ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2019; 15(2):325-340. doi:10.7150/ijbs.29009 This issue Cite
Research Paper
Metformin Promotes the Survival of Random-Pattern Skin Flaps by Inducing Autophagy via the AMPK-mTOR-TFEB signaling pathway
Hongqiang Wu1,2,3*, Jian Ding1,2,3*, Shihen Li1,2,3, Jinti Lin1,2,3, Renhao Jiang1,2,3, Chen Lin1,2,3, Li Dai1,2,3, Chenglong Xie1,2,3, Dingsheng Lin1,2,3, Huazi Xu1,2,3, Weiyang Gao1,2,3 ![]() , Kailiang Zhou1,2,3
, Kailiang Zhou1,2,3 ![]()
1. Department of Orthopaedics, The Second Affiliated Hospital and Yuying Children's Hospital of Wenzhou Medical University, Wenzhou 325027, China
2. Zhejiang Provincial Key Laboratory of Orthopaedics, Wenzhou 325027, China
3. The Second Clinical Medical College of Wenzhou Medical University, Wenzhou 325027, China
*These authors contributed equally to this work.
Abstract

Random-pattern skin flaps are widely used to close defects in reconstructive and plastic surgeries; however, they are vulnerable to necrosis, particularly in the distal portion of the flap. Here, we examined the effects of metformin on flap survival and the mechanisms underlying these effects. Following metformin treatment, the survival area, blood flow, and number of microvessels present in skin flaps were increased on postoperative day 7, whereas tissue edema was reduced. In addition, metformin promoted angiogenesis, inhibited apoptosis, relieved oxidative stress, and increased autophagy in areas of ischemia; these effects were reversed by autophagy inhibitors 3-methyladenine (3MA) or chloroquine (CQ). Either 3MA or CQ reversed the metformin-induced increase in flap viability. Moreover, metformin also activated the AMPK-mTOR-TFEB signaling pathway in ischemic areas. Inhibitions of AMPK via Compound C (CC) or AMPK shRNA adeno-associated virus (AAV) vector resulted in the downregulation of the AMPK-mTOR-TFEB signaling pathway and autophagy level in metformin-treated flaps. Taken together, our findings suggest that metformin improves the survival of random-pattern skin flaps by enhancing angiogenesis and suppressing apoptosis and oxidative stress. These effects result from increased autophagy mediated by activation of the AMPK-mTOR-TFEB signaling pathway.
Keywords: Random-pattern skin flaps, Metformin, Autophagy, AMPK-mTOR-TFEB signaling pathway
Introduction
Random-pattern skin flaps are frequently used in reconstructive surgeries to close defects caused by trauma, congenital disorders, cancer-related deformities, or diabetes mellitus [1, 2]. However, random skin flaps, particularly those used to treat large areas, are vulnerable to necrosis in the distal flap due to a lack of sufficient arterial blood supply or adequate venous outflow [3]. This complication limits their clinical application, as it restricts the length-to-width ratio of flaps to 1.5-2 [4]. When random skin flaps are established without retention of the vessel pedicle, they undergo significant ischemia followed by neovascularization starting at the flap pedicle and progressing towards the distal end of the flap [1, 5]. After neovascularization, reperfusion and restoration of the blood supply contribute to ischemia-reperfusion injury and necrosis in the distal flap [6]. Given the well-defined mechanisms underlying flap failure, a wide range of strategies have been developed to alleviate ischemic necrosis in random skin flaps including reduction of oxidative stress, promotion of angiogenesis, and inhibition of apoptosis [1, 5, 7, 8].
Autophagy, the degradation and recycling of dysfunctional proteins and organelles in autophagic vesicles, is crucial for promoting angiogenesis [9, 10], alleviating oxidative stress [11], and suppressing apoptosis [12]. Sitagliptin-induced activation of autophagy has been shown to preserve the angiogenic function of endothelial progenitor cells and to improve diabetic ischemia angiogenesis in a diabetic hind limb ischemia model [9]. Recent study has shown that autophagy plays an important role in disc degeneration by alleviating mitochondrial dysfunction and cell apoptosis during periods of oxidative stress [13]. In addition, upregulation of autophagy alleviates ischemia-reperfusion injury and inhibits excessive apoptosis induced by intestinal ischemia-reperfusion injury [14]. Taken together, these data suggest that induction of autophagy may be a useful strategy for enhancing the viability of random flaps via the promotion of angiogenesis and inhibition of apoptosis and oxidative stress.
Metformin (MET) is an important first-line treatment for type 2 diabetes due to its ability to prevent vascular complications [15, 16]. However, the benefits of MET extend well beyond diabetes, offering significant therapeutic benefits for diabetic cardiomyopathy, spinal cord injury, and Parkinson's disease because of its ability to activate autophagy [17-19]. MET improved angiogenesis by augmenting the expression of vascular endothelial growth factor (VEGF) A and inhibiting apoptosis in an acute hyperglycemia-chemical hypoxia model [20, 21]. MET has also been shown to ameliorate the survival of random-pattern skin flaps in a rat model [22]; however, the therapeutic effects of MET in random-pattern skin flaps and the mechanisms underlying these effects remain poorly understood.
Previous study has shown that the adenosine monophosphate-activated protein kinase (AMPK)-mammalian target of rapamycin (mTOR)-transcription factor EB (TFEB) pathway serves as a novel regulator of cell fate determination during differentiation via TFEB-dependent regulation of autophagic flux [23]. Specifically, activation of AMPK under low-nutrient conditions contributes to inhibition of mTOR activity and allows sufficient levels of dephosphorylated TFEB to enter the nucleus [23]. The activated TFEB are capable of binding Coordinated Lysosomal Expression and Regulation (CLEAR) sequence elements to induce transcription of its target numerous genes, including those related to autophagosome and lysosome activity [24-26]. MET was shown to regulate the AMPK-mTOR pathway in a mouse model of endotoxemia-induced inflammatory lung injury [27]; however, whether MET induces autophagy via the AMPK-mTOR-TFEB signaling pathway in random skin flaps is not known. Here, we examined whether MET-induced autophagy promotes angiogenesis and inhibits oxidative stress and apoptosis in a mouse model of random skin flaps via the AMPK-mTOR-TFEB signaling pathway.
Materials and Methods
Animals and groups
One hundred and twenty-six healthy C57BL/6 mice (male, 20-30g) were provided by Experimental Animal Center of Wenzhou Medical University (License no. SCXK [ZJ] 2005-0019). The animal operation and treatment conformed to the Guide for the Care and Use of Laboratory Animals of China National Institutes of Health. All animal experiments were approved by the Animal Research Committee of Wenzhou Medical University (wydw2017-0022). All mice were divided randomly into seven groups: a Control group (n=24), a MET group (n=24), a MET+3MA group (n=18), a MET + chloroquine (CQ) group (n=18), a MET+Compound C (CC) group (n=18), a MET+adeno-associated virus (AAV)- Scramble control group (MET+ Scramble control, n=12), and a MET +AAV- AMPK short hairpin RNA group (MET+AMPK shRNA, n=12). The animals were housed individually in standard experimental cages and allowed free access to food and water at appropriate temperature (25 °C).
Reagents and antibodies
Metformin (C4H11N5·HCl; purity ≥ 98%), H&E Staining Kit, DAB developer, and pentobarbital sodium were provided by Solar bio Science & Technology (Beijing, China). Dorsomorphin (Compound C: C24H25N5O; purity≥98.14%) was purchased from Med Chem Express (866405-64-3; Monmouth Junction, NJ, USA); the primary antibody against Cadherin 5 was acquired from Boster Biological Technology (A02632-2; Wuhan, China). The primary antibody against GAPDH was purchased from Biogot Technology (AP0063; Shanghai, China). Primary antibodies against VEGF, superoxide dismutase 1 (SOD1), vacuolar protein sorting 34 (VPS34), matrix metalloproteinase 9 (MMP9), heme oxygenase 1 (HO1), cathepsin D (CTSD), caspase 3 (CAPS3), Histone-H3, and mTOR were acquired from Protein tech Group (19003-1-AP, 10269-1-AP, 12452-1-AP, 10375-2-AP, 10701-1-AP, 21327-1-AP, 19677-1-AP, 17168-1-AP, and 20657-1-AP; Chicago, IL, USA). Primary antibodies against cytochrome C (CYC), Bax, AMPK, p-AMPK, p-mTOR, and endothelial nitric oxide synthase (eNOS) were obtained from Cell Signaling Technology (4272S, 2772S, 5832S, 2535S, 5536S, and 32027S; Beverly, MA, USA). The primary antibody against SQSTM1/p62 and CD34 were purchased from Abcam (ab56416, ab81289; Cambridge, UK). Primary antibodies against Microtubule-associated proteins 1A/1B light chain 3 (LC3B), 3-methyladenine (C6H7N5; purity≥98.00%, 3MA) and chloroquine (C18H26ClN3·2H3PO4; purity≥98.5%, CQ) were provide by Sigma-Aldrich Chemical Company (L7543, M9281, and C6628; Milwaukee, WI, USA). Primary antibody against TFEB was acquired from Bethyl Laboratories (A303-673A-M; Montgomery, TX, USA). Horseradish peroxidase (HRP)-conjugated IgG secondary antibody was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Fluorescein isothiocyanate (FITC)-conjugated IgG secondary antibody was obtained from Boyun Biotechnology (Nanjing, China), and 4',6-Diamidino-2-phenylindole (DAPI) solution was purchased from Beyotime Biotechnology (Jiangsu, China). The BCA Kit and NE-PER™ Nuclear and Cytoplasmic Extraction Reagents were acquired from Thermo Fisher Scientific (Rockford, IL, USA). The ECL Plus Reagent Kit was purchased from PerkinElmer Life Sciences (Waltham, MA, USA).
