ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2019; 15(2):369-385. doi:10.7150/ijbs.28422 This issue Cite
Research Paper
ING4 suppresses hepatocellular carcinoma via a NF-κB/miR-155/FOXO3a signaling axis
Fuliang Qian1*, Qingqing Hu2*, Yali Tian3*, Jie Wu2, Dapeng Li2, Min Tao2, Lei Qin4 ![]() , Bairong Shen1
, Bairong Shen1 ![]() , Yufeng Xie2
, Yufeng Xie2 ![]()
1. Center for Systems Biology, Soochow University, Suzhou 215006, China
2. Department of Oncology, the First Affiliated Hospital of Soochow University, Suzhou 215006, China
3. Department of Oncology, Suzhou Science & Technology Town Hospital, Suzhou 215153, China
4. Department of General Surgery, the First Affiliated Hospital of Soochow University, Suzhou 215006, China
*These authors contributed equally to this work.
Abstract

The tumor suppressor ING4 has been shown to be reduced in human HCC. The alteration of ING4 contributes to HCC progression. However, its effect in HCC and the potential mechanism is largely unclear. Herein, we found that downregulation of ING4 in HCC tumor tissues was closely associated with cancer staging, tumor size and vascular invasion. Lentivirus-mediated ING4 overexpression significantly inhibited proliferation, migration and invasion, and induced cell cycle G1 phase arrest and apoptosis in MHCC97H human HCC cells. Moreover, overexpression of ING4 dramatically suppressed MHCC97H tumor cell growth and metastasis to lung in vivo in athymic BALB/c nude mice. Mechanistic studies revealed that overexpression of ING4 markedly increased expression of FOXO3a both at the mRNA and protein level as well as enhanced nuclear level and transcriptional activity of FOXO3a in MHCC97H tumor cells. In addition, ING4 repressed transcriptional activity of NF-κB and expression of miR-155 targeting FOXO3a. Knockdown of ING4 exhibited opposing effects in MHCC97L human HCC cells. Interestingly, knockdown of FOXO3a attenuated not only ING4-elicited tumor suppression but also ING4-mediated regulatory effect on FOXO3a downstream targets, confirming that FOXO3a is involved in ING4-directed tumor-inhibitory effect in HCC. Overexpression of miR-155 attenuated ING4-induced upregulation of FOXO3a, whereas inhibition of miR-155 blunted ING4 knockdown-induced reduction of FOXO3a. Furthermore, inhibition of NF-κB markedly impaired ING4 knockdown-induced upregulation of miR-155 and downregulation of FOXO3a. Taken together, our study provided the first compelling evidence that ING4 can suppress human HCC growth and metastasis to a great extent via a NF-κB/miR-155/FOXO3a pathway.
Keywords: hepatocellular carcinoma (HCC), inhibitor of growth 4 (ING4), forkhead box 3a (FOXO3a), nuclear factor-kappa B (NF-κB), microRNA-155 (miRNA/miR-155)
Introduction
HCC, a major type of primary liver cancer, is the fifth most commonly diagnosed human malignancy and the second leading cause of cancer-related mortality in the world[1]. The incidence of HCC increased dramatically worldwide especially in China during the last 20 years[2,3]. Despite recent improvements in the diagnosis and treatment strategies for HCC, the prognosis of HCC patient is currently still far from satisfactory. The poor prognosis largely results from late detection of HCC and lack of effective therapies for advanced and recurred HCC. Alteration of signaling pathways induced by dysfunction of oncogenes and antioncogenes is one of the major pathogenic mechanisms of HCC[4]. Hence, a detailed understanding of molecular mechanisms involved in the pathogenesis and progression of HCC is urgently needed, which may help to identify potential therapeutic targets and develop effective therapies for HCC.
FOXO subfamily of forkhead transcription factors, including FOXO1a/FOXO1/FKHR, FOXO3a/FOXO3/FKHRL1, FOXO4/AFX and FOXO6 four members, has been shown to participate in various biological processes such as cell cycle control, differentiation, apoptosis, stress response, DNA repair, metabolism and longevity[5,6]. Among them, FOXO3a has been considered a bona fide tumor suppressor. Emerging evidence has demonstrated that the function of FOXO3a is inactivated by oncogenic stimuli in a broad spectrum of cancers, which is associated with carcinogenesis and cancer progression[7-9]. Activation of FOXO3a can trigger tumor cell cycle arrest and apoptosis via transcriptional activation or repression of downstream target genes implicated in regulating cell cycle and apoptosis[8]. FOXO3a also suppresses tumor angiogenesis by downregulating VEGF[10,11] and eNOS[12]. Moreover, FOXO3a reverses tumor cell EMT and inhibits cancer metastasis through directly transactivating E-cadherin[13] as well as indirectly downregulating EMT-TFs[13-15] and β-catenin signaling[16]. Interestingly, various anticancer therapeutics have been found to achieve their therapeutic effects through activation of FOXO3a[8,9,17]. FOXO3a aberration confers cancer therapy resistance and activation of FOXO3a can be effective in enhancing therapy sensitization[9,17]. Therefore, FOXO3a represents a potential therapeutic target in human cancers.
The ING family contains ING1, ING2, ING3, ING4 and ING5 five members, belonging to the family of type Ⅱ tumor suppressor genes[18,19]. ING4, an important member of ING family, has been found to function as a powerful tumor suppressor[20,21]. Accumulating data has shown that ING4 function is frequently downregulated or lost by genetic/epigenetic mechanisms and ubiquitin-mediated proteolysis in various types of human cancers, leading to cancer initiation and progression[18-22]. Restoration of ING4 inhibits tumor cell growth by induction of cell cycle arrest and apoptosis in a p53-dependent or -independent manner[20,21,23-25]. ING4 also induces tumor growth suppression via promoting toxic autophagy[26]. Of note, ING4 can enhance tumor cell sensitivity to chemotherapy[27-29] and radiotherapy[30,31]. Additionally, ING4 suppresses tumor cell spreading, migration and invasion through interacting with liprin α1[32] and repressing MMP-2, MMP-9 and EMT-TF Snail1[25,33,34]. Furthermore, ING4 attenuates tumor angiogenesis by downregulation of IL-6 and IL-8 proangiogenic factors via inhibition of NF-κB[35] and HIF-1α pathways[36]. Thus, ING4 exhibits tumor-suppressive activities via multiple pathways.
Both FOXO3a and ING4 are downregulated in human HCC and negatively correlate with histological grade, tumor size and prognosis[37,38]. However, the relationship of ING4 and FOXO3a, whether FOXO3a is involved in ING4-induced tumor inhibition, and related regulatory mechanism in HCC is unclear. In this study, we assessed the ING4 expression in human HCC cells and clinical tissues, examined the effects of ING4 on HCC cell growth and metastasis by lentivirus-mediated ING4 overexpression and knockdown, and elucidated the molecular mechanism.
Materials and Methods
HCC tissue samples
A total of 78 pairs of paraffin-embedded HCC tissues and adjacent non-tumor liver tissues (at least 6 cm away from the tumor) were obtained from randomly selected 78 patients who have undergone surgery and received no chemotherapy or radiotherapy at the First Affiliated Hospital of Soochow University (Suzhou, Jiangsu, China) and the Third Affiliated Hospital of the Second Military Medical University (Shanghai, China) from January 2013 to December 2015. The research was approved by the Ethics Committee of the First Affiliated Hospital of Soochow University. Pathological staging were reviewed independently by two experienced pathologists according to the 7th edition of the AJCC Staging Manual[39]. The clinicopathological characteristics of these patients were summarized based upon medical history.
