ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2019; 15(2):253-264. doi:10.7150/ijbs.27420 This issue Cite
Research Paper
Adiponectin Suppresses Human Pancreatic Cancer Growth through Attenuating the β-Catenin Signaling Pathway
Jinghui Jiang1,*, Yingchao Fan1,*, Wei Zhang1, Yuling Shen1,2, Tingting Liu1, Ming Yao1, Jianren Gu1, Hong Tu1 ![]() , Yu Gan1
, Yu Gan1 ![]()
1. State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Renji Hospital, Shanghai Jiao Tong University School of Medicine, 25/Ln. 2200 Xietu Road, Shanghai 200032, China.
2. Department of Otorhinolaryngology-Head and Neck Surgery, Renji Hospital, Shanghai Jiao Tong University School of Medicine, 1630 Dongfang Road, Shanghai 200127, China.
* These authors contributed equally to this work.
Abstract

Adipokines are emerging as a link between obesity and obesity-related cancers, including pancreatic cancer. Adiponectin is an abundant adipokine with pleiotropic beneficial roles in metabolic disorders. Low adiponectin levels are commonly observed in human obesity and have been associated with increased pancreatic cancer risk in prospective epidemiologic studies. Here, we investigated the direct effect of adiponectin on human pancreatic cancer in vitro and in vivo. Our results showed that adiponectin treatment significantly inhibited the proliferation of human pancreatic cancer cells. Knockdown of adiponectin receptors completely eliminated the antiproliferation effect of adiponectin and markedly promoted the growth of human pancreatic cancer xenografts in nude mice. Further analysis revealed that adiponectin blocked the phosphorylation/inactivation of GSK-3β, suppressed the intracellular accumulation of β-catenin, reduced the expression of cyclin D1, and consequently caused cell cycle accumulation at the G0-G1 phase in pancreatic cancer cells. Adiponectin-mediated attenuation of cell proliferation was abrogated by the GSK-3β inhibitor. In addition, a microarray analysis revealed that adiponectin also downregulated the expression of TCF7L2, a coactivator of β-catenin, at the transcriptional level in pancreatic cancer cells. These results indicated that the protective role of adiponectin against human pancreatic cancer might be attributed to its attenuating effect on the β-catenin signaling pathway. Taken together, our findings support a causal link between hypoadiponectinemia and increased pancreatic cancer risk, and suggest that activating adiponectin signaling could be a novel therapeutic strategy for obesity-related pancreatic cancer.
Keywords: adiponectin, pancreatic cancer, cell proliferation, GSK-3β, β-catenin
Introduction
Pancreatic cancer is one of the most fatal cancers with 5-year relative survival rate of less than 8% [1]. The estimated deaths from pancreatic cancer reached 330,000 around the world [2]. Pancreatic cancer already ranks as the sixth most common cause of death from cancer in China [3], and the third most common cause in the United States [1]. Unfortunately, the pathogenesis of pancreatic cancers is still insufficiently known, causing a lack of effective treatment for this deadly disease. Therefore, it is urgent to accelerate the mechanistic research underlying the development of pancreatic cancer.
There is sufficient evidence to support that obesity is associated with an increased risk of various cancers, including pancreatic cancer [4, 5]. Adverse associations of obesity with survival among pancreatic cancer patients have also been demonstrated [6]. Emerging evidence shows that adipokines secreted by adipose tissue are closely involved in obesity-related cancers [7]. Adiponectin is one of the major adipokines secreted by mature adipocytes and plays a vital role in modulating a number of metabolic and inflammatory processes [8-10]. Notably, the serum level of adiponec tin is significantly reduced in obese individuals [11]. A low serum level of adiponectin has been closely connected with a high risk of various obesity-related cancers [12-15]. In particular, several prospective studies have demonstrated a significant association of low circulating adiponectin levels with an increased risk of pancreatic cancer, suggesting that abnormal adiponectin signaling may be an important link between obesity and pancreatic cancer [16, 17].
Adiponectin activates downstream signaling pathways through binding to its specific receptors (AdipoRs) including two main isoforms AdipoR1 and AdipoR2 [18-20]. Several tumor tissues have been shown to express AdipoRs, indicating that adiponectin signaling may exert vital effects on tumors. It was reported that adiponectin suppressed colorectal cancer cell proliferation via AdipoR1 and AdipoR2 [21]. Similar results have also been reported in other cancers, such as gastric cancer and prostate cancer [20]. However, the direct impact of adiponectin on human pancreatic cancer remains unelucidated. The observation that pancreatic cancer tissue exhibits positive expression of AdipoR1 and AdipoR2 [22] greatly encouraged us to investigate the functional role of adiponectin in pancreatic cancer as well as its underlying molecular mechanisms.
In this study, we investigated the influence of adiponectin on human pancreatic cancer via a series of in vitro and in vivo experiments. Our results revealed that the growth of pancreatic cancer was significantly suppressed by adiponectin signaling, suggesting a protective role of adiponectin against pancreatic cancer. Furthermore, we also explored the potential downstream pathways upon the activation of adiponectin signaling in pancreatic cancer cells to uncover the molecular mechanisms underlying this tumor-inhibitory effect.
Materials and Methods
Cell culture and treatment
Human pancreatic cancer BxPC-3 cells and CFPAC-1 cells were obtained from the American Type Culture Collection (Manassas, VA) and maintained in RPMI 1640 and IMDM containing 10% FBS (Life Technologies, Gaithersburg, MD), respectively. Cells were incubated in equipment at a constant temperature and humidity with 5% carbon dioxide in air, and were passaged on reaching 80% confluence.
Human pancreatic cancer cells were treated with recombinant human adiponectin (BioVendor, Brno, Southern Moravia, Czech Republic) at a concentration of 40 μg/ml or otherwise noted for indicated durations, and then were subjected to further analysis. To detect the protein phosphorylation level of Akt and GSK-3β, BxPC-3 cells were treated with or without adiponectin (40 μg/ml) in the absence of serum for 12h followed by 1h serum stimulation. Next, the cells were lysed in RIPA buffer (Cell Signaling Technology, Boston, MA) followed by western blot analyses. To identify the role of GSK-3β, cells were treated with TWS119, a specific GSK-3βinhibitor (Selleck Chemicals, Houston, TX) at 10μM/Lin the absence or presence of adiponectin. To determine whether proteasome mediated the effect of adiponectin on β-catenin, cells were pre-treated with proteasome inhibitor MG-132 (Selleck Chemicals, Houston, TX) at 10 μM for 1 h, followed by adiponectin treatment.
Generation of stable adiponectin- overexpressing cells and AdipoR1/AdipoR2- knockdown cells
The full-length human ADIPONECTIN gene (GeneChem, Shanghai, China) was inserted into the expression lentivector pCDH-CMV-MCS (System Biosciences, Mountain View, CA) between the NheI and BamHI sites. The primers for cloning are listed in Table S1. There combinant lentivirus carrying the human ADIPONECTIN gene were generated as described before [10], and subsequently used to infect BxPC-3 cells or CFPAC-1 cells to generate stable adiponectin-overexpressing pancreatic cancer cells (BxPC-3-adiponectin or CFPAC-1-adiponectin). Moreover, the control cells (BxPC-3-VC cells or CFPAC-1-VC cells) were generated by transduction with an empty virus.