Flap animal model
Mice were anaesthetized by intraperitoneal injection of 50 mg/kg pentobarbital sodium 1% (w/v). Before surgery, dorsal fur was removed with an electric shaver and depilatory cream. A random-pattern, caudally based dorsal flap (size: 1.5 × 4.5 cm2) was elevated in the mouse dorsum (in the same position in each mouse) beneath the pannculus carnosus as previously reported [28]. After flap elevation, the flaps were inset immediately to the original position and sutured with 4-0 non-absorbable suture. The flap area was separated into three equal zones: proximal (Area I), intermediate (Area II), and distal (Area III).
AAV vector packaging
The AAV-AMPK shRNA (mouse Prkaa1) was constructed and packaged by Vigene Biosciences (Shandong, China). The shRNA sequence of protein kinase, AMP-activated, alpha 1 catalytic subunit (Prkaa1) was synthesized and cloned into pAV-U6-shRNA-CMV-EGFP plasmid to produce pAV-U6-shRNA (Prkaa1)-CMV-EGFP. AAV9-U6-shRNA (Prkaa1)-CMV-EGFP was produced by transfection of AAV-293 cells with pAV-U6-shRNA (Prkaa1)-CMV-EGFP, adenovirus helper plasmid (Ad helper), and AAV Rep/Cap expression plasmid. With a similar process, AAV9-U6-shRNA (scramble)-CMV-EGFP were produced as Scramble control. Viral particles were purified by iodixanol gradient method. The titer of AAV9-U6-shRNA (Prkaa1)-CMV-EGFP and AAV9-U6-shRNA (scramble)-CMV-EGFP was 1.243 × 1012, 1.22 × 1012 genomic copies per ml, respectively, determined by quantitative PCR.
Drugs and AAV vectors administration
The MET group was treated with 200mg/kg metformin by daily intraperitoneal injections for 7 days before operation, and then continued to receive the metformin until the mice were euthanized. The Control group received equal volumes of saline from preoperative 7 days and until the mice were euthanized. The MET+3MA group, MET+CQ and MET+CC group received 3MA (15 mg/kg), CQ (60 mg/kg) or CC (15 mg/kg) 30 minutes before metformin administration (dose) every time [27, 29, 30]. At 14 days before the operation of flap model, mice in the MET+ Scramble control group and the MET+AMPK shRNA groups were received 6 ul viral vectors in PBS with 5 × 109 packaged genomic particles total by subcutaneous injections for 3 Area via a microsyringe; the AVV-treated mice also were received the metformin in the same administration with that of the MET group.
Flap macroscopic evaluation
During the 7 days, the survival area of each flap was evaluated macroscopically in appearance, color, and hair conditions. On postoperative day (POD) 7, high photographs of the random flap were obtained to evaluate the flap viability. All photographs were processed by Image-Pro Plus imaging software (ver.6.0; Media Cybemetics) to determine the survival area, and the percentage of viable area was determined as follows: extent of viable area ×100%/total area (viable and ischemic).
Laser doppler blood flow (LDBF) measurement
LDBF measurement was performed to observe vascular flow and blood supply in the whole area of random skin flaps. On POD 7, six mice in each group were scanned with a laser doppler instrument (Moor Instruments, Axminster, UK) in a warm and quiet environment under anesthesia. The protocol of LDBF measurement was according to previous study [31]. Vascular flow and blood supply were visualized with the LDBF strong signal (green, yellow and red in color). The blood flow of skin flap was quantified with moor LDI Review software (ver.6.1; Moor Instruments). The measurement in each mouse was repeated three times and the mean value was used.
Tissue edema measurement
Water content reflects tissue edema of flap. On POD 7, six flap tissue specimens were weighed and then dehydrated in an autoclave at 50°C. We weighed all specimens every day until the weight was stabilized for 2 days. The percentage of water content was calculated as: Tissue water content = ([wet weight - dry weight]/wet weight) × 100%.
Hematoxylin and eosin (H&E) staining
Six samples (1 cm ×1 cm) of central tissue from each flap Area II were obtained and executed after sacrifice. The extracted skin flap samples were first fixed in 4% paraformaldehyde for 24 hours and embedded in paraffin wax for transverse sectioning. Sections with 4 µm of thickness were prepared by a microtome and mounted on poly-L-lysine-coated slides for H&E staining. We calculated the number of microvessels per unit area (/mm2) under a light microscope (×200 magnification, Olympus Corp, Tokyo, Japan), which indicated the microvascular density.
Immunohistochemistry (IHC)
Six sections of the middle part of Area II in each group were deparaffinized in xylene and rehydrated through a graded set of ethanol baths. After washing, sections were blocked with 3% (v/v) H2O2 and treated with 10.2 mM sodium citrate buffer for 20 min at 95°C. After blocking with 10% (w/v) bovine serum albumin phosphate buffered saline for 10 min, the sections were incubated with antibody against CD34 (1:100), VEGF (1:300), Cadherin5 (1:100), CASP3 (1:200), SOD1 (1:100), and CTSD (1:100) overnight at 4°C. Finally, the sections were incubated with HRP-conjugated secondary antibody and counterstained with hematoxylin. Flap tissues were imaged at × 200 magnification using a DP2-TWAN image-acquisition system (Olympus Corp, Tokyo, Japan). Images were calculated with Image-Pro Plus software (Media Cybernetics, Rockville, MD, USA) for integral absorbance quantitation of VEGF-, Cadherin5-, CASP3-, SOD1- and CTSD- and CD34-positive blood vessels counting. Six random fields of three random sections from each tissue sample were measured for counting.
Immunofluorescence staining
Six section specimens of Area II in each group were deparaffinized in xylene and rehydrated through a graded set of ethanol baths. After washing, the sections were treated with 10.2 mM sodium citrate buffer for 20 min at 95 °C. Then the sections were permeabilized with 0.1% (v/v) PBS-Triton X-100 for 30 min. After blocking in 10% (v/v) bovine serum albumin in PBS for 1 h, slides were incubated at 4 °C overnight with a primary antibody against TFEB (1:100), LC3II (1:200). Then the slides were washed three times for 10 min at room temperature and incubated with FITC-conjugated second antibody for 1 h at room temperature. All images were evaluated under a fluorescence microscope (Olympus, Tokyo, Japan). The percentage of TFEB- and LC3-positive cells in the dermal layer was calculated. Six random fields of three random sections from each tissue sample were used.
Western blotting
On POD 7, skin samples (n=6, 0.5 cm × 0.5 cm) from the middle of Area II were dissected and stored -80℃ before Western blotting. Six samples in each group were processed by extracting proteins with a lysis buffer. Another six samples in each group were processed by extracting cytoplasmic protein and nuclear protein with NE-PER™ Nuclear and Cytoplasmic Extraction Reagents. Protein concentrations were calculated using the BCA assay. Proteins were separated by 12% polyacrylamide gel electrophoresis and subsequently transferred to PVDF membranes (Roche Applied Science, Indianapolis, IN, USA). After blocking with 5% (w/v) nonfat milk for 2 hours at room temperature, the membranes were incubated overnight with the following primary antibodies at 4℃ overnight: VEGF (1:1000), MMP-9 (1:1000), Cadherin 5 (1:1000), HO-1 (1:1000), eNOS (1:1000), SOD1 (1:1000), Bax (1:1000), CYC (1:1000), CASP3 (1:1000), Beclin1 (1:1000), p62 (1:1000), LC3II (1:1000), VPS34 (1:1000), CTSD (1:1000), AMPK (1:1000), p-AMPK (1:1000), mTOR (1:1000), p-mTOR (1:1000), TFEB (1:1000), GAPDH (1:1000), and Histone-H3 (1:1000). Then the membranes were incubated with goat-anti-rabbit or goat-anti-mouse secondary antibodies for 2 hours at room temperature. The immunoreactive proteins were visualized using the ECL Plus Reagent Kit. Finally, the band intensity was quantified using Image Laboratory 3.0 software (Bio-Rad Laboratories Inc, Hercules, CA, USA).
Statistical analyses
Statistical analyses were performed using SPSS software version 19.0 (SPSS, Chicago, IL). All data were expressed as mean ± Standard Error of Mean (SEM). The data from the four groups were analyzed using an independent-sample t-test and one-way ANOVA. A value of p < 0.05 was considered to represent a statistical significance.
