ning G, Yoshida-Komiya H, Deng C, O'Malley BW. The steroid receptor coactivator src-3 (p/cip/rac3/aib1/actr/tram-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc Natl Acad Sci U S A. 2000;97:6379-84
27. Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2:541-6
28. Ranganathan P, Jayakumar C, Manicassamy S, Ramesh G. Cxcr2 knockout mice are protected against dss-colitis-induced acute kidney injury and inflammation. Am J Physiol Renal Physiol. 2013;305:F1422-7
29. Zheng L, Baumann U, Reymond JL. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Research. 2004;32:e115
30. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The nlrp3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32:379-91
31. Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298-306
32. McCauley HA, Guasch G. Three cheers for the goblet cell: Maintaining homeostasis in mucosal epithelia. Trends Mol Med. 2015;21:492-503
33. Watkins PE, Warren BF, Stephens S, Ward P, Foulkes R. Treatment of ulcerative colitis in the cottontop tamarin using antibody to tumour necrosis factor alpha. Gut. 1997;40:628-33
34. Melmed GY, Targan SR. Future biologic targets for ibd: Potentials and pitfalls. Nat Rev Gastroenterol Hepatol. 2010;7:110-7
35. Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573-621
36. Ohtake K, Koga M, Uchida H, Sonoda K, Ito J, Uchida M. et al. Oral nitrite ameliorates dextran sulfate sodium-induced acute experimental colitis in mice. Nitric Oxide. 2010;23:65-73
37. Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A. 2008;105:15064-9
38. Kraus S, Arber N. Inflammation and colorectal cancer. Curr Opin Pharmacol. 2009;9:405-10
39. Fre S, Huyghe M, Mourikis P, Robine S, Louvard D, Artavanis-Tsakonas S. Notch signals control the fate of immature progenitor cells in the intestine. Nature. 2005;435:964-8
40. van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H. et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435:959-63
41. Zecchini V, Domaschenz R, Winton D, Jones P. Notch signaling regulates the differentiation of post-mitotic intestinal epithelial cells. Genes Dev. 2005;19:1686-91
42. Shroyer NF, Wallis D, Venken KJ, Bellen HJ, Zoghbi HY. Gfi1 functions downstream of math1 to control intestinal secretory cell subtype allocation and differentiation. Genes Dev. 2005;19:2412-7
43. Shroyer NF, Helmrath MA, Wang VY, Antalffy B, Henning SJ, Zoghbi HY. Intestine-specific ablation of mouse atonal homolog 1 (math1) reveals a role in cellular homeostasis. Gastroenterology. 2007;132:2478-88
44. Gregorieff A, Stange DE, Kujala P, Begthel H, van den Born M, Korving J. et al. The ets-domain transcription factor spdef promotes maturation of goblet and paneth cells in the intestinal epithelium. Gastroenterology. 2009;137(e1-3):1333-1345
45. Noah TK, Kazanjian A, Whitsett J, Shroyer NF. Sam pointed domain ets factor (spdef) regulates terminal differentiation and maturation of intestinal goblet cells. Exp Cell Res. 2010;316:452-65
46. Katz JP, Perreault N, Goldstein BG, Lee CS, Labosky PA, Yang VW. et al. The zinc-finger transcription factor klf4 is required for terminal differentiation of goblet cells in the colon. Development. 2002;129:2619-28
47. Zheng H, Pritchard DM, Yang X, Bennett E, Liu G, Liu C. et al. Klf4 gene expression is inhibited by the notch signaling pathway that controls goblet cell differentiation in mouse gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol. 2009;296:G490-8
48. Chitilian JM, Thillainadesan G, Manias JL, Chang WY, Walker E, Isovic M. et al. Critical components of the pluripotency network are targets for the p300/cbp interacting protein (p/cip) in embryonic stem cells. Stem Cells. 2014;32:204-15
49. Yan J, Yu CT, Ozen M, Ittmann M, Tsai SY, Tsai MJ. Steroid receptor coactivator-3 and activator protein-1 coordinately regulate the transcription of components of the insulin-like growth factor/akt signaling pathway. Cancer Res. 2006;66:11039-46
50. Yan J, Erdem H, Li R, Cai Y, Ayala G, Ittmann M. et al. Steroid receptor coactivator-3/aib1 promotes cell migration and invasiveness through focal adhesion turnover and matrix metalloproteinase expression. Cancer Res. 2008;68:5460-8
51. Ghaleb AM, Laroui H, Merlin D, Yang VW. Genetic deletion of klf4 in the mouse intestinal epithelium ameliorates dextran sodium sulfate-induced colitis by modulating the nf-kappab pathway inflammatory response. Inflamm Bowel Dis. 2014;20:811-20
Author contact
![]() Corresponding authors: Chundong Yu, State Key Laboratory of Cellular Stress Biology, Innovation Center for Cell Biology, School of Life Sciences, Xiamen University, Xiamen, Fujian, China. Tel: 86-592-2182013, E-mail: cdyuedu.cn; Weihua Li, Department of Cardiology, The First Affiliated Hospital of Xiamen University, Xiamen, China. Email: liweihuaxmcom.
Corresponding authors: Chundong Yu, State Key Laboratory of Cellular Stress Biology, Innovation Center for Cell Biology, School of Life Sciences, Xiamen University, Xiamen, Fujian, China. Tel: 86-592-2182013, E-mail: cdyuedu.cn; Weihua Li, Department of Cardiology, The First Affiliated Hospital of Xiamen University, Xiamen, China. Email: liweihuaxmcom.
Received 2018-7-17
Accepted 2018-10-3
Published 2018-11-3