The gene specific targeting sequence for RNAi-mediated knockdown ofAdipoR1 (5'-GCAAAGACTATGATGTTAA) or AdipoR2 (5'-GTGTAGAAGTTGAGAAGAA) was inserted into the shRNA-expressing lentivector pGreenPuro (System Biosciences). The recombinant lentiviruses carrying siAdipoR1 (Lv-siAdipoR1) and siAdipoR2 (Lv-siAdipoR2) were packaged, respectively. Next, BxPC-3 cells or CFPAC-1 cells were infected with Lv-siAdipoR1 and Lv-siAdipoR2 simultaneously to generate stable AdipoRs-knockdown cells (BxPC-3-siAdipoR1/2 cells or CFPAC-1- siAdipoR1/2 cells). The corresponding pancreatic cancer cells were transduced with a recombinant lentivirus carrying the counterpart scramble sequence (scRNA) to generate the control cells (BxPC-3-scRNA cells or CFPAC-1-scRNA cells).
Cell proliferation assay
Cells were plated in a 96-well plate at a concentration of 4000 cells per well. After attachment to the wall, the cells were cultured in FBS-free medium (RPMI 1640 or IMDM) for 12h and then were treated with or without adiponectin in the corresponding culture medium containing 10% or 2% FBS. At 0, 24, 48 and 72h after treatment, the culture medium was replaced by 100 µl of fresh medium containing CCK-8 reagent (Dojindo, Tokyo, Japan), followed by incubation at 37°C for 2 h. Absorbance was measured at a wavelength of 450 nm using a microplate reader.
Western blot
Cells were scraped into lysis buffer, and lysates were then quantitated using a BCA Protein Assay kit (Thermo Scientific, Rockford, IL). Equal quantities of proteins were added to the gel for electrophoresis and transferred to polyvinylidenedifluoride membranes. Various primary antibodies were used to determine the expressions of target proteins, including adiponectin, AdipoR2, TCF7L2, cyclinD1 (Proteintech, Chicago, IL), GSK-3β, pGSK-3β (Ser9), Akt, p-Akt (Ser473) (Cell Signaling Technology), AdipoR1, β-catenin (SAB), cyclinA2 (Cell Signaling Technology), cyclinE1 (Cell Signaling Technology), p21 (Abcam) and β-actin (Sigma-Aldrich, St. Louis, MO). The protein-antibody complexes were detected using a Pierce SuperSignal West Pico chemiluminescent substrate (Thermo Scientific). Bands of interest were quantified by densitometry using Image J software.
Xenograft model
All animal experiments were performed according to the protocols approved by the Medical Experimental Animal Care Commission at the Shanghai Cancer Institute. The male athymic BALB/c nude mice (5-6 weeks old) used in this study were maintained under specific pathogen-free conditions and were provided free access to food and water. Mice were randomly assigned to two groups, and subcutaneously inoculated with BxPC-3-scRNAcells or BxPC-3-siAdipoR1/2cells (1×107 cells/mouse) in the flank region. The tumor size was measured weekly starting 2 weeks after inoculation, and the tumor volumes were calculated as follows: ½×length×width2. All mice were euthanized on day 42 after inoculation, and the tumors were excised and weighed.
Immunohistochemical analysis
Tumor tissues were fixed in 10% buffered formalin, embedded in paraffin, and sectioned at 5 µm using routine methods. For immunohistochemical staining, sections were incubated with the anti-PCNA antibody (Proteintech) overnight at 4°C. PCNA staining was visualized using the ImmPress HRP Reagent kit (Vector Laboratories, Burlingame, CA, USA) with NovaRed as a substrate (Vector Laboratories). Hematoxylin was used as a counterstain for the cell nucleus. For the quantification analysis, we counted the number of total cells and PCNA-positive cells per 400× microscope field with 3random fields per mouse. The data are expressed as the percentage of PCNA positive cells, which was calculated as [(positive cell number) / (total cell number)] ×100.
Cell cycle analysis
Forty-eight or seventy-two hours after being treated with adiponectin, BxPC-3 or CFPAC-1 cells were trypsinized and then washed with PBS. Next, the cells were transferred into ice-cold ethanol (70%) at 4°C for 24h, followed by staining with propidium iodide containing RNase A for 30 min at37°C. The cell cycle was analyzed using FACSCelesta flow cytometer (BD Biosciences, Franklin Lakes, NJ), and the percentages of cells in G0-G1, S, and G2-Mphasewere evaluated using the software FCS Express 4.0(De Novo Software, glendale, CA).
Quantitative real-time PCR
Total RNA was extracted with TRIzol (Invitrogen, Carlsbad), and cDNA was prepared by using a FastQuant RT kit (with gDNase) (Tiangen Biotech, Beijing,China). Quantitative real-time PCR analysis was performed using FastStart Universal SYBR Green Master (Roche Diagnostics, Mannheim, Germany) on a StepOne Plus real-time PCR system (Applied Biosystems, Foster City). The relative expression levels of target genes were normalized using Gapdh as an internal control. The primers used in this experiment are listed in Table S1. The data are presented as the mean ± SEM of three independent experiments.
Microarray profiling and bioinformatics analysis
BxPC-3 cells were treated with adiponectin as described above. Total RNAs from adiponectin-treated cells or control cells from three independent experiments were pooled and sent to Shanghai OE Biotech Co, Ltd. (Shanghai, China), for microarray analysis using an Agilent Whole Human Genome Gene Expression 4×44K Microarray (Agilent Technologies). The slide was scanned using an Agilent scanner, and the output images were digitalized by the Feature Extraction software (Agilent Technologies, Santa Clara, CA). The normalization of raw data was then performed using GeneSpring software (version 12.0, Agilent Technologies). The genes were considered to be differentially expressed if their fold changes were more than 2 or less than 0.5 in the comparison of adiponectin-treated cells versus control cells.
The normalized expression data of the differentially expressed genes were imported into the TIGR MeV software (available at http://www.tm4.org/ mev.html) for hierarchical clustering analysis. Simultaneously, the differently expressed genes were uploaded to the web-accessible functional annotation tool from the Database for Annotation, Visualization, and Integrated Discovery (DAVID) version 6.7 (http://david.abcc.ncifcrf.gov) for Gene Ontology (GO) enrichment analysis. The GO terms with P <0.05 was considered to be enriched.
Statistical analysis
The results are presented as the mean±SEM. Statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software, Inc., San Diego, CA). The significance of differences between groups was assessed by Student's t test or by analysis of variance (ANOVA) with Student-Newman-Keuls tests for multiple comparisons. A value of P<0.05 (two-tailed) was considered statistically significant.