Results
MET enhances the viability of random skin flaps
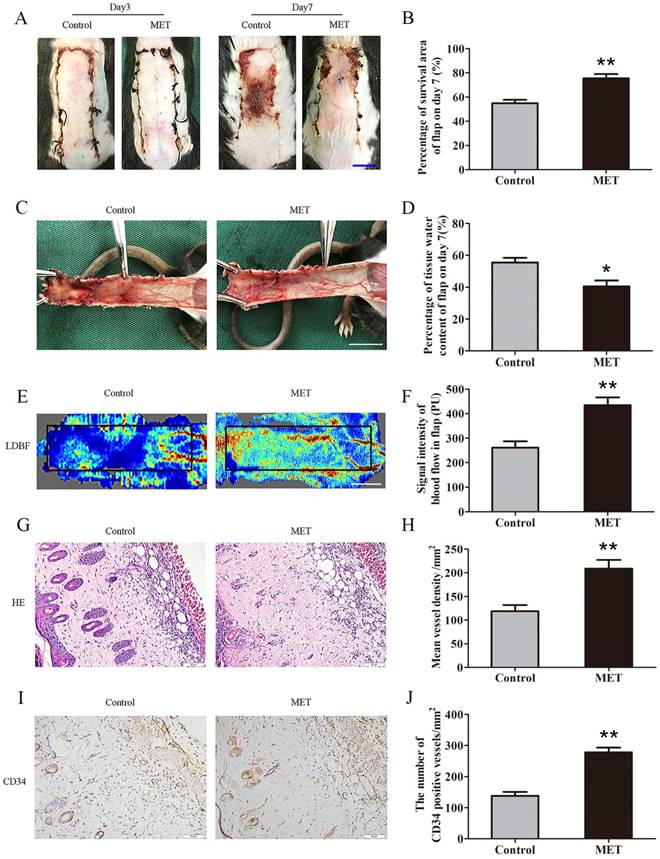
By POD 3, the random skin flaps in both groups were pale and swollen without obvious necrosis in Area III. There were no significant differences in the flap survival area between the two groups (Fig. 1A). On POD 7, each group exhibited survival in Area I, whereas Area III had become darker with necrosis spreading to Area II, along with scabbing and hardening (Fig. 1A). The mean survival area of the group treated with MET was significantly larger than that of the Control group (75.43 ± 3.54% and 54.91 ± 2.78%, respectively; p = 0.001; Fig. 1B). In the Control group, the distal portion of the flaps was significantly edematous with subcutaneous venous blood congestion (Fig. 1C). These changes were less apparent in the MET group. The water content of tissue was lower in the MET group than in the Control group (40.50 ± 3.69% and 55.48 ± 2.93%, respectively; p = 0.010; Fig. 1D). Furthermore, from observation of LDBF images, the MET group showed more obvious signal intensity of blood flow than that of the Control group (Fig. 1E). After data quantification, the signal intensity of blood flow was significantly different between the MET group and the Control group (434.42 ± 32.18PU and 261.42 ± 26.02PU, respectively; p = 0.004; Fig. 1F). The H&E staining observation showed the MET group generated a greater number of microvessels (209.00 ± 18.26/mm2) compared with the Control group (118.69 ± 13.10/mm2; p = 0.001; Fig. 1G&H). Finally, IHC showed that the number of CD34-positive vessels was also greater in the MET group than the Control group (278.35 ± 14.69/mm2 and 137.76 ± 13.08/mm2, respectively; p < 0.001; Fig. 1I&J).
MET promotes angiogenesis in random skin flaps
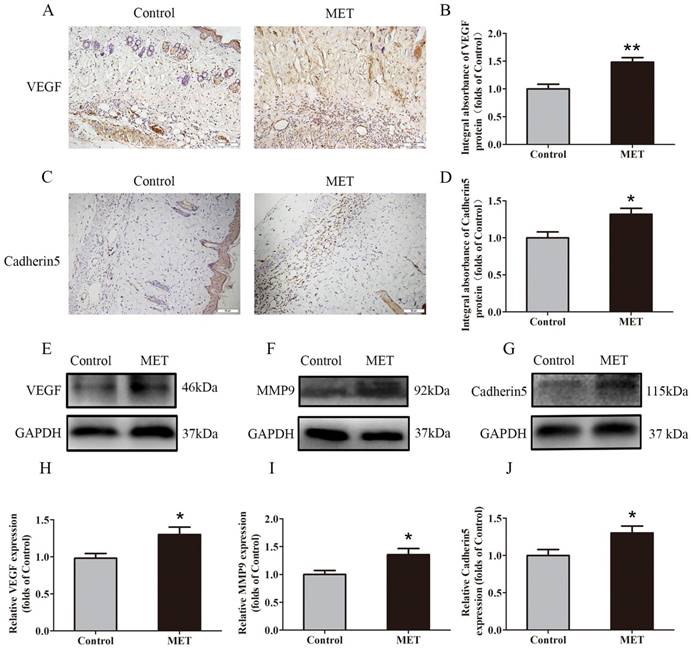
IHC for VEGF and Cadherin 5 expression was performed to quantify the capacity of neovascularization in the Control and MET groups. VEGF was detected in vessels and stromal cells in Area II and at a higher level in the MET group than in the Control group (p = 0.002; Fig. 2A&B). Cadherin5, the protein which mainly expressed in similar cells and tissue (Fig. 2C), was also higher in the MET group than in the Control group (p = 0.018; Fig. 2D). Western blotting revealed significantly upregulated VEGF, MMP9, and Cadherin 5 levels in the MET group compared with controls (p = 0.021, p = 0.021 and p = 0.033, respectively; Fig. 2E-J).
MET reduces apoptosis in random skin flaps
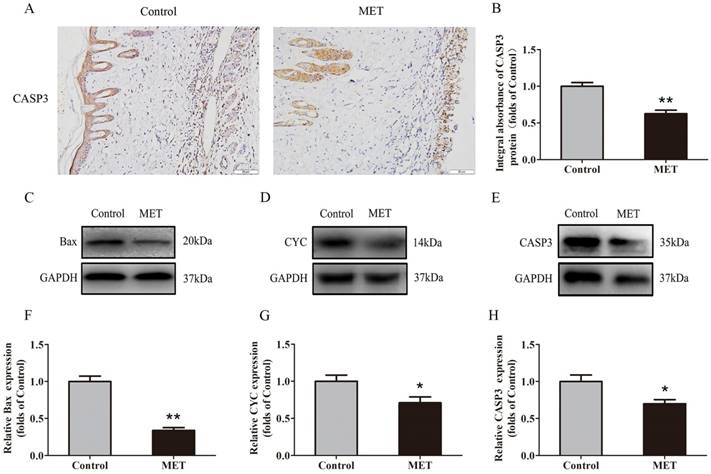
IHC was conducted to determine CASP3 level in the dermis layer of area II in the Control and MET groups. Lower CASP3 level was detected in vessels and stromal cells of the MET group than the Control group (Fig. 3A), with a lower integral absorbance in the MET group (p < 0.001; Fig. 3B). Western blotting was performed to detect the expression of Bax, CYC and CASP3 in the ischemic flap tissues (Fig. 3C-E). The results showed that the optical density values of Bax, CYC and CASP3 were significantly down-regulated in the MET group compared with the Control group (p < 0.001, p = 0.029, and p = 0.016 respectively; Fig. 3F-H).
MET inhibits oxidative stress in random skin flaps
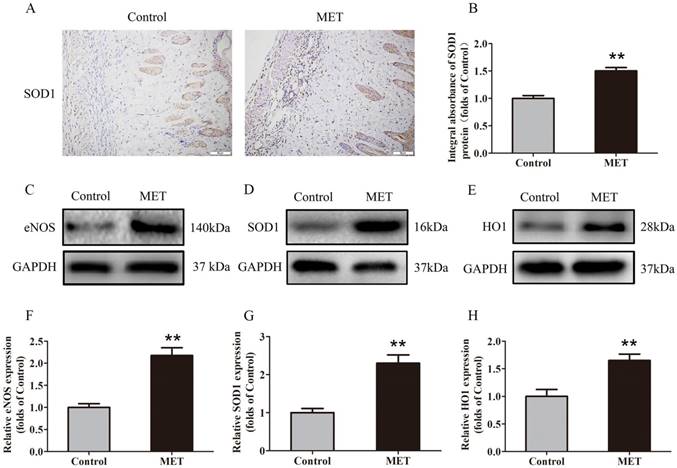
IHC was carried out for SOD1 expression to indicate oxidative stress level in area II of flaps. As shown in Fig. 4A, higher level of SOD1 was observed in vessels and stromal cells of the MET group, compared with the Control group. The integral absorbance of SOD1 was larger in the MET group than in the Control group (p < 0.001; Fig. 4B). Western blotting was also conducted for the expression of SOD1, eNOS and HO1 in the flaps (Fig. 4C-E). The results showed that the protein levels of SOD1, eNOS and HO1 were also increased in the MET group compared with the Control group (p < 0.001, p < 0.001 and p = 0.003, respectively; Fig. 4E-H).