Cell lines, vectors, reagents and mice
The HL-7702 human liver cell line and the 293T human embryonic kidney cell line were purchased from the Cell Bank, Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). The MHCC97H and MHCC97L human HCC cell lines were kindly provided by Dr. Yang Xu (Liver Cancer Institute and Zhongshan Hospital, Fudan University, Shanghai, China). The above-mentioned cells were grown in DMEM (HyClone, Logan, UT, USA) supplemented with 10% FBS (Gibco, Gaithersburgh, MD, USA) and antibiotics (100 U/ml penicillin-streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). The blank lentiviral plasmid pLenti6.3/IRES/GFP (BSD) and the lentiviral packing plasmids including pLP1, pLP2 and VSVG were purchased from Novobio Science & Technology (Shanghai, China). The recombinant lentiviral plasmid pLenti6.3/ING4/IRES/GFP harboring a humanized ING4 gene was constructed in our laboratory[34]. The scrambled siRNA GFP lentivirus (puromycin) (LVshcontrol) and the ING4-set siRNA/shRNA/RNAi GFP lentivirus (puromycin) (LVshING4) were purchased from abm (Richmond, BC, Canada). Lipofectamine 2000 was purchased from Invitrogen (Carlsbad, CA, USA). The polybrene and the enhanced infection solution were purchased from GeneChem (Shanghai, China). The BSD was purchased from Yeasen (Shanghai, China). The puromycin was purchased from Gibco (Gaithersburgh, MD, USA). The CCK-8 was purchased from Dojindo Molecular Technologies (Gaithersburg, MD, USA). The MammoCultTM basal medium and the MammoCultTM proliferation supplement were purchased from STEMCELL Technologies (Vancouver, BC, Canada). The PI cell cycle detection kit was purchased from Nanjing KeyGen Biotechnology (Nanjing, Jiangsu, China). The Annexin-V-PE/7-AAD apoptosis detection kit was purchased from BD Biosciences (Franklin Lakes, NJ, USA). The 24-well Transwell (pore size: 8 μm), the Matrigel and the PVDF membrane were purchased from Millipore (Billerica, MA, USA). The MiniBEST universal RNA extraction kit was purchased from TaKaRa (Dalian, Liaoning, China). The RevertAid RT reverse transcription kit was purchased from Thermo Fisher Scientific (Waltham, MA, USA). The FastStart Universal SYBR Green Master (Rox) was purchased from Roche Applied Science (Penzberg, Upper Bavaria, Germany). The miScript Ⅱ RT kit, the miScript SYBR Green PCR kit and the HiPerFect transfection reagent were purchased from Qiagen (Hilden, Nordrhein-Westfalen, Germany). The primers were purchased from Sangon Biotechnology (Shanghai, China). The mammalian cell lysis kit and the nuclear extraction kit were purchased from Sigma (St Louis, MO, USA). The BCA protein assay kit was purchased from Beyotime Biotechnology (Beijing, China). The SuperEnhanced chemiluminescence detection kit was purchased from Applygen Technology (Beijing, China). Luciferase Assay System was purchased from Promega (Madison, WI, USA). The DAB and BCIP/NBT substrates and the HRP-conjugated anti-rabbit IgG (Cat. No. BM3894) secondary antibody were purchased from Boster Biological Technology (Wuhan, Hubei, China). The rabbit anti-ING4 (aa162-195) (Cat. No. LS-C344210) antibody was purchased from LifeSpan BioSciences (LSBio) (Seattle, WA, USA). The rabbit anti-FOXO3a (clone No. D19A7) (Cat. No. 12829), anti-p27 (clone No. D69C12) (Cat. No. 3686), anti-Cyclin D1 (clone No. 92G2) (Cat. No. 2978), anti-Bim (clone No. C34C5) (Cat. No. 2933), anti-Puma (clone No. D30C10) (Cat. No. 12450), anti-FasL (Cat. No. 4273), anti-TRAIL (clone No. C92B9) (Cat. No. 3219), anti-β-catenin (clone No. D10A8) (Cat. No. 8480), anti-NF-κB p65 (clone No. D14E12) (Cat. No. 8242), anti-NF-κB p105/p50 (clone No. D4P4D) (Cat. No. 13586), anti-β-actin (clone No. 13E5) (Cat. No. 4970) and anti-Histone H3 (clone No. 3H1) (Cat. No. 9717) primary antibodies and the HRP-conjugated anti-rabbit IgG (Cat. No. 7074) secondary antibody were purchased from Cell Signaling Technology (CST) (Danvers, MA, USA). The AP-conjugated anti-DIG antibody (Cat. No. 200-052-156) was purchased from Jackson ImmunoResearch (West Grove, PA, USA). The FHRE-Luc luciferase reporter plasmid was purchased from addgene (Cambridge, MA, USA). The TCF/LEF1-Luc and NF-κB-Luc luciferase reporter plasmids were purchased from Genomeditech (Shanghai, China). The 5'-DIG- and 3'-DIG-labeled miR-155 miRCURY LNA Detection probe was purchased from Exiqon (Vedbaek, Denmark). The FOXO3a (FKHRL1) siRNA and the control siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The miR-155 mimics and miRNA mimics NC and the miR-155 inhibitor and miRNA inhibitor NC were purchased from RiboBio (Guangzhou, Guangdong, China). NF-κB inhibitor JSH-23 was purchased from Selleck Chemicals (Houston, TX, USA). The 4-week-old female athymic BALB/c nude mice were purchased from Shanghai Laboratory Animal Center (Shanghai, China) and maintained in the animal facility at Soochow University (Suzhou, Jiangsu, China) according to the animal research committee's guidelines of Soochow University.
Preparation of lentiviruses
The LVING4 lentivirus expressing humanized ING4 and GFP and the LV blank lentivirus (used as a control) were prepared as previously reported[34]. The biological titer (TU/ml) of lentiviruses was assessed by calculating the number of GFP-expressing 293T cells after lentiviral infection under fluorescence microscopy. The ratio of infectious lentivirus (TU) to target cells is called MOI.
Generation of stable cell lines
The MHCC97H human HCC cells were infected with LVING4 or LV at a MOI of 20 plus 10 μg/ml of polybrene in enhanced infection solution in a 24-well culture plate. The MHCC97L human HCC cells were infected with LVshING4 or LVshcontrol as described above. 72 hours after infection, the MHCC97H cells were selected with 10 μg/ml of BSD, whereas the MHCC97L cells were selected with 2 μg/ml of puromycin. The transgene efficiency of BSD-resistant MHCC97H including MHCC97H-LVING4 (termed MHCC97H-ING4) and MHCC97H-LV (termed MHCC97H-mock, used as a control), and puromycin-resistant MHCC97L including MHCC97L-LVshING4 (termed MHCC97L-shING4) and MHCC97L-LVshcontrol (termed MHCC97L-shcontrol, used as a control) cell derivatives were detected by fluorescence microscopic and flow cytometric analysis of GFP, respectively. The overexpression and knockdown efficiency of ING4 was further analyzed by Western blot.
CCK-8 assay
The ING4-overexpressed MHCC97H or ING4-silenced MHCC97L HCC cells and the corresponding control cells were seeded at a density of 1×104 cells/200 μl culture medium per well in 96-well culture plates. Cell proliferation ability was evaluated at day 1, 2, 3 and 4 after cell culture using a CCK-8 according to the company's protocols. The OD of each well was measured at 450 nm in an automatic microplate reader. Cell growth curves were plotted with OD value vs culture time.
Colony formation assay
The ING4-overexpressed MHCC97H or ING4-silenced MHCC97L HCC cells and the corresponding control cells were dispensed into 6-well culture plates at a density of 200 cells/2 ml culture medium per well. After 2 weeks' incubation, the colonies were fixed with 4% paraformaldehyde and stained by crystal violet. The clonogenic ability of tumor cells was then analyzed.
Tumor sphere assay
The ING4-overexpressed MHCC97H or ING4-silenced MHCC97L HCC cells and the corresponding control cells were seeded at a density of 4×103 cells/2 ml complete MammoCultTM medium (MammoCultTM basal medium plus 10% MammoCultTM proliferation supplement) per well into 6-well ultra-low adherent plates. One week after incubation, the number of tumor spheres was counted.
Cell cycle analysis
The ING4-overexpressed MHCC97H or ING4-silenced MHCC97L HCC cells and the corresponding control cells were cultured, harvested, washed and fixed in ice-cold 70% ethanol at 4 oC overnight. They were then washed, treated with 500 U/ml RNase A at 37 oC for 30 min and stained with 50 μg/ml PI in the dark for 30 min. Finally, the cells were washed and the cell cycle profiles were analyzed by flow cytometry.
Annexin-V-PE/7-AAD apoptosis assay
The ING4-overexpressed MHCC97H or ING4-silenced MHCC97L HCC cells and the corresponding control cells were cultured, collected, washed and resuspended in 1×Annexin V binding buffer at a density of 1×105 cells/100 μl. The 100 μl each of above cell suspension were then incubated with Annexin V-PE (5 μl) and 7-AAD (5 μl) at room temperature for 15 minutes and analyzed by flow cytometry.
Transwell migration and invasion assays
In a Transwell migration assay, the ING4-overexpressed MHCC97H or ING4-silenced MHCC97L HCC cells and the corresponding control cells (2×104 cells/100 μl serum-free DMEM medium) were directly added to the upper chamber of 24-well/8 μm pore size Transwell filters. In a Transwell invasion assay, the above cells (2×105 cells/100 μl serum-free DMEM medium) were seeded onto the upper chamber of Transwell filters which were precoated with Matrigel. The lower chamber was filled with 500 μL of DMEM medium supplemented with 20% FBS. After 24 hours of incubation, tumor cells on the upper surface of filter were removed and cells migrating or invading into the bottom side of insert were fixed by 4% paraformaldehyde, stained with crystal violet, photographed and counted in 5 randomly selected ×200 high-power fields by other investigators that were blinded to the group allocation. The migratory and invasive ability of tumor cells was then analyzed.