Results
Adiponectin inhibited the proliferation of human pancreatic cancer cells through its receptors in vitro
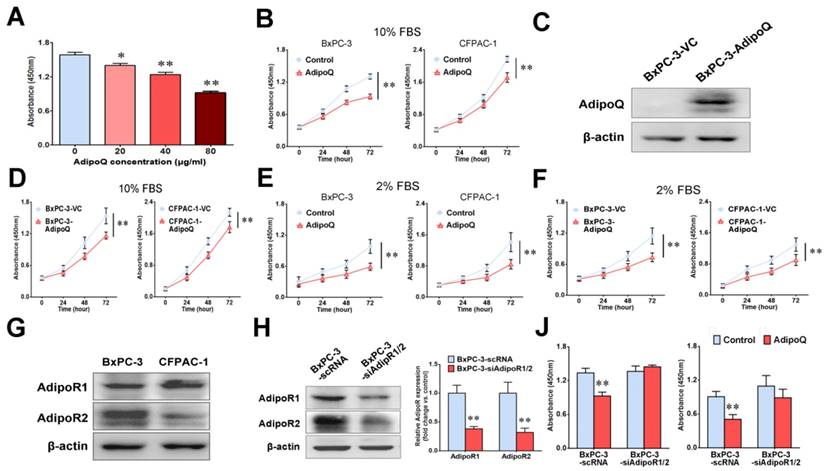
We first investigated the in vitro effects of adiponectin on human pancreatic cancer cells.BxPC-3 or CFPAC-1 cells were treated with recombinant human adiponectin followed by the detection of cell proliferation, migration and invasion. Our data showed that adiponectin treatment significantly inhibited the proliferation of both BxPC-3 cells (P< 0.01) and CFPAC-1 cells (P< 0.01) in a dose-dependent manner (Fig. 1A&B), but had no influence on their migration and invasion in vitro (Fig. S1). To further confirm the antiproliferative effect of adiponectin on pancreatic cancer cells, we overexpressed adiponectin in BxPC-3 and CFPAC-1 cells using lentiviral vectors (Fig. 1C). Similarly, the forced expression of adiponectin in BxPC-3 cells and CFPAC-1 cells suppressed their growth significantly (both P< 0.01) (Fig. 1D). Moreover, in order to reduce the impact of a discrete amount of endogenous adiponectin presented in FBS, we repeated the experiments under alow-serum condition with 2% FBS. Compared to the normal medium with 10% FBS, exogenous adiponectin treatment caused a more pronounced inhibition of cell growth under low-serum condition in both BxPC-3 and CFPAC-1 cells (Fig. 1E). Similar trends were also observed in adiponectin-overexpressing BxPC-3 and CFPAC-1 cells (Fig. 1F).
Adiponectin exerts its cellular function through binding to their specific receptors [18]. There are two isoforms of the adiponectin receptors, AdipoR1 and AdipoR2, both of which possess seven transmembrane domains with a cytoplasmic N-terminal region and an extracellular C-terminal region [18].As proven by western blot, these receptors were positively expressed in both BxPC-3 andCFPAC-1 cells (Fig. 1G). To evaluate the functional role of AdipoR1/2in the adiponectin-induced inhibitory effect on pancreatic cancer cells, we next knocked down the expression of both adiponectin receptors in BxPC-3 cells (BxPC-3-siAdipoR1/2) via lentivirus-mediated shRNAs targeting AdipoR1 andAdipoR2 (Fig. 1H). The results of the cell proliferation assay showed that knockdown of AdipoR1/2 abolished the antiproliferative effect induced by adiponectin in BxPC-3 cells in the presence of either 10% or 2% FBS in culture medium (Fig. 1J).
Taken together, these results indicated that adiponectin exerted a significant inhibitory effect on the in vitro proliferation of human pancreatic cancer cells via its receptors.
Knockdown of adiponectin receptors promoted the growth of pancreatic cancer in vivo
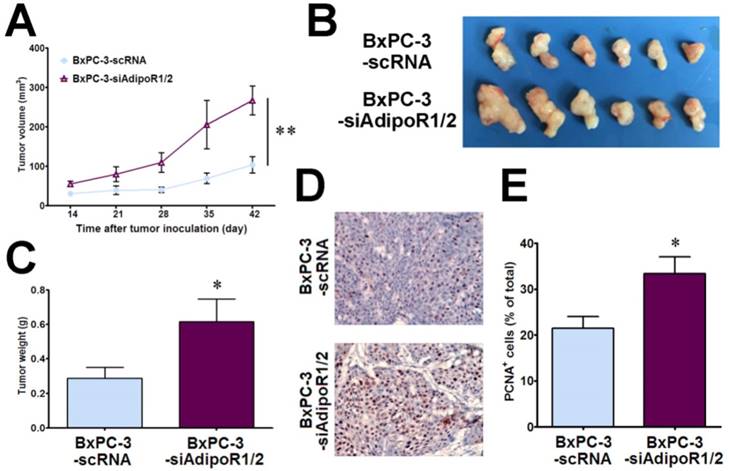
To determine whether adiponectin signaling inhibits pancreatic cancer growth in vivo, we subcutaneously inoculated nude mice with BxPC-3-siAdipoR1/2cells and control cells (BxPC-3-scRNA), respectively. The tumor growth was monitored up to 6 weeks after inoculation. As shown in Fig. 2A, the BxPC-3-siAdipoR1/2 tumors had a drastically faster growth rate than the controls (P< 0.01). Compared with the control tumors, the weights of BxPC-3-siAdipoR1/2 tumors were increased by more than 2-fold (0.29±0.06g vs 0.62±0.13g, P< 0.05) at the time of euthanasia (Fig. 2B &C). These data suggested that the knockdown of adiponectin receptors accelerated the growth of pancreatic cancer in vivo.
Given the role of adiponectin in inhibiting the proliferation of pancreatic cancer cells in vitro, we detected the expression of PCNA, a marker of cell proliferation, in tumor tissues using immunohistochemistry. There was a significant increase in PCNA-positive cells in BxPC-3-siAdipoR1/2 tumors (P< 0.05), demonstrating that blocking adiponectin signaling promoted the proliferation of pancreatic cancer cells in vivo (Fig. 2D&E). These results suggested that, consistent with the results of the in vitro experiments, adiponectin-stimulated signaling via binding to its receptors inhibited the growth of pancreatic cancer cells in vivo.
Adiponectin induced G1accumulation in pancreatic cancer cells via suppressing cyclin D1 expression
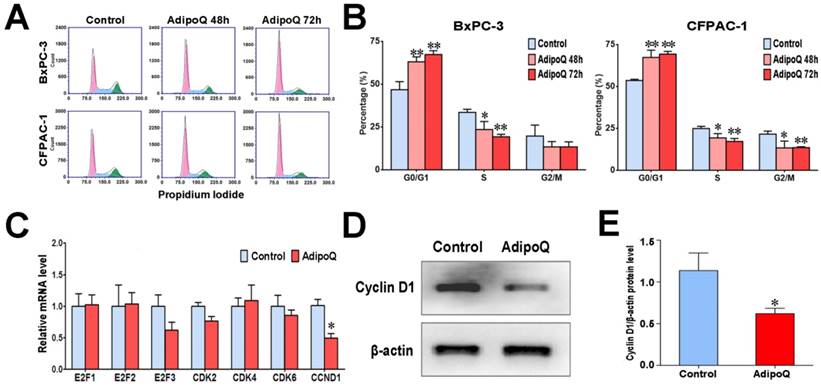
Given the antiproliferative effect of adiponectin in pancreatic cancer cells, we further assessed its influence on cell cycle. After adiponectin treatment for 48h or 72h, BxPC-3cells were subjected to cell cycle analysis by flow cytometry. As shown in Fig. 3A & B, the proportion of cells in G0/G1 phase was overtly increased after adiponectin treatment for both 48h and 72h, while those in S phase and G2/M phase were reduced, suggesting G1 accumulation in BxPC-3 cells in response to adiponectin treatment. Similar results were also observed in CFPAC-1 cells (Fig. 3A&B). These data indicated that adiponectin suppressed pancreatic cancer cell proliferation through preventing the cells from entering the S phase.