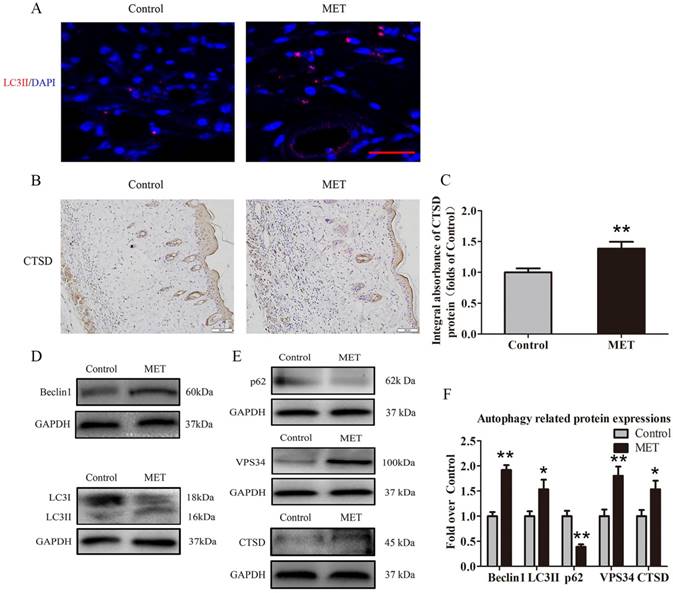
MET activates autophagy in random skin flaps
To verify activation of autophagy in flaps, we evaluated the levels of autophagosomal proteins Beclin1, VPS34, and LC3II, as well as autolysosome-related protein CTSD and autophagic substrate protein p62. As shown in Fig. 5A, autophagosomes were labeled as LC3II punctate dots (red), and nuclei were labeled with DAPI (blue) in the dermis of ischemic flaps; the MET group exhibited higher numbers of LC3II-positive cells compared with the Control group. Moreover, higher level of CTSD was observed in vessels and stromal cells of the MET group, compared with the Control group (Fig. 5B). The integral absorbance of CTSD was significantly higher in the MET group than in the Control group (p = 0.011; Fig. 5C). Western blotting was also performed for Beclin1, VPS34, LC3II, CTSD and p62 expressions in the flaps (Fig. 4D&E). As a result, optical density values of Beclin1, LC3II, VPS34, CTSD were higher in the MET group than in the Control group (p < 0.001, p = 0.03, p = 0.005, and p = 0.028, respectively; Fig. 5F), with a lower value of p62 in the MET group (p < 0.001; Fig. 5F).
MET enhances the viability of random skin flaps. (A) Digital photographs of flaps of the Control and MET groups on POD 3 and POD 7 (scale bar, 1 cm). (B) The percentages of survival area in the Control group and the MET group were quantified and analyzed. (C) Digital photographs of the inner side of flaps in the Control and MET groups on POD7 (scale bar, 1 cm). (D) Histogram of percentage of tissue water content in each group. (E) Full field LDBF images of flaps in each group on POD 7 (scale bar, 1 cm). (F) The signal intensity of blood flow of flaps was quantified and analyzed. (G) H&E staining to show vessels in area II of flaps in the Control and MET groups (original magnifcation×200; scale bar, 50μm). (H) Histogram of percentage of MVDs in each group. (I) IHC for CD34 to present vessels of area II in the Control group and the MET group (original magnifcation×200; scale bar, 50μm). (J) Histogram of percentage of CD34-positive vessels in each group. Values are expressed as means± SEM, n= 6 per group. *p<0.05 and **p< 0.01, vs. Control group.
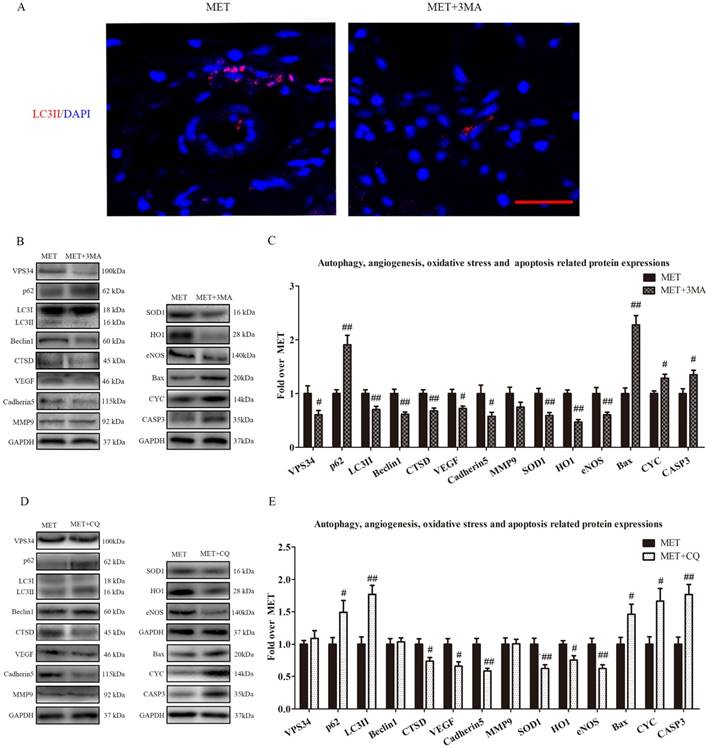
Inhibition of autophagy reverses the effects of MET on angiogenesis, apoptosis and oxidative stress
To verify that autophagy was the primary mechanism underlying MET activity, we next examined the effects of MET on random-pattern skin flaps treated with the autophagy inhibitors 3MA or CQ. Then the levels of autophagy, angiogenesis, apoptosis, and oxidative stress were compared among the MET-, MET+3MA- and MET+CQ-treated groups. As shown in Fig. 6A, the number of LC3II-positive cells was decreased in the dermis of MET+3MA mice compared with those receiving MET alone. Western blotting revealed the expressions of Beclin1, LC3II, VPS34, CTSD were lower in the MET+3MA group than in the MET group (p = 0.002, p = 0.008, p = 0.037, and p = 0.005, respectively; Fig. B&C), with a higher level of p62 in the MET+3MA group (p = 0.001; Fig. B&C); the expression of VEGF, Cadherin 5, eNOS, SOD1, and HO1 was significantly downregulated in the MET+3MA group (p = 0.011, p = 0.034, p = 0.008, p = 0.004, and p < 0.001; respectively; Fig. 6B&C), whereas that the expression of CYC, Bax, and CASP3 were remarkably upregulated in the MET+3MA group (p = 0.008, p < 0.001, and p = 0.017, respectively; Fig. 6B&C), compared with the MET group; but MMP9 expression was not different between the two groups (p = 0.121; Fig. 6B&C). As shown in Fig. 6 C&D, Western blotting showed that the CQ treatment significantly increased the expression of LC3II and p62 (p = 0.001 and p = 0.042, respectively), and decreased CTSD expression (p = 0.027), in the MET+CQ group, but not changed the levels of Beclin1 and VPS34 (p = 0.739 and p = 0.522, respectively); CQ also downregulated the level of VEGF, Cadherin 5, eNOS, SOD1, and HO1 (p = 0.011, p = 0.002, p = 0.006, p = 0.009, and p = 0.017, respectively), and upregulated the expression of CYC, Bax, and CASP3 (p = 0.015, p = 0.035 and p = 0.002, respectively), but not changed the expression of MMP9 (p = 0.952) in MET+CQ group.
MET promotes angiogenesis in random skin flaps. (A&C) IHC for VEGF and Cadherin 5 expressions in the ischemic flaps of the Control and MET groups (original magnification × 200; scale bar, 50μm). (B&D) The optical density values of VEGF and Cadherin 5 were quantified and analyzed in each group. (E-G) Western blotting for VEGF, MMP9, and Cadherin 5 expressions in the ischemic flaps of the Control and MET groups. The gels have been run under the same experimental conditions, and cropped blots are used here. (H-J) Optical density values of VEGF, MMP9, and Cadherin 5 were quantified and analyzed in each group. Values are shown as means± SEM, n= 6 per group. *p< 0.05 and **p< 0.01, vs. Control group.
MET reduces apoptosis in random skin flaps. (A) IHC for CASP3 expression in the ischemic flaps of the Control and MET groups (original magnification × 200; scale bar, 50μm). (B) The optical density values of CASP3 were quantified and analyzed in each group. (C-E) Western blotting for Bax, CYC, and CASP3 expressions in the ischemic flaps of the Control and MET groups. The gels have been run under the same experimental conditions, and cropped blots are used here. (F-H) Optical density values of Bax, CYC and CASP3 were quantified and analyzed in each group. Values are presented as means± SEM, n= 6 per group. *p< 0.05 and **p< 0.01, vs. Control group.
MET inhibits oxidative stress in random skin flaps. (A) IHC for SOD1 expression in the ischemic flaps of the Control and MET groups (original magnification × 200; scale bar, 50μm). (B) The optical density values of SOD1 were quantified and analyzed in each group. (C-E) Western blotting for eNOS, SOD1, and HO1 expressions in the ischemic flaps of the Control and MET groups. The gels have been run under the same experimental conditions, and cropped blots are used here. (F-H) Optical density values of eNOS, SOD1, and HO1 were quantified and analyzed in each group. Values are exhibited as means± SEM, n= 6 per group. **p< 0.01, vs. Control group.
MET activates autophagy in random skin flaps. (A) Autophagosomes (red) in cells in Area II of flaps in the Control and MET groups by Immunofluorescence staining for LC3II (scale bar, 25μm). (B) IHC for CTSD expression in the ischemic flaps of the Control and MET groups (original magnification × 200; scale bar, 50μm). (C) The optical density values of CTSD were quantified and analyzed in each group. (D&E) Western blotting for Beclin1, LC3II, p62, VPS34, and CTSD expressions in the ischemic flaps of the Control and MET groups. The gels have been run under the same experimental conditions, and cropped blots are used here. (F) Optical density values of Beclin1, LC3II, p62, VPS34, and CTSD were quantified and analyzed in each group. Values are expressed as means± SEM, n= 6 per group. *p< 0.05 and **p< 0.01, vs. Control group.