Xenograft mouse models
The BALB/c nude mice were subcutaneously inoculated with ING4-overexpressed MHCC97H or ING4-silenced MHCC97L HCC cells and corresponding control cells (2×106 cells/100 μl PBS/mouse) (6 mice/group). Tumor growth in vivo was monitored by other investigators that were blinded to the group allocation. Tumor volume was measured with a caliper and calculated by the formula, tumor size=ab2/2, where a is the larger of the two dimensions and b is the smaller. The tumor-bearing mice were sacrificed 4 weeks after tumor cell inoculation and the xenografted tumors were then removed and weighted. In another in vivo lung metastasis model, the nude mice (6 mice/group) were intravenously injected with the above-mentioned cells (2×106 cells/200 μl PBS/mouse) through tail vein. The mice were killed 4 weeks after tumor cell injection and the lung tissues were removed, fixed in 10% neutral formalin and embedded in paraffin. The lung metastasis nodules of HCC were analyzed by HE staining. The tumor metastasis nodules were then counted by other investigators that were blinded to the group allocation at 5 randomly selected ×40 low-power fields of each section under microscopy.
Western blot analysis
For preparation of total proteins, the HL-7702, MHCC97H and MHCC97L cells; and the ING4-overexpressed MHCC97H or ING4-silenced MHCC97L HCC cells and corresponding control cells (1×107 cells/1 ml lysis buffer) were lysed in lysis buffer supplemented with a protease inhibitor cocktail using a mammalian cell lysis kit. The nuclear proteins derived from ING4-overexpressed MHCC97H or ING4-silenced MHCC97L HCC cells and corresponding control cells were isolated using a nuclear extraction kit. The protein concentrations were measured by a BCA protein assay kit. The lysates were loaded (50 µg/lane) and separated in 12% SDS-PAGE and transferred to PVDF membranes. The membranes were then blocked with 5% fat-free milk in PBS containing 0.1% Tween-20. The membranes transferred by total lysates derived from HL-7702, MHCC97H and MHCC97L cells were subsequently incubated with a primary antibody anti-ING4 (1:1000) or anti-β-actin (1:1000) (used as a loading control) at 4 oC overnight. The membranes transferred by total lysates derived from ING4-overexpressed MHCC97H or ING4-silenced MHCC97L HCC cells and corresponding control cells were incubated with primary antibodies including anti-ING4 (1:1000), anti-FOXO3a (1:1000), anti-p27 (1:1000), anti-Cyclin D1 (1:1000), anti-Bim (1:1000), anti-Puma (1:1000), anti-FasL (1:1000), anti-TRAIL (1:1000), anti-β-catenin (1:1000), anti-NF-κB p65 (1:1000), anti-NF-κB p105/p50 (1:1000) and anti-β-actin (1:1000) (used as a loading control), respectively. The membranes transferred by nuclear lysates were incubated with primary antibodies including anti-FOXO3a (1:1000), anti-β-catenin (1:1000), anti-NF-κB p65 (1:1000), anti-NF-κB p105/p50 (1:1000) and Histone H3 (1:1000) (used as a loading control), respectively. Followed by incubation with a secondary antibody HRP-conjugated anti-rabbit IgG (CST, 1:3000) for 1 hour, the protein signaling on the membranes was detected using a SuperEnhanced chemiluminescence detection kit and the protein bands were then visualized after their exposure to X-ray films.
Real-time qRT-PCR analysis
The total RNAs of ING4-overexpressed MHCC97H or ING4-silenced MHCC97L HCC cells and corresponding control cells (2×106 cells) were extracted using a MiniBEST universal RNA extraction kit. The RNAs were reversely transcribed to first-strand cDNAs by a RevertAid RT reverse transcription kit and a miScript Ⅱ RT kit, respectively. The RevertAid RT-derived cDNAs were then subjected to qPCR analysis of transcriptional expression of FOXO3a (FOXO3a-F: 5'-TCT TCA GGT CCT CCT GTT CCT G-3' and FOXO3a-R: 5'-GGA AGC ACC AAA GAA GAG AGA AG-3' for amplifying 132 bp) using a FastStart Universal SYBR Green Master (Rox) following manufacturer's instructions. The β-actin (β-actin-F: 5'-CTC ACC ATG GAT GAT GAT ATC GC-3' and β-actin-R: 5'-AGG AAT CCT TCT GAC CCA TGC-3' for amplifying 163 bp) was used an internal control. The miScript Ⅱ RT-derived cDNAs were subjected to qPCR analysis of expression of miR-96, miR-155, miR-182, miR-551b and miR-592 (U6 used as an internal control) using a miScript SYBR Green PCR kit following company's protocols. The expression level was normalized to the internal control and calculated by a 2-∆∆CT method as previously described[40].
Luciferase reporter assays
The ING4-overexpressed MHCC97H or ING4-silenced MHCC97L HCC cells and corresponding control cells were transfected with FHRE-Luc, TCF/LEF1-Luc and NF-κB-Luc luciferase reporter plasmids using Lipofectamine 2000, respectively. 48 hours after transfection, the cells were harvested, lysed and luciferase activity was detected by a Luciferase Assay System according to manufacturer's instructions. The effect of ING4 on the transcriptional activity of FOXO3a, β-catenin and NF-κB in HCC cells was then analyzed.
FOXO3a siRNA knockdown assay
The MHCC97H-ING4 HCC cells were transfected with 100 nM human FOXO3a siRNA (siFOXO3a) or control siRNA (sicontrol) using a HiPerFect transfection reagent following company's instructions. After 48 hours of transfection, the siFOXO3a- or sicontrol-transfected MHCC97H-ING4 cells and the untransfected MHCC97H-ING4 or MHCC97H-mock cells were then subjected to in vitro and in vivo functional assays as well as Western blot analysis of FOXO3a, p27, Cyclin D1, Bim, Puma, FasL, TRAIL and β-catenin.
MiR-155 mimics/inhibitor assay
The MHCC97H-ING4 HCC cells were transfected with 200 nM miR-155 mimics or miRNA mimics NC using a HiPerFect transfection reagent following company's protocols. The MHCC97L-shING4 HCC cells were transfected with 200 nM miR-155 inhibitor or miRNA inhibitor NC. After 48 hours of transfection, the miR-155 mimics- or miR-155 mimics NC-transfected MHCC97H-ING4 cells and the untransfected MHCC97H-ING4 or MHCC97H-mock cells; and the miR-155 inhibitor- or miR-155 inhibitor NC-transfected MHCC97L-shING4 cells and the untransfected MHCC97L-shING4 or MHCC97L-shcontrol cells were then subjected to qRT-PCR and Western blot analysis of FOXO3a.
NF-κB inhibition assay
The MHCC97L-shING4 HCC cells were pretreated with NF-κB inhibitor JSH-23 (10 μM) or DMSO without JSH-23 in culture medium for 1 hour. Then the JSH-23-treated and DMSO-treated MHCC97L-shING4 cells and the untreated MHCC97L-shING4 and MHCC97L-shcontrol cells were cultured in fresh culture medium. After another 48 hours of incubation, the above cells were subjected to qRT-PCR analysis of miR-155 and FOXO3a, respectively.
Immunohistochemistry and in situ hybridization analyses
The above formalin-fixed and paraffin-embedded HCC and adjacent non-tumor liver tissue samples were cut into 4 μm-thick sections, respectively. The sections were then deparaffinized, rehydrated, microaved in 0.01 M citrate buffer (pH=6.0) for antigen retrieval, treated with 3% H2O2 for quenching of endogenous peroxidase activity, and then blocked with goat serum. Subsequently, the sections were incubated with rabbit anti-ING4 (1:25), anti-FOXO3a (1:200) or anti-NF-κB p65 (1:100) primary antibody in a humidity chamber overnight at 4 oC. HRP-conjugated anti-rabbit IgG secondary antibody (Boster, 1:1000) was then incubated for 1 hour at room temperature and immunostaining signal was detected by DAB. Finally, the slides were counterstained with HE and coverslipped. The percentage of positive tumor cells and the intensity of immunostaining were used to gain the IHC scoring, respectively. The percentage of positive tumor cells was assigned to 5 categories: ≤5% (0), 5-25% (1), 25-50% (2), 50-75% (3), and ≥75% (4). The staining intensity was scored as follows: negative (0), weak (1), moderate (2), and strong (3). The percentage of positive tumor cells and the staining intensity were then added to produce a weighted score for each specimen. The IHC scores were finally grouped as (-), 0-1; (+), 2-3; (++), 4-5; and (+++), 6-7. It was considered as high expression when the final weighted scores were ≥4 (++, +++). In addition, the expression of miR-155 in the sections were analyzed by in situ hybridization using 5'-DIG- and 3'-DIG-labeled miR-155 miRCURY LNA Detection probe (50 nM) according to company's instructions. After hybridization, the sections were incubated with AP-conjugated anti-DIG antibody (1:400) followed by reaction with BCIP/NBT. The signal of hybridization was then analyzed.