To explain the G1 accumulation induced by adiponectin, we detected the expression level of several important genes involved in G1/S transition following adiponectin treatment in BxPC-3 cells. Compared to control cells, adiponectin significantly decreased the mRNA expression of cyclin D1 gene (CCND1), but had no effect on other regulators of G1/S transition (Fig. 3C). The decreased expression of cyclin D1 induced by adiponectin was further confirmed at the protein level by western blot (Fig. 3D&E). Cyclin D1has been proven to promote the transition from G1 phase to S phase and is mandatory for cell proliferation [23, 24]. It has been shown that the overexpression of cyclin D1 in various cancer cells, including pancreatic cancer cells, shortens the G1 phase [25, 26]. It is thus likely that adiponectin suppressed the expression of cyclin D1, resulting in G1accumulation, to exert its negative role in the proliferation of pancreatic cancer cells.
Adiponectin inhibited the proliferation of human pancreatic cancer cells through its receptors in vitro. A, BxPC-3 cells were left untreated or were treated with AdipoQ at the indicated concentrations for 72 h, and then the cell proliferation was determined via a CCK-8 assay. The results are expressed as the absorbance at 450 nm. Note that adiponectin inhibited the proliferation of BxPC-3 cells in a dose-dependent manner. B, Cell proliferation assay was also performed on BxPC-3 and CFPAC-1 cells viaCCK-8 assay after the cells were treated with AdipoQ(40 μg/ml) in the presence of 10% FBS for the indicated time durations. Untreated cells were used as controls. C and D, BxPC-3 and CFPAC-1 cells were infected with a recombinant lentivirus carrying the human adiponectin gene (adiponectin-overexpressing cells: BxPC-3-AdipoQ and CFPAC-1-AdipoQ) or an empty virus (control cells: BxPC-3-VC and CFPAC-1-VC). C,A western blot analysis of the expression of adiponectin in BxPC-3-AdipoQ and BxPC-3-VC cells. D, The cell proliferation of the infected BxPC-3 and CFPAC-1 cells was determined via CCK-8 assay in the presence of 10% FBS. E, Cell proliferation assay was performed on BxPC-3 and CFPAC-1 cells via CCK-8 assay after the cells were treated with AdipoQ (40 μg/ml) in the presence of 2% FBS for the indicated time durations. Untreated cells were used as controls. F, The cell proliferation of the infected BxPC-3 and CFPAC-1 cells was determined via CCK-8 assay in the presence of 2% FBS. G,The protein expression of adiponectin receptors AdipoR1 and AdipoR2 in BxPC-3 and CFPAC-1 cells was analyzed by western blot. H, A western blot analysis of the expression of adiponectin receptors in the RNAi-mediated AdipoR-knockdown cells (BxPC-3-siAdipoR1/2) and control cells (BxPC-3-scRNA).J, Both BxPC-3-siAdipoR1/2 and BxPC-3-scRNA cells were left untreated or were treated with adiponectin (40 μg/ml)for 72 h in the presence of 10% FBS (Left) or 2% FBS (Right), and then the cell proliferation was determined via a CCK-8 assay. AdipoQ, adiponectin. Data are expressed as the mean ± SEM of 3-4 independent experiments. *P< 0.05; **P< 0.01.
Knockdown of AdipoRs promotes the growth of human pancreatic cancer in vivo. RNAi-mediated AdipoR-knockdown human pancreatic cancer cells (BxPC-3-siAdipoR1/2) or control cells (BxPC-3-scRNA) were subcutaneously injected into nude mice (n = 6 per group) A, The tumor volume for each mouse was measured once a week starting 14 days after tumor inoculation. B, The tumor-bearing mice were euthanized on day 42 after inoculation, and the subcutaneous pancreatic tumors were dissected and photographed. C, The tumor weight was compared between the BxPC-3-siAdipoR1/2 and BxPC-3-scRNA groups. D, Representative immunohistochemical staining of PCNA in the BxPC-3-siAdipoR1/2 tumors and BxPC-3-scRNA tumors (original magnification,×200). E, Quantitative analysis of PCNA positive tumor cells. AdipoQ, adiponectin. Data are expressed as the mean ± SEM. *P< 0.05; **P< 0.01.
Adiponectin induced G1accumulationin pancreatic cancer cells possibly via suppressing cyclin D1 expression. A and B, The BxPC-3 cells and CFPAC-1 cells were left untreated or were treated with adiponectin(40 μg/ml)for 48h or 72h, and then subjected to cell cycle analysis by flow cytometry. A, Representative flow cytometry panels of the cell cycle analysis of adiponectin-treated BxPC-3 or CFPAC-1 cells and control cells (from at least three independent experiments) B, The comparison of the percentage of cells in different cell-cycle phases between adiponectin-treated cells and corresponding control cells. C, Quantitative real-time PCR analysis of the mRNA levels of the indicated genes in adiponectin-treated BxPC-3 cells and control cells. D, A western blot analysis of the protein expression of cyclin D1 in adiponectin-treated BxPC-3 cells and control cells. E, Quantification results of cyclin D1 protein levels. Similar results were seen in 3 independent experiments. AdipoQ, adiponectin. Data are expressed as the mean ± SEM of 3-4 independent experiments. *P< 0.05; **P< 0.01.
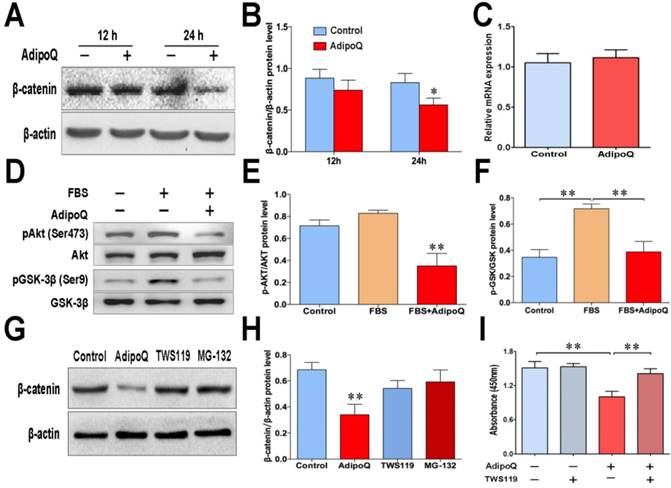
Adiponectin down-regulated the cellular β-catenin levels via the GSK-3β pathway in pancreatic cancer cells. A, The BxPC-3 cells and were left untreated or were treated with adiponectin(40 μg/ml) for the indicated time durations, and then subjected to western blot analysis of the protein levels of β-catenin. B, Quantification results of Fig. 4A. C, Quantitative real-time PCR analysis of the mRNA levels of β-catenin in adiponectin-treated BxPC-3 cells and control cells. D, western blot analysis of the levels of Akt serine-473 phosphorylation and GSK-3β serine-9 phosphorylation in adiponectin-treated BxPC-3 cells and control cells following serum stimulation. E and F, Quantification results of p-AKT and p-GSK-3β protein levels. G, BxPC-3 cells were treated with adiponectin alone for the time indicated durations or in combination with the GSK-3β inhibitor TWS119 (10μM) for 72 h. In parallel, BxPC-3 cells were pre-treated with proteasome inhibitor MG-132 (10μM) for 1h and cultured with adiponectin, then subjected to western blot analysis of the protein levels of β-catenin. H, Quantification result of β-catenin protein levels. I, BxPC-3 cells were treated with adiponectin alone or in combination with the GSK-3β inhibitor TWS119 for 72 h, and then the cell proliferation was determined via a CCK-8 assay. The western blot images shown here are representative results of 3 independent experiments. AdipoQ, adiponectin. Data are expressed as the mean ± SEM of 3-4 independent experiments. **P< 0.01.