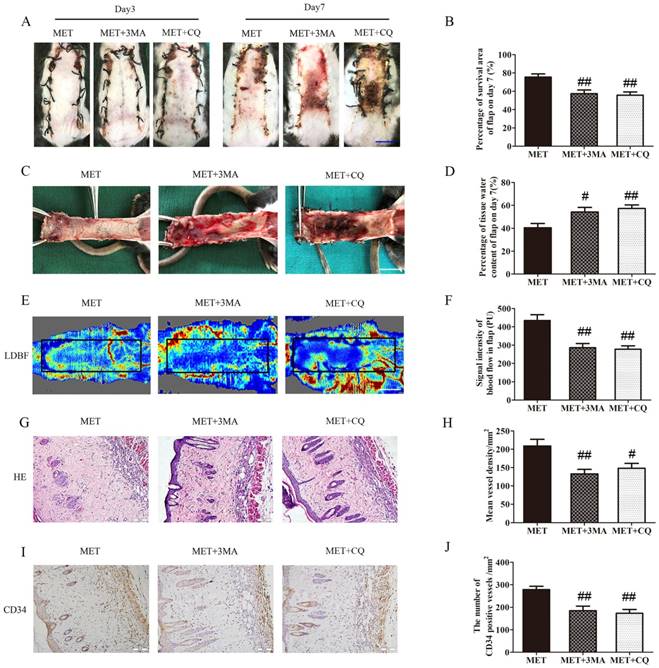
Inhibition of autophagy reverses the effects of MET on flap vitality
The effects of 3MA or CQ on the vitality of the flaps treated with MET were further examined in the next step. As shown in Fig. 7A&B, there was no significant difference in the flap survival area among the MET, MET+3MA and MET+CQ groups on POD 3; on POD 7, the flap survival area was decreased in both MET+3MA and MET+CQ groups compared with the MET group (57.44 ± 3.92%, 55.75 ± 3.52% and 75.43 ± 3.54%, respectively; p = 0.003 and p = 0.002, respectively;). In the inner side of flap, edema with subcutaneous venous blood congestion was more apparent in the MET+3MA and MET+CQ groups, compared with the MET group (Fig. 7C). After data quantification, the water content of tissue was higher in both MET+3MA and MET+CQ groups than in the MET group (54.29 ± 3.96%, 57.30 ± 3.14% and 40.50 ± 3.69%, respectively; p = 0.016 and p = 0.005, respectively; Fig. 7D). Furthermore, from observation of LDBF images, both MET+3MA and MET+CQ groups showed less obvious signal intensity of blood flow than the MET group (Fig. 7E). After data quantification, the signal intensity blood of flow was significantly weaker in the MET+3MA and MET+CQ groups (286.27 ± 22.81PU, and 277.82 ± 18.57PU, respectively) than the MET group (434.42 ± 32.18PU; p = 0.001 and p = 0.001, respectively; Fig. 7F). The H&E staining observation showed that the MET+3MA and MET+CQ groups (132.83 ± 12.34/mm2 and 148.03 ± 13.35/mm2) generated a smaller number of microvessels compared with the MET group (209.00 ± 18.26/mm2; p = 0.003 and p = 0.011, respectively; Fig. 7G&H). Finally, IHC showed that the number of CD34-positive vessels was also fewer in the MET+3MA and MET+CQ groups (184.72 ± 20.50/mm2 and 173.03 ± 17.09/mm2) than the MET group (278.35 ± 14.69/mm2; p = 0.002 and p = 0.001, respectively; Fig. 7I&J).
Inhibition of autophagy reverses the effects of MET on angiogenesis, apoptosis and oxidative stress. (A) Autophagosomes (red) in cells in Area II of flaps in the MET and MET+3MA groups by Immunofluorescence staining for LC3II (scale bar, 25μm). (B&D) The expressions of autophagy-related protein VPS34, p62, LC3II, Beclin1, and CTSD; angiogenesis-related protein VEGF, Cadherin 5, and MMP9; oxidative stress-related protein SOD1, HO1, and eNOS and apoptosis-related protein Bax, CYC, and CASP3 in each group, were assessed by Western blotting. The gels have been run under the same experimental conditions, and cropped blots are used here. (C&E) Optical density values of VPS34, p62, LC3II, Beclin1, CTSD, VEGF, Cadherin 5, MMP9, SOD1, HO1, eNOS, Bax, CYC, and CASP3 expressions in the two groups. Values are expressed as means± SEM, n= 6 per group. #p< 0.05 and ##p< 0.01, vs. MET group.
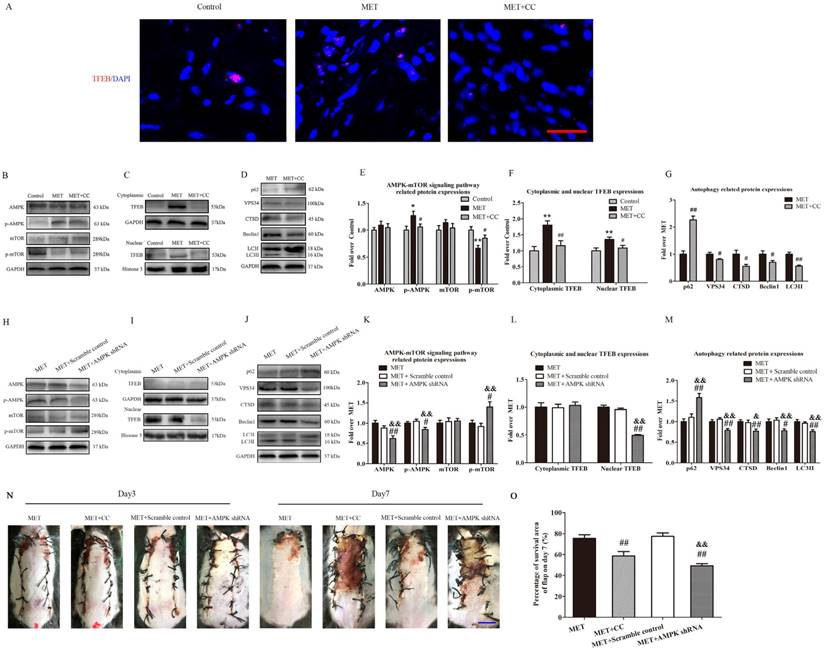
MET activates autophagy via the AMPK-mTOR-TFEB signaling pathway
To investigate the role of the AMPK-mTOR-TFEB signaling pathway in MET-associated autophagy, the MET-treated flaps were treated with CC or AMPK shRNA AAV vector to the AMPK-mTOR-TFEB signaling pathway, and then detected for levels of autophagy and flap vitality. As shown in Fig. 8A, TFEB (red) translocation to nuclear (blue) in cells of flaps was increased in the MET group, compared with the Control group; the CC treatment decreased the nuclear translocation of TFEB in the MET+CC group. Western blotting revealed that the levels of phosphorylated AMPK and nuclear TFEB were upregulated (p = 0.016, p = 0.008), whereas that phosphorylated mTOR was downregulated (p = 0.001) in the MET group compared with the Control group; the CC treatment reversed the effects of MET on AMPK and mTOR phosphorylation and TFEB nuclear translocation (p = 0.049, p = 0.041, p = 0.039; Fig. 8B&E; C&F). Moreover, as shown in Fig. 8 D&G, Western blotting showed the expression of Beclin1, LC3II, VPS34, CTSD was lower in the MET+CC group than in the MET group (p = 0.049, p < 0.001, p = 0.03, and p = 0.018, respectively), with a higher level of p62 in the MET+CC group (p < 0.001). These data suggested MET activated AMPK-mTOR-TFEB signaling pathway in ischemic flaps, and the CC-induced inhibition of the signaling pathway activation resulted in the downregulation of autophagy in the MET+CC group. On the other hand, AAV- AMPK shRNA injection significantly decreased AMPK phosphorylation and TFEB nuclear translocation, and increased mTOR phosphorylation in flaps with the treatment of MET (p = 0.037, p < 0.001, p = 0.011; Fig. 8H&K; I&L). In addition, expressions of Beclin1, LC3II, VPS34, CTSD were lower in the MET+AMPK shRNA group than in the MET group (p = 0.013, p = 0.001, p = 0.003, and p = 0.008, respectively), with a higher level of p62 (p < 0.001, Fig. 8J&M). And there is no significant difference of AMPK-mTOR-TFEB signaling pathway and autophagy- related proteins expression between the MET group and the MET+Scramble control group (Fig. 8 H-M). Finally, the vitality of random skin flap was assessed in each group. As shown in Fig. 8M&N, there was no significant difference in the flap survival area among the MET, MET+CC, MET+Scramble control, and MET+AMPK shRNA groups on POD 3; on POD 7, the flap survival area was decreased in both MET+CC and MET+AMPK shRNA groups compared with the MET group (58.55±10.50% and 49.26±5.14%, respectively; p = 0.002 and p < 0.001, respectively); There was no apparent difference of the flap survival area between the MET group and the MET+Scramble control group on POD 7.
Inhibition of autophagy reverses the effects of MET on flap vitality. (A) Digital photographs of flaps of the MET, MET+3MA and MET+CQ groups on POD 3 and POD 7 (scale bar, 1 cm). (B) The percentages of survival area in the MET group, the MET+3MA group and the MET+CQ group were quantified and analyzed. (C) Digital photographs of the inner side of flaps in the MET, MET+3MA and MET+CQ groups on POD7 (scale bar, 1 cm). (D) Histogram of percentage of tissue water content in each group. (E) Full field LDBF images of flaps in each group on POD 7 (scale bar, 1 cm). (F) The signal intensity of blood flow of flaps was quantified and analyzed. (G) H&E staining to show vessels in area II of flaps in the MET, MET+3MA and MET+CQ groups (original magnifcation×200; scale bar, 50μm). (H) Histogram of percentage of MVDs in each group. (I) IHC for CD34 to present vessels of area II in the MET, MET+3MA and MET+CQ groups (original magnifcation×200; scale bar, 50μm). (J) Histogram of percentage of CD34-positive vessels in each group. Values are expressed as means± SEM, n= 6 per group. #p<0.05 and ##p< 0.01, vs. MET group.