Statistical analyses
All statistical analyses were carried out with Mann-Whitney U test, Pearson's χ2 test, Student t test and one-way or two-way repeated measures analysis of variance (ANOVA) using SPSS13.0 software (SPSS, Chicago, IL, USA). A value of p<0.05 was considered statistically significant.
Results
ING4 is reduced in human HCC and correlated with clinicopathological features of HCC
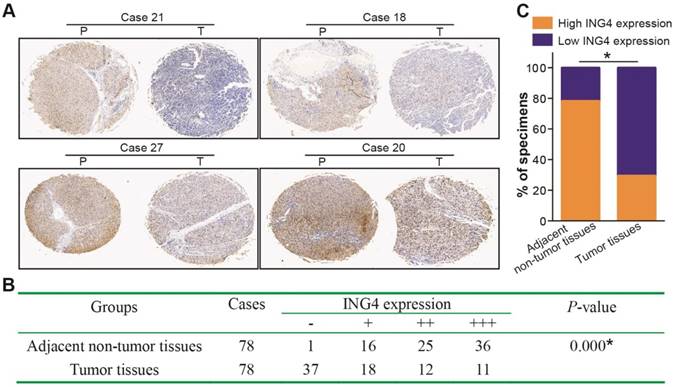
Immunohistochemistry analysis of 78 paired paraffin-embedded human HCC and adjacent non-tumor clinical tissue sections (Figure 1) showed that low expression of ING4 in tumor tissues occurred in 55 cases (37 cases scored “-” and 18 cases scored “+”) (70.5%), whereas high expression of ING4 only occurred in 23 cases (12 cases scored “++” and 11 cases scored “+++”) (29.5%). Among matched adjacent non-tumor tissues, 61 cases (78.2%) displayed high expression of ING4 (25 cases scored “++” and 36 cases scored “+++”), while 17 cases (21.8%) exhibited low expression of ING4 (1 case scored “-” and 16 cases scored “+”). Consistent with a previous report [38], our results demonstrated that the expression of ING4 in HCC tumor tissues was significantly lower than that in adjacent normal tissues (p<0.05). On the basis of ING4 expression in HCC tumor tissues, 78 patients with HCC were divided into two groups: low-expression group (“-” or “+”, n=55) and high-expression group (“++” or “+++”, n=23). To evaluate the clinical significance of ING4 in HCC, the relationship between ING4 expression in HCC tumor tissues and clinicopathological variables was analyzed. As shown in Table 1, ING4 expression was inversely correlated with AJCC cancer staging, tumor size and vascular invasion (p<0.05). Our data indicated that downregulation of ING4 may contribute to human HCC progression and metastasis.
Lentivirus-mediated ING4 overexpression or knockdown in HCC cells
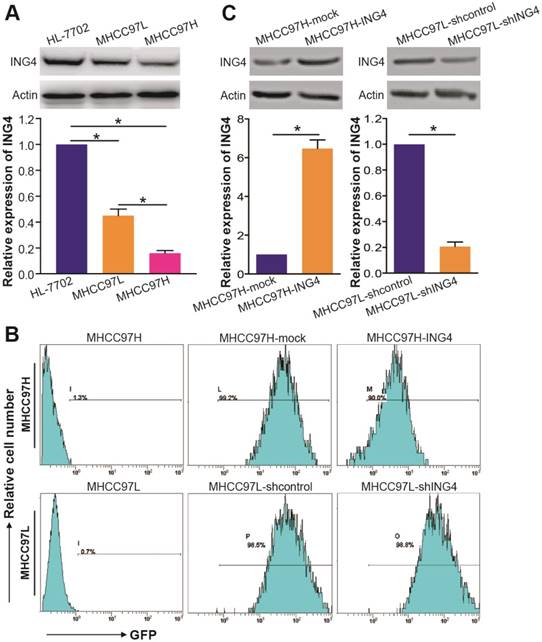
To generate ING4-overexpressed/silenced stable HCC cell lines for functional studies, we measured the expression level of ING4 in MHCC97H and MHCC97L human HCC cell lines. Among them, MHCC97H is a highly malignant and metastatic HCC cell line whose genetic background is similar to a relative low-metastasis HCC cell line MHCC97L[41].
ING4 is downregulated in human HCC clinical tissues. Immunohistochemistry analysis of ING4 in HCC tissue specimens. A. The representative pictures (HCC tumor tissues, Case 21, -; Case 18, +; Case 27, ++; and Case 20, +++) of immunohistochemical staining. T: HCC tumor tissue, and P: adjacent non-tumor tissue. B. The IHC scoring of ING4 in 78 paired HCC tumor and adjacent non-tumor tissues. *, p<0.05 compared with adjacent non-tumor tissue, Mann-Whitney U test, n=78 cases. C. The percentage of high and low ING4 expression in HCC tumor and adjacent non-tumor tissues. *, p<0.05 compared with adjacent non-tumor tissue, Pearson's χ2 test, n=78 cases. Data shown were representative of two independent experiments.
The relationship of ING4 expression with HCC clinicopathological features (Pearson's χ2 test)
| Variables | High ING4 expression (n=23) | Low ING4 expression (n=55) | P-value |
|---|---|---|---|
| Gender | |||
| Female | 11 | 26 | 0.9644 |
| Male | 12 | 29 | |
| Age (years) | |||
| ≤60 | 10 | 30 | 0.3726 |
| >60 | 13 | 25 | |
| HbsAg | |||
| Negative | 8 | 21 | 0.7770 |
| Positive | 15 | 34 | |
| Cirrhosis | |||
| Absent | 9 | 22 | 0.9430 |
| Present | 14 | 33 | |
| Serum AFP (ng/ml) | |||
| ≤20 | 8 | 24 | 0.4685 |
| >20 | 15 | 31 | |
| Tumor size (cm) | |||
| ≤5 | 15 | 20 | 0.0195* |
| >5 | 8 | 35 | |
| Tumor number | |||
| Single | 18 | 43 | 0.9938 |
| Multiple | 5 | 12 | |
| Vascular invasion | |||
| Absent | 16 | 18 | 0.0028* |
| Present | 7 | 37 | |
| TNM stage | |||
| Early (I/II) | 14 | 17 | 0.0137* |
| Late (III/IV) | 9 | 38 |
Compared with HL-7702 immortal non-cancerous human liver cell line control, the expression of ING4 is significantly downregulated in MHCC97H and MHCC97L HCC cell lines (Figure 2A) (p<0.05). Notably, ING4 was detected at a lower level in MHCC97H cell line than MHCC97L cell line (Figure 2A) (p<0.05), which was consistent with the clinical relationship of ING4 expression level and HCC pathological features. Therefore, we employed MHCC97H and MHCC97L HCC cell lines for the following gain-of-function and loss-of-function researches by lentivirus-mediated ING4 overexpression and shRNA knockdown, respectively. Fluorescence microscopic (data not shown) and flow cytometric (Figure 2B) analyses showed that almost all (more than 90%) of the ING4- or ING4 shRNA-expressing lentivirus-transduced and corresponding control lentivirus-transduced MHCC97H or MHCC97L HCC cells exhibited GFP expression, indicating high transgene efficiency. The lentivirus-mediated overexpression or knockdown of ING4 in MHCC97H or MHCC97L HCC cells was then determined by Western blot analysis. As shown in Figure 2C, the MHCC97H HCC cells showed a marked increase in the expression of ING4 after lentivirus-directed ING4 gene transfer, whereas the MHCC97L HCC cells displayed a remarkable reduction in the expression of ING4 after transduced by ING4 shRNA compared with that transduced by shcontrol (p<0.05). Our results revealed that lentivirus-mediated ING4-transgenic MHCC97H and ING4-silenced MHCC97L HCC cell lines were successfully generated.
ING4 inhibits HCC cell growth and induces cell cycle G1 phase arrest and apoptosis
To examine the effect of ING4 on human HCC cell growth, the in vitro proliferation ability of MHCC97H-ING4 vs MHCC97H-mock and MHCC97L-shING4 vs MHCC97L-shcontrol tumor cells was determined by a CCK-8 assay. As shown in Figure 3A, the growth curve in the MHCC97H-ING4 group was much lower than that in the LV-transduced MHCC97H-mock control group (p<0.05), demonstrating that forced expression of ING4 obviously inhibited MHCC97H tumor cell growth in vitro. In contrast, knockdown of ING4 significantly promoted MHCC97L tumor cell growth compared with the shcontrol-transduced MHCC97L-shcontrol control group (Figure 3A) (p<0.05). Consistently, MHCC97H-ING4 cells formed smaller and less colonies (Figure 3B) or tumor spheres (Figure 3C) than MHCC97H-mock cells (p<0.05). The colonies or tumor spheres that grew from MHCC97L-shING4 cells were larger and more than MHCC97L-shcontrol cells (Figure 3B and C) (p<0.05). Our data indicated that ING4 suppresses clonogenicity and self-renewal activity of human HCC cells. To further assess whether the suppressive effect of ING4 on in vitro growth of HCC cells could be reproduced in vivo, we monitored human HCC subcutaneously xenografted tumor growth of MHCC97H-ING4 vs MHCC97H-mock and MHCC97L-shING4 vs MHCC97L-shcontrol in athymic BALB/c nude mice.