In addition to cyclin D1, protein levels of other cell cycle regulatory proteins such as cyclin A2, cyclin E1 and p21 were also determined by western blot. No significant changes were found between adiponectin-treated and untreated cells (Fig. S2). Because CDKN2A/p16 mutation is frequently observed in pancreatic cancer, we therefore evaluated the impact of adiponectin in PANC1 pancreatic cancer cells which carries deficient p16. Similar to the results shown in CFPAC-1 and BxPC-3 cells, adiponectin treatment significantly inhibited the cell proliferation (Fig. S3A), and reduced the mRNA level of CCND1 (Fig. S3B). These results indicated that the impact of adiponectin on cell growth inhibition and the important role of cyclin D1 in regulating adiponectin-induced G1 accumulation may have a broad implication regardless of whether p16 is defective or not.
Adiponectin downregulated the cellular β-caten in levels via the Akt/GSK3-β pathway in pancreatic cancer cells
Cyclin D1 is a well-known target of β-catenin, which is involved in the development of many cancers [27]. To elucidate the signaling pathway that participated in the adiponectin-induced decrease in cyclin D1 expression, we further detected the expression level of β-catenin in response to adiponectin treatment in BxPC-3 cells. As shown in Fig. 4A&B, the protein level of β-catenin was evidently decreased 24 h after adiponectin treatment. However, there was no change in the β-catenin mRNA expression at the same time point (Fig. 4C), suggesting that adiponectin decrease the β-catenin protein via promoting its degradation.
The protein level of β-catenin is mostly controlled by the proteasomal degradation through its phosphorylation, which can be performed by GSK-3β [28]. Previous studies have shown that Akt signaling negatively regulates GSK-3β activity by phosphorylating Ser-9 [29, 30], and thus impedes the phosphorylation and degradation of β-catenin. In BxPC-3 cells, GSK-3β was inactivated in response to serum stimulation, as demonstrated by the GSK-3βSer9 phosphorylation (Fig. 4D). However, adiponectin treatment obviously suppressed the serum-stimulated phosphorylation of GSK-3β (Fig. 4D). The suppression of GSK-3β phosphorylation was associated with a remarkable decrease in Akt serine473 phosphorylation following adiponectin treatment (Fig. 4D&E). These results suggested a suppressive role of adiponectin on regulating the inhibitory phosphorylation of GSK-3β in human pancreatic cancer cells.
To confirm that adiponectin promoted the proteasome-mediated degradation of β-catenin via regulation of the GSK-3β pathway, we performed the experiments with GSK-3β inhibitor TWS119 and proteasome inhibitor MG-132. Results showed that although β-catenin in BxPC-3 cells was significantly reduced by adiponectin treatment, such reduction could be completely reversed by TWS119 or MG-132 (Fig. 4G&H).
To further verify the functional role of the GSK-3β signaling pathway in the adiponectin-induced inhibition of pancreatic cancer cell growth, BxPC-3 cells were treated with the GSK-3β specific inhibitor TWS119in the presence or absence of adiponectin, the cell proliferation assay showed that TWS119 significantly reversed the growth inhibitory effect of adiponectin on BxPC-3 cells (Fig. 4I). Taken together, our results suggested that adiponectin promoted the degradation of β-catenin via suppression of the inhibitory phosphorylation of GSK-3β, which contributed to its antiproliferative effect in pancreatic cancer cells.
Adiponectin inhibited the expression of β-catenin-associated transcription factor TCF7L2 in pancreatic cancer cells
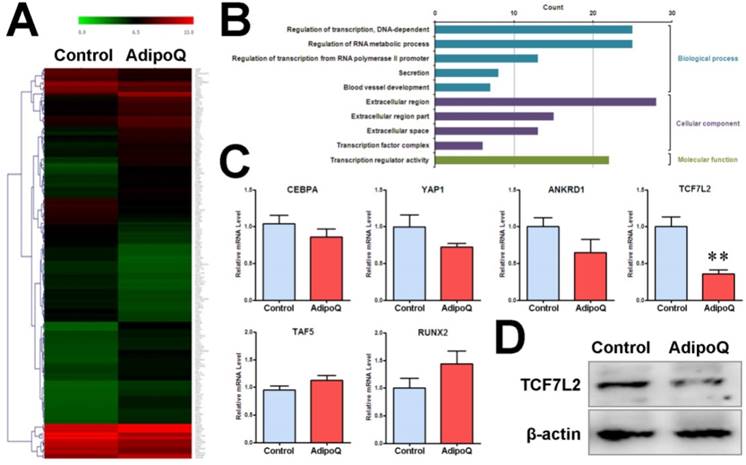
In parallel, we performed a genome-wide microarray analysis of the mRNA expression profile in BxPC-3 cells after adiponectin treatment for 48h. As a result, a total of 180 genes were found to be differentially expressed (fold change ≥ 2) in response to adiponectin treatment, including 102 upregulated genes and 78 down-regulated genes (Fig. 5A). GO-based enrichment analysis was used to classify the adiponectin-induced gene expression changes in pancreatic cancer cells under three different subontologies: biological process, cellular component, and molecular function. The analysis for GO biological process and molecular function subontologies revealed that the differentially expressed genes were mainly enriched in “Regulation of transcription, DNA-dependent”, “Regulation of transcription from RNA polymerase II promoter”, and “Transcription regulator activity”(P<0.05 and count >10) (Fig. 5B). These results supported a regulatory effect of adiponectin signaling on transcription activity in pancreatic cancer cells. The results of the analysis for GO cellular component subontologies showed that six differentially expressed genes were involved in the transcriptional complex, comprising TCF7L2, TAF5, YAP1, CEBPA, RUNX2 and ANKRD1 (Table 1). Consistent with the microarray results, the results of quantitative PCR revealed the same change in the above six genes. In particular, as shown in Fig. 5C, the mRNA level of TCF7L2 was significantly reduced after adiponectin treatment. TCF7L2is a coeffector of β-catenin and can interact with β-catenin to promote target genes activation, including cyclin D1, by recruiting transcriptional complexes [31]. Consistent with the mRNA level, the protein level of TCF7L2 was substantially reduced after adiponectin treatment (Fig. 5D), suggesting that TCF7L2 may participate in the function of adiponectin.