MET activates autophagy via AMPK-mTOR-TFEB signaling pathway. (A) Nuclear translocation of TFEB (red) in cells of flaps in the Control, MET and MET+CC groups by Immunofluorescence staining for TFEB (scale bar, 25μm). (B-D) The expressions of AMPK-mTOR-TFEB signaling pathway-related protein AMPK, p-AMPK, mTOR, p-mTOR, cytoplasmic TFEB, nuclear TFEB in the Control, MET and MET+CC groups, and autophagy-related protein p62, VPS34, CTSD, Beclin1, and LC3II in the MET and MET+CC groups, as assessed by Western blotting. The gels have been run under the same experimental conditions, and cropped blots are used here. (E-G) Optical density values of AMPK, p-AMPK, mTOR, p-mTOR, cytoplasmic TFEB, nuclear TFEB, p62, VPS34, CTSD, Beclin1, and LC3II expressions in each group. (H-J) The expressions of AMPK, p-AMPK, mTOR, p-mTOR, cytoplasmic TFEB, nuclear TFEB, p62, VPS34, CTSD, Beclin1, and LC3II in the MET, MET+Scramble control and MET+AMPK shRNA groups, as assessed by Western blotting. The gels have been run under the same experimental conditions, and cropped blots are used here. (K-M) Optical density values of AMPK, p-AMPK, mTOR, p-mTOR, cytoplasmic TFEB, nuclear TFEB, p62, VPS34, CTSD, Beclin1, and LC3II expressions in each group. (N) Digital photographs of flaps of the MET, MET+CC, MET+Scramble control and MET+AMPK shRNA groups on POD 3 and POD 7 (scale bar, 1 cm). (O) The percentages of survival area in the MET, MET+CC, MET+Scramble control and MET+AMPK shRNA groups were quantified and analyzed. Values are expressed as means± SEM, n= 6 per group. *p< 0.05 and **p< 0.01, vs. Control group; #p< 0.05 and ##p< 0.01, vs. MET group; &p< 0.05 and &&p< 0.01, vs. MET+Scramble control group.
Discussion
MET is a common oral hypoglycemic agent used to treat type 2 diabetes and prevent vascular complications [15, 16]. Recent studies examining the therapeutic effects of MET have demonstrated its efficacy in a wide range of conditions, including diabetic cardiomyopathy [17], acute hyperglycemia-chemical hypoxia [20, 21], and spinal cord injury [18]. MET treatment has also been shown to improve flap survival in a random skin flap model [22]; however, the mechanisms underlying these effects remain poorly understood. The data presented here showed that MET promotes the survival of random skin flaps via increased neovascularization of the affected areas, driven by increases autophagy, while inhibiting oxidative stress and apoptosis. Furthermore, our study found that MET induced autophagy via its regulation of the AMPK-mTOR-TFEB signaling pathway.
MET promoted angiogenesis by augmenting the expression of VEGFA in an acute hyperglycemia-chemical hypoxia model [20, 21], as well as in mice after middle cerebral artery occlusion via regulation of the AMPK-eNOS signaling pathway [32]. In our study, hematoxylin and eosin and IHC staining for CD34 revealed a remarkably increased number of microvessels in the dermis of random skin flaps treated with MET. Further analysis using LDBF demonstrated that the blood flow of flaps was improved in the MET group. These findings indicated that treatment with MET increases the survival of flaps by elevating the number of microvessels and restoring blood supply. Angiogenesis involves the destruction of pre-existing cell connections, mitosis, sprouting of endothelial cells, and maturation of new capillaries. Throughout the process of angiogenesis, VEGF serves as the initiating signal and induces endothelial cell sprouting [33]. MMP9 stimulates the release of VEGF and contributes to the angiogenic process [34]. Cadherin 5 plays an important role in migration and positional changes of angiogenic endothelial cells [35]. Our data showed strong inductions of MMP9, VEGF, and Cadherin 5 protein expression in response to MET treatment. Moreover, increased expression of Cadherin 5 in dermal flaps demonstrated by Western blotting was validated by IHC. Together, these data show that MET enhances the survival and neovascularization of random skin flaps via increased expression of MMP9, VEGF and Cadherin 5.
Generation of random-pattern skin flaps creates a region of acute ischemia, with the resulting ischemia-induced oxidative stress and cell apoptosis closely related to flap survival [4, 36, 37]. Previous studies have shown that MET protects the fatty liver from acute oxidative stress-related mitochondrial injury and cell death via three contributory mechanisms: increased antioxidant enzyme activity, decreased mitochondrial ROS production in complex I, and reduced post-ischemic inflammation [38]. Ischemic damage and oxidative stress lead to significant changes in gene expression [39], including increases in ischemia-related genes, such as HO-1 and eNOS, and oxidative stress-related genes, such as SOD1. Upregulation of these genes significantly attenuates ischemic and oxidative conditions and is necessary for antioxidant activity [39-42]. Western blotting demonstrated increases in SOD1, eNOS, and HO-1 levels in the dermis of random skin flaps by MET treatment. Increased SOD1 expression in ischemic flaps was also evident in IHC analyses. Taken together, these data showed that treatment with MET improves the survival of random skin flaps by inhibiting oxidative stress.
Oxidative stress plays a vital role in the progression of ischemia-reperfusion injuries, which can result in extensive cell apoptosis if left untreated [37]. MET has been shown to protect cells from apoptosis in a variety of ischemic diseases such as acute hyperglycemia-chemical hypoxia, ischemic brain damage, and stroke [21, 43, 44]. Ischemia-reperfusion injury-induced apoptosis ordinarily accompanies high levels of ROS production [45]. Excessive ROS levels can increase the permeability of the inner mitochondrial membrane and disturb the balance of Bcl-2-like anti-apoptotic factors and Bax-like pro-apoptotic factors, resulting in the release of pro-apoptotic proteins, such as CYC, into the intermembrane space [46]. Release of CYC activates CASP3, which can induce apoptosis [47]. Accordingly, we examined Bax, CYC, and CASP3 levels to assess the extent of cell apoptosis occurring in response to various treatments. Western blotting showed that Bax, CYC, and CASP3 expression was inhibited in the MET group. IHC analysis of CASP3 expression also showed a significant decrease in the MET group relative to the Control group, indicating that MET attenuates the level of apoptosis in ischemic flaps.
Macroautophagy (hereafter named autophagy) plays a vital role in cell survival and maintenance by degrading and recycling damaged organelles, toxic agents and long-lived proteins through an autophagosomal - lysosomal pathway [48]. Autophagy is particularly important in vascular diseases, having been shown to preserve the angiogenic function of endothelial progenitor cells, as well as reduce cardiac hypertrophy, apoptosis, and fibrosis in a mouse model of chronic ischemic remodeling [9, 49]. Recently, MET was shown to induce autophagy as well as protect against ischemic brain injury [50], suggesting potential benefits for use in random-pattern skin flaps, although such an application had not been investigated. To our knowledge, this is the first report of MET-mediated activation of autophagy in the context of random skin flaps. Upon initiation of autophagy, the levels of the autophagosomal proteins Beclin1, VPS34, and LC3II and the autolysosome-related protein CTSD are increased, while that of autophagic substrate protein p62, used to monitor autophagic flux, is decreased [51-53]. In the present study, immunofluorescence and IHC revealed significant increases in LC3II and CTSD levels in the MET group. Similar increases in Beclin1, VPS34, LC3II, and CTSD, and a decrease in p62, protein levels in the MET group relative to the Control group were also evident by Western blotting. Together, these observations suggest that MET upregulates autophagy in random-pattern skin flaps.
Like many cellular functions, autophagy is a double-edged process. On the one hand, activation of autophagy plays an essential role in the prevention of disease, particularly vascular conditions [54]. However, on the other hand, autophagy serves as a distinct cell death mechanism independent of apoptosis [55]. To verify the role of MET-induced autophagy in random skin flaps, the autophagy inhibitor 3MA or CQ was administered together with MET. Inhibition of autophagy by 3MA or CQ reversed the effects of MET on survival and blood flow, as well as the MET-induced decrease in tissue edema and increase in microvessel density. Moreover, both 3MA and CQ decreased the levels of angiogenesis-related proteins VEGF and Cadherin 5, suggesting MET is able to promote angiogenesis in random skin flaps by activating autophagy. Previous study of disc degeneration showed that activation of autophagy alleviates mitochondrial dysfunction and cell apoptosis under oxidative stress [13]. Interestingly, both 3MA and CQ significantly increased the apoptosis-related proteins Bax, CYC, and CASP3, while reducing the oxidative stress-related proteins eNOS, HO-1, and SOD1. These changes suggest that MET inhibits cell apoptosis and alleviates oxidative stress by activating autophagy. In the context of flap survival, MET-induced autophagy enhances the survival of random skin flaps via promotion of angiogenesis and suppression of oxidative stress and apoptotic processes.