Lentivirus-directed ING4 overexpression or knockdown in human HCC cells. A. Western blot analysis of ING4 in HCC cells. The lysates of MHCC97H and MHCC97L HCC cells and HL-7702 normal liver cells were immunoblotted with anti-ING4 or anti-β-actin (a loading control) antibody. The representative pictures of Western blot were shown. *, p<0.05 compared with HL-7702; *, p<0.05 compared with MHCC97L, one-way repeated measures ANOVA, n=6 replicates per sample. B. Flow cytometric analysis of GFP expression. C. Western blot analysis of lentivirus-mediated ING4 overexpression or knockdown. The lysates of MHCC97H-ING4 vs MHCC97H-mock and MHCC97L-shING4 vs MHCC97L-shcontrol HCC cells were immunoblotted with anti-ING4 or anti-β-actin (a loading control) antibody. The representative pictures of Western blot were shown. MHCC97H: *, p<0.05 compared with MHCC97H-mock; MHCC97L: *, p<0.05 compared with MHCC97L-shcontrol, Student t test, n=6 replicates per sample. The expression level of ING4 in these Western blot assays was normalized to β-actin (ING4/β-actin) and then expressed as a ratio or fold of respective control, with 1 being the value for HL-7702, MHCC97H-mock or MHCC97L-shcontrol control. Data shown were representative of three independent experiments.
ING4 suppresses cell growth through inducing G1 phase arrest and apoptosis in human HCC cells. A. CCK-8 assay. MHCC97H: *, p<0.05 compared with MHCC97H-mock at day 2, 3 and 4; MHCC97L: *, p<0.05 compared with MHCC97L-shcontrol at day 2, 3 and 4, respectively, two-way repeated measures ANOVA, n=6 replicates per condition. B. Colony formation assay. The representative pictures of colonies were shown and the relative clonogenic ability was calculated. MHCC97H: *, p<0.05 compared with MHCC97H-mock; MHCC97L: *, p<0.05 compared with MHCC97L-shcontrol, Student t test, n=6 replicates per condition. C. Tumor sphere assay. The representative pictures of tumor spheres were shown and the number of tumor spheres was counted. MHCC97H: *, p<0.05 compared with MHCC97H-mock; MHCC97L: *, p<0.05 compared with MHCC97L-shcontrol, Student t test, n=6 replicates per condition. D-F. Subcutaneous xenograft mouse model. The tumor volume (D) was measured after implantation of tumor cells. MHCC97H: *, p<0.05 compared with MHCC97H-mock at week 2, 3 and 4; MHCC97L: *, p<0.05 compared with MHCC97L-shcontrol at week 2, 3 and 4, respectively, two-way repeated measures ANOVA, n=6 replicates per condition. The xenografted tumors were removed (E) 4 weeks after tumor cell's implantation and tumor weight (F) was then measured. MHCC97H: *, p<0.05 compared with MHCC97H-mock; MHCC97L: *, p<0.05 compared with MHCC97L-shcontrol, Student t test, n=6 replicates per condition. G. Flow cytometric analysis of cell cycle profile. The percentage of each cell cycle (G0/G1, S and G2/M phase) distribution in the total cell population was presented. MHCC97H G0/G1 or S: *, p<0.05 compared with MHCC97H-mock; MHCC97L G0/G1 or S: *, p<0.05 compared with MHCC97L-shcontrol, Student t test, n=6 replicates per condition. H. Flow cytometric analysis of apoptosis. The percentage of apoptotic cells including early and late apoptotic cells in the total cell population was presented. MHCC97H: p<0.05 compared with MHCC97H-mock; MHCC97L (serum-starved): *, p<0.05 compared with MHCC97L-shcontrol, Student t test, n=6 replicates per condition. Data shown were representative of three independent experiments.
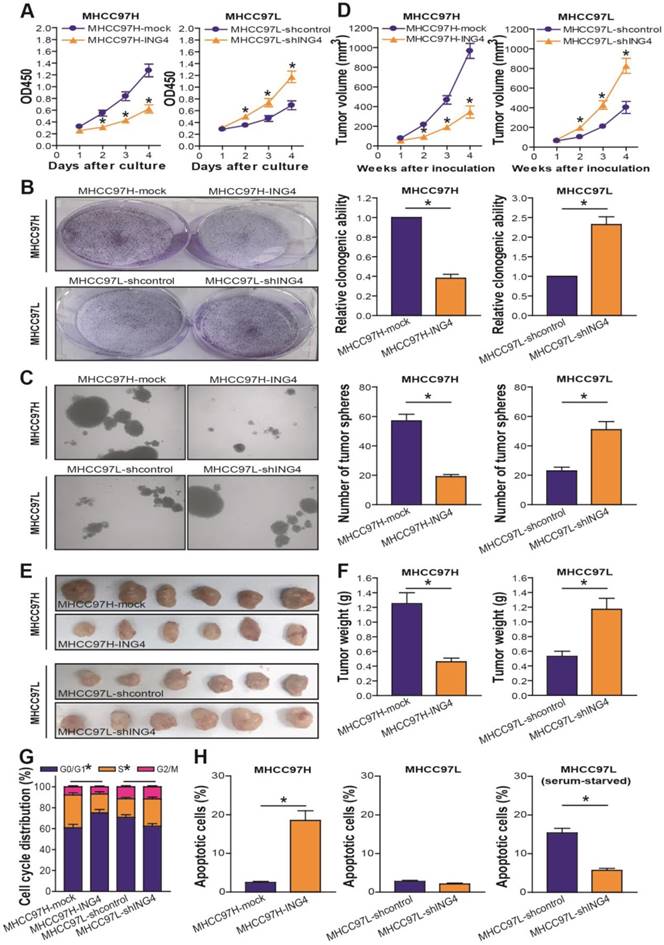
As shown in Figure 3D-F, overexpression of ING4 obviously suppressed MHCC97H tumor cell growth in vivo, whereas knockdown of ING4 remarkably facilitated MHCC97L tumor cell growth (p<0.05). Our data demonstrated that ING4 is also capable of repressing HCC cell growth in vivo. To assess whether ING4 inhibits HCC cell growth via induction of cell cycle alteration and apoptosis, flow cytometric analysis was applied to detect the cell cycle distribution and the apoptotic cells in MHCC97H-ING4 vs MHCC97H-mock and MHCC97L-shING4 vs MHCC97L-shcontrol tumor cells. As shown in Figure 3G, overexpression of ING4 impeded the entry of G1 to S phase and induced G1 phase arrest in MHCC97H tumor cells, whereas knockdown of ING4 induced a reduction of cells on G1 phase as well as an accumulation of cells on S phase in MHCC97L tumor cells (p<0.05). Furthermore, forced expression of ING4 obviously induced apoptosis in MHCC97H tumor cells (Figure 3H) (p<0.05). Additionally, knockdown of ING4 could induce resistance of MHCC97L tumor cells to apoptosis in a serum-starved condition (Figure 3H) (p<0.05). Taking these results together, ING4 can suppress HCC cell growth via inhibition of cell cycle G1 to S phase transition and induction of apoptosis.
ING4 suppresses HCC cell migration, invasion and distant lung metastasis
To analyze the association of ING4 with metastatic potential of HCC cells, Transwell chamber migration and invasion assays were conducted to examine the in vitro migratory and invasive ability of MHCC97H-ING4 vs MHCC97H-mock and MHCC97L-shING4 vs MHCC97L-shcontrol tumor cells. As shown in Figure 4A and B, the migratory capacity of MHCC97H-ING4 tumor cells was dramatically impaired compared with MHCC97H-mock control group (p<0.05). Knockdown of ING4 significantly boosted the migration of MHCC97L tumor cells (Figure 4A and B) (p<0.05).
ING4 inhibits HCC cell migration and invasion in vitro and lung metastasis in vivo. A, B. Transwell migration assay. The representative photos of Transwell migration assay of the indicated cells were shown (A). The relative migratory ability was calculated (B). MHCC97H: *, p<0.05 compared with MHCC97H-mock; MHCC97L: *, p<0.05 compared with MHCC97L-shcontrol, Student t test, n=6 replicates per condition, n=5 observations per replicate. C, D. Transwell invasion assay. The representative photos of Transwell invasion assay of the indicated cells were shown (C). The relative invasive ability was calculated (D). MHCC97H: *, p<0.05 compared with MHCC97H-mock; MHCC97L: *, p<0.05 compared with MHCC97L-shcontrol, Student t test, n=6 replicates per condition, n=5 observations per replicate. E, F. In vivo lung metastasis assay. The representative photos of HE staining of lung tissues were shown (E). The tumor metastasis nodules in the lungs were counted according to HE staining (F). MHCC97H: *, p<0.05 compared with MHCC97H-mock; MHCC97L: *, p<0.05 compared with MHCC97L-shcontrol, Student t test, n=6 replicates per condition, n=5 sections per sample, n=5 observations per section. Data shown were representative of three independent experiments.