Fold changes of the differentially expressed genes involved in transcriptional complex from microarray data
| Gene Symbol | Gene Name | Log2 Ratio (AdipoQ vsControl) |
|---|---|---|
| CEBPA | CCAAT/enhancer binding protein (C/EBP), alpha | -2.889 |
| YAP1 | Yes-associated protein 1 | -2.763 |
| ANKRD1 | Ankyrin repeat domain 1 (cardiac muscle) | -2.023 |
| TCF7L2 | Transcription factor 7-like 2 (T-cell specific, HMG-box) | -2.370 |
| TAF5 | TAF5 RNA polymerase II, TATA box binding protein (TBP)-associated factor | 2.129 |
| RUNX2 | Runt-related transcription factor 2 | 2.298 |
Discussion
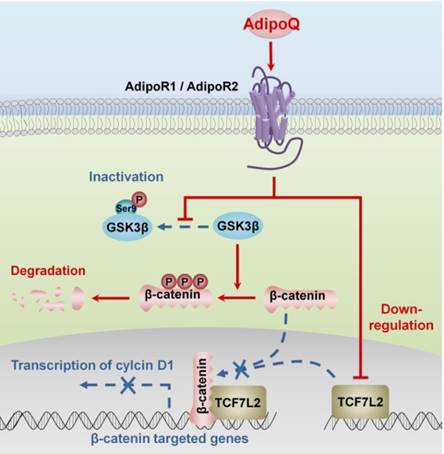
Obesity is one of the common risk factors for various cancers including pancreatic cancer [4]. Obesity-induced adipokine imbalance has been considered an important molecular connection between obesity and cancers [7]. Adiponectin, one of the most abundant adipokines, is significantly decreased in obese individuals [5]. Moreover, long-term prospective studies have demonstrated an association of hypoadiponectinemia with increased pancreatic cancer risk, suggesting a protective role of adiponectin against this fatal cancer [16, 17]. In this study, we provided solid in vitro and in vivo evidence supporting a direct inhibitory effect of this adipokine on the growth of human pancreatic cancer. Importantly, we characterized the molecular mechanisms of adiponectin-induced growth inhibition in pancreatic cancer cells. Our results suggested that adiponectin negatively regulated the β-catenin signaling via promoting the GSK-3β-mediated degradation of β-catenin and downregulating the expression of the β-catenin coeffector TCF7L2 in pancreatic cancer cells. As a consequence, the transcription of β-catenin target genes, such as cyclin D1, was disturbed, resulting in the growth inhibition of pancreatic cancer cells (Fig. 6).
Adiponectin inhibited the expression of β-catenin-associated transcription factor TCF7L2 in pancreatic cancer cells. A and B, The pooled RNA sample from the adiponectin-treated BxPC-3 cells or control cells was subjected to microarray analysis of the mRNA expression profile. A total of 180 genes were identified to be differentially expressed at the mRNA level in response to adiponectin treatment. A, Hierarchical clustering of the 180 differentially expressed genes. The colored images are presented as described; the green and red colors indicate low and high expression levels, respectively. B, GO annotation of differentially expressed genes in BxPC-3 cells induced by adiponectin treatment. The overrepresented GO terms (P< 0.05)are ranked by count number under different GO subontologies (biological process, cellular component, and molecular function) and only top 5 GO biological process terms are shown here. C, quantitative real-time PCR analysis of the mRNA expression changes of six differentially expressed genes involved in the “transcription factor complex”. Note that all six genes were confirmed to be changed in the same direction as that identified by the microarray and that the β-catenin-associated transcription factor TCF7L2 was significantly down-regulated by adiponectin treatment. D, A western blot analysis of the protein expression of TCF7L2 in adiponectin-treated BxPC-3 cells and control cells. Similar results were seen in 3 independent experiments. AdipoQ, adiponectin. Data are expressed as the mean ± SEM of 3 independent experiments. **P< 0.01.
Proposed mechanisms of adiponectin-induced growth inhibition of pancreatic cancer. The binding of adiponectin to its receptors AdipoR1 and AdipoR2 on pancreatic cancer cells inhibits the phosphorylation of GSK-3β, which promotes the degradation of β-catenin via enhancing its phosphorylation. Moreover, the activation of adiponectin signaling suppresses the expression of β-catenin-associated transcription factor TCF7L2 in pancreatic cancer cells. Both of these effects lead to the transcriptional inhibition of β-catenin-targeted genes, such as cyclin D1.In consequence, the proliferation of pancreatic cancer cells is inhibited. AdipoQ, adiponectin.
Adiponectin has been shown to play a role in inhibiting cancer progression in various cancers [12, 32, 33]. Regarding human pancreatic cancer, a previous study has reported an in vitro inhibitory effect of adiponectin on the proliferation of Panc-1 and MiaPaCa-2 cells [34]. However, the in vivo role of adiponectin in inhibiting the growth of human pancreatic cancer has not been determined. Our study not only confirmed the adiponectin-induced proliferation inhibition in another two human pancreatic cancer cell lines (BxPC-3 and CFPAC-1) but also showed, for the first time, that blocking adiponectin signaling could promote the growth of human pancreatic cancer xenografts in nude mice. Our findings presented in this study support that activating adiponectin signaling, such as administrating the small molecule agonist of AdipoRs AdipoRon [31], may be a promising strategy for the treatment of pancreatic cancer. Interestingly, studies investigating the in vivo effect of adiponectin on murine pancreatic cancer cells have reported conflicting results. Kato et al and Messaggio et al showed that loss of adiponectin causes increased growth of murine pancreatic tumors inoculated orthotopically [34, 35], supporting our results obtained from the subcutaneous tumor model of human pancreatic cancer. In contrast, Huang et al reported a growth-promoting effect of adiponectin on murine pancreatic cancer, showing a decreased growth of subcutaneous tumors in adiponectin deficient mice [36]. These discrepant in vivo results with respect to murine pancreatic cancer have been explained by the location of the inoculated tumor [36]. Future studies using orthotopic cancer models are required to further confirm the role of adiponectin in inhibiting human pancreatic cancer.
Adiponectin commonly exerts its biological activity through binding to its receptors AdipoR1 and AdipoR2 [37]. The presence of both AdipoR1 and AdipoR2 has been demonstrated in human pancreatic cancer tissues [22]. Consistently, we found that both BxPC-3 cells and CFPAC-1 cells expressed these AdipoRs. Knockdown of AdipoRs abolished the growth-inhibiting effect induced by adiponectin. It is noted that the effect of adiponectin was not parallel to the expression level of AdipoR1/2 in our knockdown experiment. This is probably due to the endogenous adiponectin existing in FBS. Because the blood concentration of adiponectin is very high (5-10μg/ mLin human and mouse), the binding sites of Adipo R1/R2 may be already saturated with the binding of endogenous adiponectin, especially in the medium with 10% FBS. Under such circumstances, adiponectin treatment could not exert further inhibition effect on cell growth. The important function of AdipoRs in regulating cell growth has also been shown in a previous study that treatment with an AdipoR agonist could increase the adiponectin-induced growth inhibition in pancreatic cancer cells [34]. Most recently, AdipoRs were found to be downregulated in pancreatic cancer tissues compared to adjacent normal acinar tissues [34], supporting the impaired adiponectin signaling in human pancreatic cancer. Nevertheless, AdipoRs expressed by pancreatic cancer cells appear to be sufficient enough to respond to AdipoQ stimulation [34].