AMPK is a key regulator of autophagy activated under the conditions of nutrient or energy starvation or oxidative stress; and in this system, changes in cellular energy result in a decrease in the ATP/ADP ratio and activation of AMPK [56, 57]. The activation of AMPK can repress mTOR activity both directly, by phosphorylating raptor, and indirectly, via activation of tuberous sclerosis complex-2 [58]. This inhibition of mTOR signaling by AMPK further enhances autophagy [59]. From a mechanistic standpoint, when activation of mTOR is suppressed, TFEB is activated by dephosphorylation and then translocated into the nucleus [23]. Numerous lysosomal and autophagy genes, such as LC3, Beclin1, autophagy-related gene 5 (ATG5), CTSD, and lysosomal-associated membrane proteins 2 (LAMP2), with one or more 10-base-pair motifs (GTCACGTGAC) termed as CLEAR elements are recognized by the TFEB which in turn promotes gene transcription [24, 25]. Therefore, the nucleus translocation of TFEB results in upregulation of autophagy level in cells [60].
MET acts as a regulator of autophagy by activating AMPK-dependent suppression of mTOR in endotoxemia-induced acute lung injury and diabetic cardiomyopathy [27, 61]. While MET can induce autophagy via activation of the AMPK-mTOR pathway, the effects of this on TFEB nuclear translocation has not been examined. Our data suggest that MET treatment upregulated both autophagy and AMPK-mTOR-TFEB signaling. Suppression of AMPK activity by CC or AMPK shRNA AAV vector was used to verify whether the AMPK-mTOR-TFEB signaling pathway also mediates the autophagic effects stimulated by MET. Interestingly, we found that either CC or AMPK shRNA AAV vector significantly attenuated the effects of MET on mTOR signaling and even reversed the effects of MET on TFEB nuclear translocation, consistent with the decrease in the nuclear translocation of TFEB in the dermis. Furthermore, both CC and AMPK shRNA AAV vector inhibited MET-induced autophagy as evidenced by a decrease in the expression of autophagy-related proteins Beclin1, VPS34, LC3II, and CTSD and an increase in autophagic substrate protein p62 expression, suggesting that the AMPK-mTOR-TFEB pathway may be important in enhancing MET-induced autophagy. Thus, the mechanistic studies presented here demonstrated that MET regulated autophagy and TFEB nuclear translocation via the AMPK-mTOR signaling pathway.
Taken together, the present study found that treatment with MET enhances the survival of random-pattern skin flaps by alleviating oxidative stress, promoting angiogenesis, and inhibiting apoptosis. These effects were associated with activation of autophagy via the AMPK-mTOR-TFEB signaling pathway.
Acknowledgements
This work was supported by grants from Natural Science Foundation of China (No. 81601705 to Kailiang Zhou, No.81873942 to Weiyang Gao, No. 81801930 to Jian Ding, No. 81572227 and No. 81873992 to Huazi Xu); Zhejiang Provincial Medicine and Health Technology Project (No. 2017KY472 to Kailiang Zhou); Science Technology Department of Zhejiang Province Project (No. 2017C33024 to Dingsheng Lin); Wenzhou Science and Technology Bureau Foundation (No.2016Y0350 to Jian Ding, Y20160039 to Dingsheng Lin).
Author contributions
Hongqiang Wu and Jian Ding wrote the manuscript text. Hongqiang Wu, Shihen Li, Jinti Lin, and Renhao Jiang, Chen Lin prepared figures and collected samples. Chenglong Xie, Dingsheng Lin analyzed data, Weiyang Gao and Kailiang Zhou designed the experiment. Huazi Xu, Weiyang Gao, Kailiang Zhou revised manuscript. All authors reviewed and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Basu G, Downey H, Guo S, Israel A, Asmar A, Hargrave B. et al. Prevention of distal flap necrosis in a rat random skin flap model by gene electrotransfer delivering VEGF165plasmid. The Journal of Gene Medicine. 2014;16:55-65
2. Wang C, Cai Y, Zhang Y, Xiong Z, Li G, Cui L. Local injection of deferoxamine improves neovascularization in ischemic diabetic random flap by increasing HIF-1alpha and VEGF expression. PloS one. 2014;9:e100818
3. Myers MB, Cherry G. Causes of necrosis in pedicle flaps. Plastic & Reconstructive Surgery. 1968;42:43
4. Zhou KL, Zhang YH, Lin DS, Tao XY, Xu HZ. Effects of calcitriol on random skin flap survival in rats. Scientific reports. 2016;6:18945
5. Seyed Jafari SM, Shafighi M, Beltraminelli H, Geiser T, Hunger RE, Gazdhar A. Improvement of Flap Necrosis in a Rat Random Skin Flap Model by In Vivo Electroporation-Mediated HGF Gene Transfer. Plast Reconstr Surg. 2017;139:1116e-27e
6. Zhang P, Feng J, Liao Y, Cai J, Zhou T, Sun M. et al. Ischemic flap survival improvement by composition-selective fat grafting with novel adipose tissue derived product - stromal vascular fraction gel. Biochemical and biophysical research communications. 2018;495:2249-56
7. Deheng C, Kailiang Z, Weidong W, Haiming J, Daoliang X, Ningyu C. et al. Salidroside Promotes Random Skin Flap Survival in Rats by Enhancing Angiogenesis and Inhibiting Apoptosis. Journal of reconstructive microsurgery. 2016;32:580-6
8. Chen L, Zhou K, Chen H, Li S, Lin D, Zhou D. Calcitriol promotes survival of experimental random pattern flap via activation of autophagy. American journal of translational research. 2017;9:3642-53
9. Dai X, Zeng J, Yan X, Lin Q, Wang K, Chen J. et al. Sitagliptin-mediated preservation of endothelial progenitor cell function via augmenting autophagy enhances ischaemic angiogenesis in diabetes. J Cell Mol Med. 2018;22:89-100
10. An Y, Liu WJ, Xue P, Ma Y, Zhang LQ, Zhu B. et al. Autophagy promotes MSC-mediated vascularization in cutaneous wound healing via regulation of VEGF secretion. Cell death & disease. 2018;9:58
11. Xu X, Shao N, Duan X, Zhang X, Zhang Y. Tacrolimus alleviates Ox-LDL damage through inducing vascular endothelial autophagy. Eur Rev Med Pharmacol Sci. 2018;22:3199-206
12. Lee J, Ha T, Park J, Lee G. Anti-cell death engineering of CHO cells: co-overexpression of Bcl-2 for apoptosis inhibition, Beclin-1 for autophagy induction. Biotechnol Bioeng. 2013;110:2195-207
13. Zhang B, Xu L, Zhuo N, Shen J. Resveratrol protects against mitochondrial dysfunction through autophagy activation in human nucleus pulposus cells. Biochemical and biophysical research communications. 2017;493:373-81
14. Li Z, Wang G, Feng D, Zu G, Li Y, Shi X. et al. Targeting the miR-665-3p-ATG4B-autophagy axis relieves inflammation and apoptosis in intestinal ischemia/reperfusion. Cell death & disease. 2018;9:483
15. Abbasi F, Chu J, McLaughlin T, Lamendola C, Leary E, Reaven G. Effect of metformin treatment on multiple cardiovascular disease risk factors in patients with type 2 diabetes mellitus. Metab Clin Exp. 2004;53:159-64
16. Katakam P, Ujhelyi M, Hoenig M, Miller A. Metformin improves vascular function in insulin-resistant rats. Hypertension. 2000;35:108-12
17. Wang G, Bi Y, Liu X, Zhao Y, Han J, Wei M. et al. Autophagy was involved in the protective effect of metformin on hyperglycemia-induced cardiomyocyte apoptosis and Connexin43 downregulation in H9c2 cells. Int J Med Sci. 2017;14:698-704
18. Guo Y, Wang F, Li H, Liang H, Li Y, Gao Z. et al. Metformin Protects Against Spinal Cord Injury by Regulating Autophagy via the mTOR Signaling Pathway. Neurochem Res. 2018
19. Yan Q, Han C, Wang G, Waddington J, Zheng L, Zhen X. Activation of AMPK/mTORC1-Mediated Autophagy by Metformin Reverses Clk1 Deficiency-Sensitized Dopaminergic Neuronal Death. Mol Pharmacol. 2017;92:640-52
20. Bakhashab S, Ahmed FW, Schulten HJ, Bashir A, Karim S, Al-Malki AL. et al. Metformin improves the angiogenic potential of human CD34(+) cells co-incident with downregulating CXCL10 and TIMP1 gene expression and increasing VEGFA under hyperglycemia and hypoxia within a therapeutic window for myocardial infarction. Cardiovascular diabetology. 2016;15:27
21. Bakhashab S, Ahmed F, Schulten HJ, Ahmed FW, Glanville M, Al-Qahtani MH. et al. Proangiogenic Effect of Metformin in Endothelial Cells Is via Upregulation of VEGFR1/2 and Their Signaling under Hyperglycemia-Hypoxia. International journal of molecular sciences. 2018:19
22. Taleb S, Moghaddas P, Rahimi Balaei M, Taleb S, Rahimpour S, Abbasi A. et al. Metformin improves skin flap survival through nitric oxide system. J Surg Res. 2014;192:686-91
23. Young N, Kamireddy A, Van Nostrand J, Eichner L, Shokhirev M, Dayn Y. et al. AMPK governs lineage specification through Tfeb-dependent regulation of lysosomes. Genes Dev. 2016;30:535-52