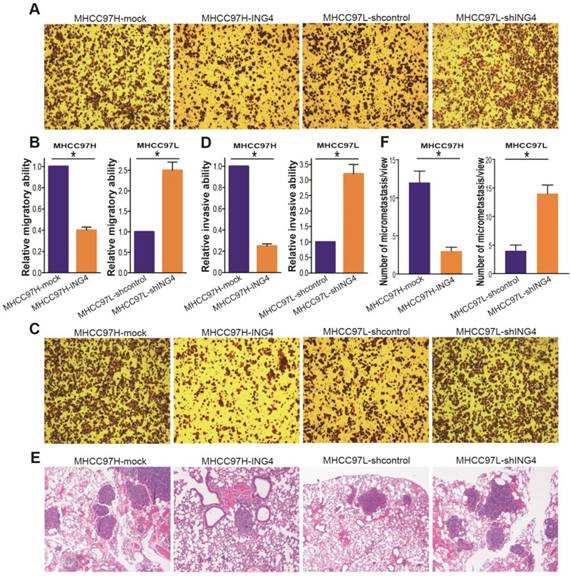
Moreover, lentivirus-mediated ING4 overexpression suppressed MHCC97H tumor cell invasion, whereas its knockdown promoted the process in MHCC97L tumor cells (Figure 4C and D) (p<0.05). To further wonder whether the correlation between ING4 expression and HCC metastatic traits in vitro could be reproduced in vivo, the ING4-overexpressed MHCC97H or ING4-silenced MHCC97L HCC cells and corresponding control cells were injected into the tail vein of athymic BALB/c nude mice. Four weeks after intravenous injection, the lung tissues of mice were removed and the lung metastasis was assessed by HE analysis. As shown in Figure 4E and F, the MHCC97H-ING4 tumor cells exhibited a significant decrease in tumor lung metastasis nodule compared with MHCC97H-mock control cells, while MHCC97L-shING4 tumor cells had much higher metastatic ability in vivo than MHCC97L-shcontrol control cells (p<0.05). These data indicated that ING4 efficiently weakens the metastasis potential of HCC cells.
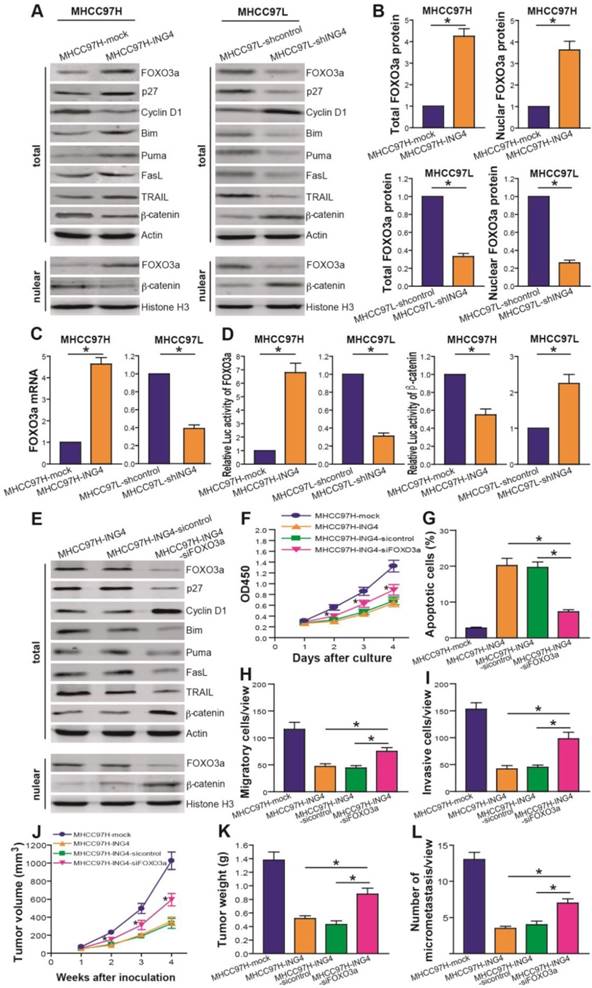
ING4 represses HCC growth and metastasis via upregulation of FOXO3a
It has been generally recognized that FOXO3a functions as an important negative mediator of HCC progression[37,42]. To address the potential mechanism responsible for ING4-induced inhibition of growth and metastasis in HCC cells, we thus asked whether FOXO3a is involved in ING4-mediated tumor-suppressive effects. Western blot analysis (Figure 5A and B) showed that compared to MHCC97H-mock control group, both the total and the nuclear levels of FOXO3a were dramatically increased in MHCC97H-ING4 group (p<0.05). Conversely, the total and nuclear levels of FOXO3a were reduced in MHCC97L-shING4 group compared with MHCC97L-shcontrol control group (Figure 5A and B) (p<0.05). qRT-PCR analysis (Figure 5C) also demonstrated that forced expression of ING4 upregulated the level of FOXO3a mRNA in MHCC97H HCC cells, whereas knockdown of ING4 downregulated it in MHCC97L HCC cells (p<0.05). Luciferase reporter assay further showed that ING4 significantly enhanced the transcriptional activity of FOXO3a (Figure 5D) (p<0.05). In addition, we found that overexpression of ING4 upregulated the expression of FOXO3a's transactive targets[8] such as p27, Bim, Puma, FasL and TRAIL as well as downregulated the expression of FOXO3a's transrepressive target Cyclin D1[8] in MHCC97H HCC cells, whereas knockdown of ING4 displayed opposing regulatory effects in MHCC97L HCC cells (Figure 5A). Moreover, ING4 inhibited the total/nuclear expression (Figure 5A) and transcriptional activity (Figure 5D) of another FOXO3a's target β-catenin[16] in MHCC97H HCC cells, while knockdown of ING4 augmented β-catenin signaling in MHCC97L HCC cells (p<0.05). Our data revealed that ING4 can positively modulate the expression and transcriptional activity of FOXO3a in human HCC cells. To evaluate the role of FOXO3a in ING4-elicted effects, we carried out FOXO3a siRNA knockdown assays in ING4-overexpressed MHCC97H-ING4 HCC cells. Our results demonstrated that knockdown of FOXO3a using siRNA remarkably attenuated the regulatory effects of ING4 on FOXO3a downstream targets in MHCC97H HCC cells (Figure 5E). More importantly, not only did knockdown of FOXO3a markedly impair ING4-triggered apoptosis and inhibition of proliferation, migration and invasion in MHCC97H HCC cells in vitro (Figure 5F-I) (p<0.05), but knockdown of FOXO3a also attenuated ING4-induced inhibition of growth and metastasis in MHCC97H HCC cells in vivo in BALB/c nude mice (Figure 5J-L) (p<0.05). Collectively, our results indicated that ING4 suppresses HCC in a FOXO3a-dependent manner.
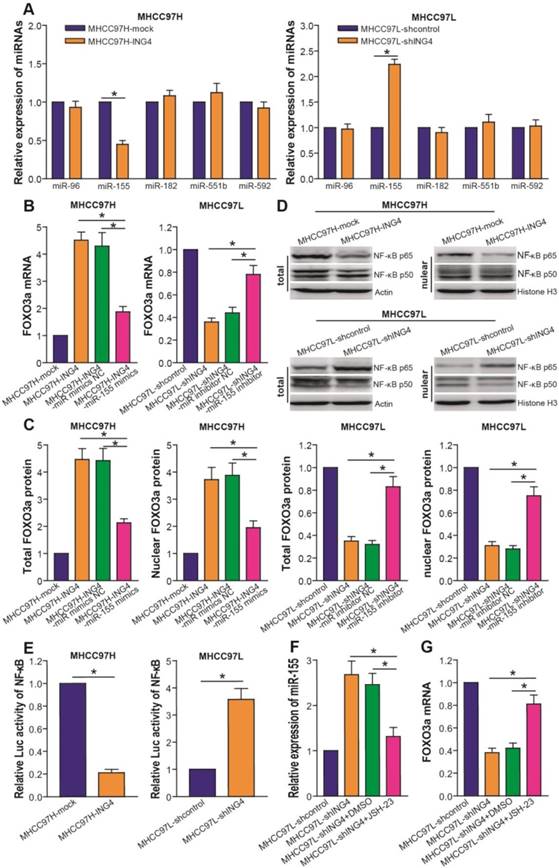
ING4 upregulates FOXO3a via a NF-κB/miR-155 pathway
Accumulating evidence[43-47] has documented that several miRNAs such as miR-96, miR-155, miR-182, miR-551b and miR-592 target FOXO3a. To address the molecular mechanism by which ING4 regulates FOXO3a and elucidate whether miRNAs targeting FOXO3a are involved in ING4-mediated regulation of FOXO3a, we analyzed the expression of above miRNAs in MHCC97H-ING4 vs MHCC97H-mock and MHCC97L-shING4 vs MHCC97L-shcontrol HCC cells. We demonstrated that ING4 significantly downregulated miR-155 but not miR-96, miR-182, miR-551b and miR-592 in MHCC97H tumor cells, whereas knockdown of ING4 upregulated miR-155 in MHCC97L tumor cells (Figure 6A) (p<0.05). MiR-155 mimics/inhibitor assay (Figure 6B and C) further showed that overexpression of miR-155 by transfection of miR-155 mimics could obviously blunt the ING4-induced upregulation of FOXO3a in MHCC97H-ING4 tumor cells, whereas inhibition of miR-155 by transfection of miR-155 inhibitor could block the ING4 knockdown-mediated downregulation of FOXO3a and rescue the expression of FOXO3a in MHCC97L-shING4 tumor cells (p<0.05), indicating that ING4 upregulates FOXO3a in HCC cells via repressing miR-155 expression. MiR-155 as a well-known NF-κB downstream target has been found to be positively regulated by a NF-κB pathway[44].