We did not observe the adiponectin expression in human pancreatic cancer cells, suggesting that the action of adiponectin in pancreatic cancer may be exerted via a paracrine or endocrine mode rather than an autocrine mode. Therefore, our in vivo experiments, in which the endocrine and/or paracrine effect of murine adiponectin on human pancreatic cancer xenografts in nude mice was blocked by silencing of human AdipoRs in cancer cells, were designed based on the hypothesis that murine adiponectin can bind to human AdipoRs and exert a similar action on human pancreatic cancer cells. This hypothesis was supported by the following observations: (i) murine adiponectin shares a high amino acid sequence identity (more than 81%) with human orthologous protein; (ii) murine adiponectin could protect human lung fibroblasts against paraquat-induced cytotoxicity, whereas AdipoR knockdown reversed this protection effect[38]; (iii) overexpression of human adiponectin in mice has been shown to improve a high calorie diet-induced metabolic disorder [39], indicating that, on the other hand, human adiponectin could also bind to murine AdipoRs and exert its biological function in mice. These findings, together with our results, are suggestive of the protein structure conservation of adiponectin and AdipoRs between humans and mice.
Importantly, we characterized the adiponectin-triggered intracellular signaling events downstream of AdipoRs in human pancreatic cancer cells, which was rarely investigated before. Our results revealed that adiponectin exerted its inhibitory effect on the growth of pancreatic cancer via modulating the GSK-3β/β-catenin signaling pathway. This conclusion is supported by our findings that adiponectin treatment substantially reduced the serum-induced phosphorylation of GSK-3β, decreased the intracellular accumulation of β-catenin, and downregulated the expression of cyclin D1. Additionally, treatment with the GSK-3β inhibitor TWS119 could attenuate the inhibitory effect of adiponectin on the proliferation of pancreatic cancer cells. Consistent with our findings, a similar regulatory effect of adiponectin on GSK-3β/β-catenin signaling has also been reported in breast cancer cells [40]. The key signaling molecules that link the adiponectin and GSK-3β/β-catenin pathways were not been identified in our study. A remarkable decrease in the phosphorylation of Akt at Ser473 was observed following adiponectin treatment in our experiments. Due to the existence of crosstalk between the PI3K/ Akt and Wnt/β-catenin signaling pathways in cancer cells [41], Akt is possibly involved in the adiponectin- induced modulation of GSK-3β/β-catenin signaling. In addition, the binding of adiponectin to its receptors canonically activates AMPK signaling through the adaptor protein APPL1 [42]. Inhibiting AMPK could alter the phosphorylation level of GSK-3β [43], suggesting AMPK as a molecular link between adiponectin signaling and GSK-3β/β-catenin signaling. Further studies are needed to determine the roles of these two signaling molecules in mediating the adiponectin-induced modulation of GSK-3β/β- catenin signaling in pancreatic cancer cells.
Moreover, we found that adiponectin treatment significantly downregulated the TCF7L2 expression at the transcription level. TCF7L2 is one of T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors, which are the major end point effectors of Wnt/β-catenin signaling [31]. TCF/LEF members have the ability to undergo a transcriptional switch from repression to activation of target genes via recruiting β-catenin in the presence of Wnt signals [31]. Since stabilization of β-catenin proteins and active Wnt signaling are usually observed in human pancreatic cancers [44], adiponectin-induced downregulation of TCF7L2appeared to further attenuate β-catenin signaling in pancreatic cancer cells. Our ongoing studies are seeking to determine the detailed mechanisms by which adiponectin inhibits the expression of TCF7L2. It is possible that TCF7L2 downregulation is a direct consequence of the attenuation of GSK-3β/β-catenin signaling in response to adiponectin due to the fact that TCF/LEF members are potential target genes of Wnt/β-catenin signaling [45]. In this case, adiponectin appears to provoke a “positive feedback loop” that enhances the attenuation of β-catenin signaling by downregulating TCF7L2 in pancreatic cancer cells.
Taken together, our present study provides not only in vitro but also in vivo evidence for the first time that adiponectin signaling could suppress the growth of human pancreatic cancer. Mechanistically, the ability of adiponectin to attenuate β-catenin signaling might play a critical role in mediating the inhibitory effects of adiponectin on pancreatic cancer growth. Our results support a causal link between hypoadiponectinemia and increased pancreatic cancer risk and suggest adiponectin as a novel therapeutic target especially for obesity-related pancreatic cancer.
Abbreviations
GSK-3β: Glycogen synthase kinase 3 beta; TCF7L2: Transcription factor 7-like 2; AdipoR: Adiponectin receptor; PCNA: Proliferating cell nuclear antigen; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; E2F1; CDK: Cyclin-dependent kinase; TAF5: TATA-box binding protein associated factor 5; RUNX2: Runt-related transcription factor 2; CEBPA: CCAAT/enhancer-binding protein alpha.
Supplementary Material
Supplementary figures and table.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81572312, 81571618) and the State Key Laboratory of Oncogenes and Related Genes (91-10-03, YSJJ-2014- 02), the Shanghai Municipal Commission of Health and Family Planning (20174Y0149), the Specialized Research Fund for the Doctoral Program of Higher Education of China (20120073120094), and the Innovation Program of Shanghai Municipal Education Commission (13YZ036).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, KD Miller, A Jemal. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7-30
2. Torre LA, F Bray, RL Siegel. et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87-108
3. Chen W, R Zheng, PD Baade. et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115-32
4. Lauby-Secretan B, C Scoccianti, D Loomis. et al. Body Fatness and Cancer — Viewpoint of the IARC Working Group. New England Journal of Medicine. 2016;375(8):794-798
5. Davoodi SH, T Malek-Shahabi, A Malekshahi-Moghadam. et al. Obesity as an Important Risk Factor for Certain Types of Cancer. Iranian Journal of Cancer Prevention. 2013;6(4):186-194
6. Ligibel JA, CM Alfano, KS Courneya. et al. American Society of Clinical Oncology Position Statement on Obesity and Cancer. Journal of Clinical Oncology. 2014;32(31):3568-3574
7. Nam SY. Obesity-Related Digestive Diseases and Their Pathophysiology. Gut and Liver. 2017;11(3):323-334