24. Lu H, Fan Y, Qiao C, Liang W, Hu W, Zhu T. et al. TFEB inhibits endothelial cell inflammation and reduces atherosclerosis. Science signaling. 2017:10
25. Pastore N, Vainshtein A, Klisch TJ, Armani A, Huynh T, Herz NJ. et al. TFE3 regulates whole-body energy metabolism in cooperation with TFEB. EMBO molecular medicine. 2017;9:605-21
26. Civiletto G, Dogan SA, Cerutti R, Fagiolari G, Moggio M, Lamperti C. et al. Rapamycin rescues mitochondrial myopathy via coordinated activation of autophagy and lysosomal biogenesis. EMBO molecular medicine. 2018;10:e8799
27. Wu K, Tian R, Huang J, Yang Y, Dai J, Jiang R. et al. Metformin alleviated endotoxemia-induced acute lung injury via restoring AMPK-dependent suppression of mTOR. Chem Biol Interact. 2018;291:1-6
28. Lee MS, Ahmad T, Lee J, Awada HK, Wang Y, Kim K. et al. Dual delivery of growth factors with coacervate-coated poly(lactic-co-glycolic acid) nanofiber improves neovascularization in a mouse skin flap model. Biomaterials. 2017;124:65-77
29. Gao Y, Wang N, Li RH, Xiao YZ. The Role of Autophagy and the Chemokine (C-X-C Motif) Ligand 16 During Acute Lung Injury in Mice. Medical science monitor: international medical journal of experimental and clinical research. 2018;24:2404-12
30. Lin C-W, Zhang H, Li M, Xiong X, Chen X, Chen X. et al. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. Journal of Hepatology. 2013;58:993-9
31. Wang L, Jin Z, Wang J, Chen S, Dai L, Lin D. et al. Detrimental effect of Hypoxia-inducible factor-1alpha-induced autophagy on multiterritory perforator flap survival in rats. Scientific reports. 2017;7:11791
32. Liu Y, Tang G, Zhang Z, Wang Y, Yang G-Y. Metformin promotes focal angiogenesis and neurogenesis in mice following middle cerebral artery occlusion. Neuroscience Letters. 2014;579:46-51
33. Bousseau S, Vergori L, Soleti R, Lenaers G, Carmen Martinez M, Andriantsitohaina R. Glycosylation as new pharmacological strategies for diseases associated with excessive angiogenesis. Pharmacol Ther. 2018
34. Bellafiore M, Battaglia G, Bianco A, Farina F, Palma A, Paoli A. The involvement of MMP-2 and MMP-9 in heart exercise-related angiogenesis. Journal of translational medicine. 2013;11:283
35. Cao J, Ehling M, März S, Seebach J, Tarbashevich K, Sixta T. et al. Polarized actin and VE-cadherin dynamics regulate junctional remodelling and cell migration during sprouting angiogenesis. Nature Communications. 2017:8
36. Kailiang Z, Yihui Z, Dingsheng L, Xianyao T. Effects of Muscone on Random Skin Flap Survival in Rats. Journal of reconstructive microsurgery. 2016;32:200-7
37. Yin Z, Ren H, Liu L, Chen W, Gan C, Jiao H. et al. Thioredoxin Protects Skin Flaps from Ischemia-Reperfusion Injury: A Novel Prognostic and Therapeutic Target. Plast Reconstr Surg. 2016;137:511-21
38. Cahova M, Palenickova E, Dankova H, Sticova E, Burian M, Drahota Z. et al. Metformin prevents ischemia reperfusion-induced oxidative stress in the fatty liver by attenuation of reactive oxygen species formation. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2015;309:G100-G11
39. Goldenberg-Cohen N, Dadon-Bar-El S, Hasanreisoglu M, Avraham-Lubin BC, Dratviman-Storobinsky O, Cohen Y. et al. Possible neuroprotective effect of brimonidine in a mouse model of ischaemic optic neuropathy. Clin Exp Ophthalmol. 2009;37:718-29
40. Biswas C, Shah N, Muthu M, La P, Fernando AP, Sengupta S. et al. Nuclear heme oxygenase-1 (HO-1) modulates subcellular distribution and activation of Nrf2, impacting metabolic and anti-oxidant defenses. The Journal of biological chemistry. 2014;289:26882-94
41. Katusic ZS, Austin SA. Endothelial nitric oxide: protector of a healthy mind. European heart journal. 2014;35:888-94
42. Dong J, Wong SL, Lau CW, Lee HK, Ng CF, Zhang L. et al. Calcitriol protects renovascular function in hypertension by down-regulating angiotensin II type 1 receptors and reducing oxidative stress. European heart journal. 2012;33:2980-90
43. Ge X, Zhu G, Geng D, Zhang H, He J, Guo A. et al. Metformin protects the brain against ischemia/reperfusion injury through PI3K/Akt1/JNK3 signaling pathways in rats. Physiol Behav. 2017;170:115-23
44. Mohammad Alizadeh E, Mahdavi M, Jenani Fard F, Chamani S, Farajdokht F, Karimi P. Metformin protects PC12 cells against oxygen-glucose deprivation/reperfusion injury. Toxicol Mech Methods. 2018:1-20
45. Zweier JL, Talukder MA. The role of oxidants and free radicals in reperfusion injury. Cardiovasc Res. 2006;70:181-90
46. Chen J, Wang YX, Dong MQ, Zhang B, Luo Y, Niu W. et al. Reoxygenation Reverses Hypoxic Pulmonary Arterial Remodeling by Inducing Smooth Muscle Cell Apoptosis via Reactive Oxygen Species-Mediated Mitochondrial Dysfunction. J Am Heart Assoc. 2017:6
47. Zhuang Z, Lian P, Wu X, Shi B, Zhuang M, Zhou R. et al. Abate Cytochrome C induced apoptosome to protect donor liver against ischemia reperfusion injury on rat liver transplantation model. American journal of translational research. 2016;8:1738-47
48. Parzych KR, Klionsky DJ. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxidants & Redox Signaling. 2014;20:460-73
49. Sciarretta S, Yee D, Nagarajan N, Bianchi F, Saito T, Valenti V. et al. Trehalose-Induced Activation of Autophagy Improves Cardiac Remodeling After Myocardial Infarction. J Am Coll Cardiol. 2018;71:1999-2010
50. Zhu J, Liu K, Huang K, Gu Y, Hu Y, Pan S. et al. Metformin Improves Neurologic Outcome Via AMP-Activated Protein Kinase-Mediated Autophagy Activation in a Rat Model of Cardiac Arrest and Resuscitation. J Am Heart Assoc. 2018:7
51. Miki Y, Tanji K, Mori F, Utsumi J, Sasaki H, Kakita A. et al. Alteration of Upstream Autophagy-Related Proteins (ULK1, ULK2, Beclin1, VPS34 and AMBRA1) in Lewy Body Disease. Brain Pathology. 2016;26:359-70
52. Bain H, Davidson Y, Robinson A, Ryan S, Rollinson S, Richardson A. et al. The role of lysosomes and autophagosomes in frontotemporal lobar degeneration. Neuropathol Appl Neurobiol. 2018
53. Jiang P, Mizushima N. LC3- and p62-based biochemical methods for the analysis of autophagy progression in mammalian cells. Methods (San Diego, Calif). 2015;75:13-8
54. Fan Y, Lu H, Liang W, Garcia-Barrio M, Guo Y, Zhang J. et al. Endothelial TFEB (Transcription Factor EB) Positively Regulates Postischemic Angiogenesis. Circ Res. 2018;122:945-57
55. Aki T, Funakoshi T, Unuma K, Uemura K. Impairment of autophagy: from hereditary disorder to drug intoxication. Toxicology. 2013;311:205-15
56. Huang Q, Wu Y, Tan H, Ong C, Shen H. A novel function of poly(ADP-ribose) polymerase-1 in modulation of autophagy and necrosis under oxidative stress. Cell Death Differ. 2009;16:264-77
57. Kim J, Kundu M, Viollet B, Guan K-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature Cell Biology. 2011;13:132-41
58. R C, LJ T, AR T, RS S, CL W, GN W. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E2950-7
59. Yang Z, Klionsky D. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124-31
60. E G, W O, M H, J S, L S. TFEB-mediated activation of the lysosome-autophagy system affects the transduction efficiency of adeno-associated virus 2. Virology. 2017;510:1-8
61. Xie Z, He C, Zou M. AMP-activated protein kinase modulates cardiac autophagy in diabetic cardiomyopathy. Autophagy. 2011;7:1254-5
Author contact
![]() Corresponding authors: Prof. Weiyang Gao, Department of Orthopaedics, The Second Affiliated Hospital and Yuying Children's Hospital of Wenzhou Medical University, 109 West Xueyuan Road, Wenzhou 325000, Zhejiang, China; Tel: +86-577-88002812; E-mail: weiyanggaoicom; Dr. Kailiang Zhou, Department of Orthopaedics, The Second Affiliated Hospital and Yuying Children's Hospital of Wenzhou Medical University, 109 West Xueyuan Road, Wenzhou 325000, Zhejiang, China; Tel: +86-577-88002815; E-mail: zhoukailiangedu.cn
Corresponding authors: Prof. Weiyang Gao, Department of Orthopaedics, The Second Affiliated Hospital and Yuying Children's Hospital of Wenzhou Medical University, 109 West Xueyuan Road, Wenzhou 325000, Zhejiang, China; Tel: +86-577-88002812; E-mail: weiyanggaoicom; Dr. Kailiang Zhou, Department of Orthopaedics, The Second Affiliated Hospital and Yuying Children's Hospital of Wenzhou Medical University, 109 West Xueyuan Road, Wenzhou 325000, Zhejiang, China; Tel: +86-577-88002815; E-mail: zhoukailiangedu.cn
Received 2018-8-5
Accepted 2018-11-30
Published 2019-1-1