ING4 represses HCC via upregulating expression and transcriptional activity of FOXO3a. A, B. Western blot analysis of FOXO3a and its targets. The total lysates of MHCC97H-ING4 vs MHCC97H-mock and MHCC97L-shING4 vs MHCC97L-shcontrol HCC cells were immunoblotted with anti-FOXO3a, anti-p27, anti-Cyclin D1, anti-Bim, anti-Puma, anti-FasL, anti-TRAIL, anti-β-catenin or anti-β-actin (a loading control) antibody. The nuclear lysates derived from above cells were immunoblotted with anti-FOXO3a, anti-β-catenin or Histone H3 (a loading control) antibody. The representative pictures of Western blot were shown (A). The total expression level of FOXO3a was normalized to β-actin (FOXO3a/β-actin); the nuclear expression level of FOXO3a was normalized to Histone H3 (FOXO3a/Histone H3), and then expressed as a ratio or fold of respective control, with 1 being the value for MHCC97H-mock or MHCC97L-shcontrol control (B). MHCC97H: *, p<0.05 compared with MHCC97H-mock; MHCC97L: *, p<0.05 compared with MHCC97L-shcontrol, Student t test, n=6 replicates per sample. C. Real-time qRT-PCR analysis of FOXO3a. The mRNA level of FOXO3a was normalized to β-actin and calculated by a 2-∆∆CT method, with 1 being the value for MHCC97H-mock or MHCC97L-shcontrol control. MHCC97H: *, p<0.05 compared with MHCC97H-mock; MHCC97L: *, p<0.05 compared with MHCC97L-shcontrol, Student t test, n=6 replicates per sample. D. Luciferase reporter analysis of transcriptional activity of FOXO3a and β-catenin. The luciferase activity was expressed as a ratio or fold of MHCC97H-mock or MHCC97L-shcontrol control, with 1 being the value for controls. MHCC97H FOXO3a or β-catenin: *, p<0.05 compared with MHCC97H-mock; MHCC97L FOXO3a or β-catenin: *, p<0.05 compared with MHCC97L-shcontrol, Student t test, n=6 replicates per sample. E. Western blot analysis after FOXO3a siRNA knockdown. The total or nuclear lysates derived from the siFOXO3a- or sicontrol-transfected MHCC97H-ING4 cells and the untransfected MHCC97H-ING4 cells were immunoblotted with a panel of antibodies as described above. The representative pictures of Western blot were shown. F. CCK-8 assay after FOXO3a siRNA knockdown. *, p<0.05 compared with MHCC97H-ING4 and sicontrol-transfected MHCC97H-ING4 at day 2, 3 and 4, respectively, two-way repeated measures ANOVA, n=6 replicates per condition. G. Apoptosis analysis after FOXO3a siRNA knockdown. *, p<0.05 compared with MHCC97H-ING4 and sicontrol-transfected MHCC97H-ING4, one-way repeated measures ANOVA, n=6 replicates per condition. H. Transwell migration assay after FOXO3a siRNA knockdown. *, p<0.05 compared with MHCC97H-ING4 and sicontrol-transfected MHCC97H-ING4, one-way repeated measures ANOVA, n=6 replicates per condition, n=5 observations per replicate. I. Transwell invasion assay after FOXO3a siRNA knockdown. *, p<0.05 compared with MHCC97H-ING4 and sicontrol-transfected MHCC97H-ING4, one-way repeated measures ANOVA, n=6 replicates per condition, n=5 observations per replicate. J, K. Subcutaneous xenograft mouse model after FOXO3a siRNA knockdown. The tumor volume (J) was measured after implantation of tumor cells. *, p<0.05 compared with MHCC97H-ING4 and sicontrol-transfected MHCC97H-ING4 at day 2, 3 and 4, respectively, two-way repeated measures ANOVA, n=6 replicates per condition. The xenografted tumors were removed 4 weeks after tumor cell's implantation and tumor weight (K) was then measured. *, p<0.05 compared with MHCC97H-ING4 and sicontrol-transfected MHCC97H-ING4, one-way repeated measures ANOVA, n=6 replicates per condition. L. In vivo lung metastasis assay after FOXO3a siRNA knockdown. *, p<0.05 compared with MHCC97H-ING4 and sicontrol-transfected MHCC97H-ING4, one-way repeated measures ANOVA, n=6 replicates per condition, n=5 sections per sample, n=5 observations per section. Data shown were representative of three independent experiments.
ING4 upregulates FOXO3a via inhibition of NF-κB/miR-155 pathway. A. Real-time qRT-PCR analysis of miRNAs. The expression levels of miR-96, miR-155, miR-182, miR-551b and miR-592 were normalized to U6 and calculated by a 2-∆∆CT method, with 1 being the value for MHCC97H-mock or MHCC97L-shcontrol control. MHCC97H miR-155: *, p<0.05 compared with MHCC97H-mock; MHCC97L miR-155: *, p<0.05 compared with MHCC97L-shcontrol, Student t test, n=6 replicates per sample. B. Real-time qRT-PCR analysis of FOXO3a after miR-155 transfection or inhibition. The mRNA level of FOXO3a was normalized to β-actin and calculated by a 2-∆∆CT method, with 1 being the value for MHCC97H-mock or MHCC97L-shcontrol control. MHCC97H: *, p<0.05 compared with MHCC97H-ING4 and miR-155 mimics NC-transfected MHCC97H-ING4; MHCC97L: *, p<0.05 compared with MHCC97L-shING4 and miR-155 inhibitor NC-transfected MHCC97L-shING4, one-way repeated measures ANOVA, n=6 replicates per sample. C. Western blot analysis of FOXO3a after miR-155 transfection or inhibition. The total or nuclear expression level of FOXO3a was normalized to β-actin (FOXO3a/β-actin) or Histone H3 (FOXO3a/Histone H3) and then expressed as a ratio or fold of respective control, with 1 being the value for MHCC97H-mock or MHCC97L-shcontrol control. MHCC97H: *, p<0.05 compared with MHCC97H-ING4 and miR-155 mimics NC-transfected MHCC97H-ING4; MHCC97L: *, p<0.05 compared with MHCC97L-shING4 and miR-155 inhibitor NC-transfected MHCC97L-shING4, one-way repeated measures ANOVA, n=6 replicates per sample. D. Western blot analysis of NF-κB p65 and p50. The representative pictures of Western blot were shown. E. Luciferase reporter analysis of transcriptional activity of NF-κB. The luciferase activity was expressed as a ratio or fold of MHCC97H-mock or MHCC97L-shcontrol control, with 1 being the value for controls. MHCC97H: *, p<0.05 compared with MHCC97H-mock; MHCC97L: *, p<0.05 compared with MHCC97L-shcontrol, Student t test, n=6 replicates per sample. F. Real-time qRT-PCR analysis of miR-155 after NF-κB inhibition. The expression level of miR-155 was normalized to U6 and calculated by a 2-∆∆CT method, with 1 being the value for MHCC97L-shcontrol. *, p<0.05 compared with MHCC97L-shING4 and DMSO-treated MHCC97L-shING4, one-way repeated measures ANOVA, n=6 replicates per sample. G. Real-time qRT-PCR analysis of FOXO3a after NF-κB inhibition. The mRNA level of FOXO3a was normalized to β-actin and calculated by a 2-∆∆CT method, with 1 being the value for MHCC97L-shcontrol. *, p<0.05 compared with MHCC97L-shING4 and DMSO-treated MHCC97L-shING4, one-way repeated measures ANOVA, n=6 replicates per sample. Data shown were representative of three independent experiments.