8. Yamauchi T, J Kamon, Y Minokoshi. et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8(11):1288-95
9. Cheng X, EJ Folco, K Shimizu. et al. Adiponectin induces pro-inflammatory programs in human macrophages and CD4+ T cells. J Biol Chem. 2012;287(44):36896-904
10. Ohashi K, JL Parker, N Ouchi. et al. Adiponectin Promotes Macrophage Polarization toward an Anti-inflammatory Phenotype. The Journal of Biological Chemistry. 2010;285(9):6153-6160
11. Kern PA, GB Di Gregorio, T Lu. et al. Adiponectin Expression From Human Adipose Tissue. Diabetes. 2003;52(7):1779
12. Kelesidis I, T Kelesidis, CS Mantzoros. Adiponectin and cancer: a systematic review. Br J Cancer. 2006;94(9):1221-5
13. Mauro L, GD Naimo, E Ricchio. et al. Cross-Talk between Adiponectin and IGF-IR in Breast Cancer. Front Oncol. 2015;5:157
14. Hebbard L, B Ranscht. Multifaceted roles of adiponectin in cancer. Best Pract Res Clin Endocrinol Metab. 2014;28(1):59-69
15. Wei EK, E Giovannucci, CS Fuchs. et al. Low Plasma Adiponectin Levels and Risk of Colorectal Cancer in Men: A Prospective Study. JNCI: Journal of the National Cancer Institute. 2005;97(22):1688-1694
16. Grote VA, S Rohrmann, L Dossus. et al. The association of circulating adiponectin levels with pancreatic cancer risk: a study within the prospective EPIC cohort. Int J Cancer. 2012;130(10):2428-37
17. Bao Y, EL Giovannucci, P Kraft. et al. A prospective study of plasma adiponectin and pancreatic cancer risk in five US cohorts. J Natl Cancer Inst. 2013;105(2):95-103
18. Brochu-Gaudreau K, C Rehfeldt, R Blouin. et al. Adiponectin action from head to toe. Endocrine. 2010;37(1):11-32
19. Dalamaga M, KN Diakopoulos, CS Mantzoros. The role of adiponectin in cancer: a review of current evidence. Endocr Rev. 2012;33(4):547-94
20. Obeid S, L Hebbard. Role of adiponectin and its receptors in cancer. Cancer Biol Med. 2012;9(4):213-20
21. Kim AY, YS Lee, KH Kim. et al. Adiponectin Represses Colon Cancer Cell Proliferation via AdipoR1- and -R2-Mediated AMPK Activation. Molecular Endocrinology. 2010;24(7):1441-1452
22. Dalamaga M, I Migdalis, JL Fargnoli. et al. Pancreatic cancer expresses adiponectin receptors and is associated with hypoleptinemia and hyperadiponectinemia: a case-control study. Cancer Causes Control. 2009;20(5):625-33
23. Sherr CJ, JM Roberts. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18(22):2699-711
24. Sherr CJ. Cancer cell cycles. Science. 1996;274(5293):1672-7
25. Fu M, C Wang, Z Li. et al. Minireview: Cyclin D1: Normal and Abnormal Functions. Endocrinology. 2004;145(12):5439-5447
26. Biliran H, Y Wang, S Banerjee. et al. Overexpression of Cyclin D1 Promotes Tumor Cell Growth and Confers Resistance to Cisplatin-Mediated Apoptosis in an Elastase-<em>myc</em> Transgene-Expressing Pancreatic Tumor Cell Line. Clinical Cancer Research. 2005;11(16):6075
27. Shang S, F Hua, ZW Hu. The regulation of beta-catenin activity and function in cancer: therapeutic opportunities. Oncotarget. 2017;8(20):33972-33989
28. Mills CN, S Nowsheen, JA Bonner. et al. Emerging roles of glycogen synthase kinase 3 in the treatment of brain tumors. Front Mol Neurosci. 2011;4:47
29. Jope RS, CJ Yuskaitis, E Beurel. Glycogen Synthase Kinase-3 (GSK3): Inflammation, Diseases, and Therapeutics. Neurochemical research. 2007;32(4-5):577-595
30. Wang C-Y, A-C Tsai, C-Y Peng. et al. Dehydrocostuslactone Suppresses Angiogenesis In Vitro and In Vivo through Inhibition of Akt/GSK-3β and mTOR Signaling Pathways. PLoS ONE. 2012;7(2):e31195
31. Mao CD, SW Byers. Cell-Context Dependent TCF/LEF Expression and Function: Alternative Tales of Repression, De-Repression and Activation Potentials. Critical reviews in eukaryotic gene expression. 2011;21(3):207-236
32. Illiano M, E Nigro, L Sapio. et al. Adiponectin down-regulates CREB and inhibits proliferation of A549 lung cancer cells. Pulmonary Pharmacology & Therapeutics. 2017;45:114-120
33. Siegel AB, A Goyal, M Salomao. et al. Serum Adiponectin is Associated with Worsened Overall Survival in a Prospective Cohort of Hepatocellular Carcinoma Patients. Oncology. 2015;88(1):57-68
34. Messaggio F, AM Mendonsa, J Castellanos. et al. Adiponectin receptor agonists inhibit leptin induced pSTAT3 and in vivo pancreatic tumor growth. Oncotarget. 2017;8(49):85378-85391
35. Kato M, K Watabe, M Tsujii. et al. Adiponectin Inhibits Murine Pancreatic Cancer Growth. Digestive Diseases and Sciences. 2014;59(6):1192-1196
36. Huang B, X Cheng, D Wang. et al. Adiponectin promotes pancreatic cancer progression by inhibiting apoptosis via the activation of AMPK/Sirt1/PGC-1alpha signaling. Oncotarget. 2014;5(13):4732-45
37. Kadowaki T, T Yamauchi. Adiponectin and Adiponectin Receptors. Endocrine Reviews. 2005;26(3):439-451
38. Yao R, Y Cao, Y-r He. et al. Adiponectin Attenuates Lung Fibroblasts Activation and Pulmonary Fibrosis Induced by Paraquat. PLoS ONE. 2015;10(5):e0125169
39. Otabe S, X Yuan, T Fukutani. et al. Overexpression of human adiponectin in transgenic mice results in suppression of fat accumulation and prevention of premature death by high-calorie diet. American Journal of Physiology-Endocrinology and Metabolism. 2007;293(1):E210-E218
40. Wang Y, JB Lam, KS Lam. et al. Adiponectin modulates the glycogen synthase kinase-3beta/beta-catenin signaling pathway and attenuates mammary tumorigenesis of MDA-MB-231 cells in nude mice. Cancer Res. 2006;66(23):11462-70
41. Fresno Vara JA, E Casado, J de Castro. et al. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004;30(2):193-204
42. Deepa SS, LQ Dong. APPL1: role in adiponectin signaling and beyond. American Journal of Physiology-Endocrinology and Metabolism. 2009;296(1):E22-E36
43. Duan J, Y Yin, J Cui. et al. Chikusetsu Saponin IVa Ameliorates Cerebral Ischemia Reperfusion Injury in Diabetic Mice via Adiponectin-Mediated AMPK/GSK-3β Pathway In Vivo and In Vitro. Molecular Neurobiology. 2016;53(1):728-743
44. Pai P, S Rachagani, P Dhawan. et al. Mucins and Wnt/β-catenin signaling in gastrointestinal cancers: an unholy nexus. Carcinogenesis. 2016;37(3):223-232
45. Cadigan KM, ML Waterman. TCF/LEFs and Wnt Signaling in the Nucleus. Cold Spring Harbor Perspectives in Biology. 2012;4(11):a007906
Author contact
![]() Corresponding authors: Hong Tu M.D., Ph.D. (tuhongorg) or Yu Gan Ph.D. (ganyuorg), State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Renji Hospital, Shanghai Jiao Tong University School of Medicine, 25/Ln. 2200 Xietu Road, Shanghai 200032, China. Tel/Fax: 86-21-64437196
Corresponding authors: Hong Tu M.D., Ph.D. (tuhongorg) or Yu Gan Ph.D. (ganyuorg), State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Renji Hospital, Shanghai Jiao Tong University School of Medicine, 25/Ln. 2200 Xietu Road, Shanghai 200032, China. Tel/Fax: 86-21-64437196
Received 2018-5-22
Accepted 2018-11-28
Published 2019-1-1