Clinical association of ING4 with FOXO3a, NF-κB p65 and miR-155 as well as FOXO3a with NF-κB p65 and miR-155. A. Immunohistochemistry and in situ hybridization analyses. The expression of ING4, NF-κB p65 and FOXO3a in HCC tumor tissue specimens was analyzed by immunohistochemistry. The expression of miR-155 in HCC tumor tissue specimens was analyzed by in situ hybridization. The representative pictures derived from four representative cases (Case 21, Case 18, Case 27 and Case 20) were shown. B. Percentage of specimens showing high or low ING4 expression in relation to the expression levels of FOXO3a, NF-κB p65 and miR-155. *, p<0.05, Pearson's χ2 test. C. Percentage of specimens showing high or low FOXO3a expression in relation to the expression levels of NF-κB p65 and miR-155. *, p<0.05, Pearson's χ2 test. Data shown were representative of three independent experiments.
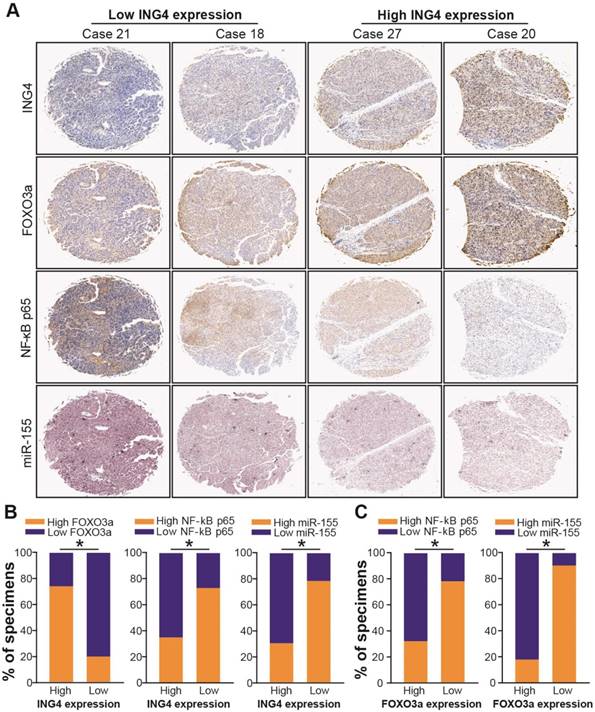
ING4 has been shown to inhibit the transcriptional activity of NF-κB[35,48,49]. To confirm whether ING4 regulates miR-155 via a NF-κB pathway, we firstly analyzed the effect of ING4 on the expression of NF-κB p65/p50 and the transcriptional activity of NF-κB by Western blot and luciferase reporter assays. As shown in Figure 6D and E, ING4 drastically suppressed the expression of NF-κB p65 but not p50 and the activation of NF-κB in MHCC97H tumor cells, whereas knockdown of ING4 promoted the activation of NF-κB in MHCC97L tumor cells (p<0.05). NF-κB inhibition assay (Figure 6F) further demonstrated that inhibition of NF-κB using JSH-23, a NF-κB activation inhibitor which can prevent nuclear translocation of NF-κB, in ING4-silenced MHCC97L-shING4 tumor cells markedly impaired the ING4 knockdown-induced upregulation of miR-155 (p<0.05), implying that ING4 suppresses miR-155 expression via inactivating NF-κB pathway. Additionally, NF-κB inhibition attenuated the ING4 knockdown-induced downregulation of FOXO3a (Figure 6G) (p<0.05). To further validate the relationships between ING4, FOXO3a, NF-κB p65 and miR-155 in human HCC clinical tissues, the expression of ING4, NF-κB p65 and FOXO3a was analyzed by immunohistochemistry (Figure 7A) and the expression of miR-155 was analyzed by in situ hybridization (Figure 7A), respectively. Consistent with above cell model data, ING4 showed a positive correlation with FOXO3a and a negative correlation with NF-κB p65 and miR-155 (Figure 7A and B) (p<0.05). Additionally, FOXO3a showed a negative correlation with NF-κB p65 and miR-155 (Figure 7A and C) (p<0.05). Taken together, ING4 is capable of upregulating FOXO3a in human HCC via a NF-κB/miR-155 pathway.
Discussion
The present study demonstrated that ING4 is dramatically reduced in human HCC and its expression level is inversely associated with AJCC cancer staging, tumor size and vascular invasion. Forced expression of ING4 not only significantly suppresses HCC cell proliferation, migration and invasion, and induces HCC cell apoptosis in vitro but also retards HCC subcutaneously xenografted tumor growth and distant lung metastasis in vivo in nude mice via upregulating FOXO3a and modulating its downstream targets, whereas knockdown of ING4 exerts opposing effects. Moreover, ING4 increases the expression and the transcriptional activity of FOXO3a by inhibition of NF-κB/miR-155 pathway.
It has been reported that FOXO3a is a promising therapeutic target for human HCC and various toxic compounds exhibits tumor-suppressive effects in HCC via FOXO3a activation[42,50]. To decipher the underlying mechanism by which ING4 suppresses HCC, we was thus interested in examining whether FOXO3a is involved in ING4-medicated tumor inhibition in HCC. Our results showed that ING4 obviously upregulated expression of FOXO3a in HCC cells both at mRNA and total/nuclear protein level, whereas knockdown of ING4 downregulated it. Luciferase reporter assay further demonstrated that ING4 enhanced the transcription activity of FOXO3a. Previous studies showed that FOXO3a induces cell cycle arrest and apoptosis by regulating target genes such as p27, Cyclin D1, Bim, Puma, FasL and TRAIL[8]. Previous studies also showed that FOXO3a impedes cancer metastasis via inactivation of β-catenin signaling by indirect transactivation of miR-34b/c targeting β-catenin[16] or direct transcriptional inhibition of β-catenin[51]. In addition, FOXO3a can directly bind to β-catenin and compete with TCF for interaction with β-catenin, therefore inhibiting β-catenin signaling[16,52]. As we expected, ING4 upregulated p27, Bim, Puma, FasL and TRAIL expression as well as downregulated Cyclin D1 expression and β-catenin signaling in HCC cells. Furthermore, knockdown of FOXO3a could significantly impair the ING4-induced tumor suppression and the ING4-medicated regulatory effect on downstream targets of FOXO3a. Our data indicated that ING4 suppresses HCC growth and metastasis in a FOXO3a-dependent fashion.
The expression and transcriptional activity of FOXO3a is regulated by at multilayer levels such as transcription, post-transcription and post-translation modifications[6,8]. miRNAs negatively regulates gene expression at a post-transcriptional level by interacting with sequences within the 3'-UTR of the target mRNA. A panel of miRNAs including miR-96, miR-155, miR-182, miR-551b and miR-592 has been shown to target FOXO3a[43-47]. Our data found that ING4 could increase the mRNA level of FOXO3a in HCC cells, which promoted us to investigate whether ING4 upregulates FOXO3a via downregulation of above miRNAs targeting FOXO3a. Interestingly, qRT-PCR analysis revealed that ING4 efficiently inhibited the expression of miR-155 in HCC cells, whereas knockdown of ING4 increased the expression of miR-155. Moreover, restoration of miR-155 significantly attenuated ING4-mediated upregulation of FOXO3a (mRNA and protein). Inhibition of miR-155 blocked the effect on decrease of FOXO3a induced by ING4 knockdown. These results suggested that ING4 upregulates FOXO3a in HCC cells via inhibiting miR-155 expression and preventing miR-155-mediated degradation of FOXO3a mRNA. It has also been shown that FOXO3a is negatively regulated via phosphorylation by AKT (Thr32, Ser253 and Ser315)[53,54], ERK (Ser294, Ser344 and Ser425)[55] and IKKβ (Ser644)[56] kinases. Phosphorylation of FOXO3a induces its nuclear exclusion, sequestration in the cytosol and degradation through the ubiquitin-proteasome pathway, thereby inhibiting the transcriptional activity of FOXO3a[53-56]. MiR-155 has been reported to upregulate PI3K/AKT signaling by targeting PTEN[57,58]. Therefore, whether post-translational regulation of FOXO3a, elicited by ING4-mediated inhibition of PI3K/AKT signaling via upregulating PTEN through inhibiting miR-155, is also implicated in ING4-directed upregulation of FOXO3a needs further investigation. Previous studies has shown that ING4 can inhibit the transcriptional activity of NF-κB by a chromatin-remolding manner and a ubiqutin-mediated degradation of NF-κB p65[35,48,49]. MiR-155 as an oncomiRNA has been found to be positively regulated by NF-κB pathway[44,59,60]. To further address whether ING4 regulates miR-155 via a NF-κB pathway, we determined the effect of ING4 on NF-κB in HCC cells. Consistent with previous findings, we found that ING4 did repress the activation of NF-κB in HCC cells, whereas knockdown of ING4 enhanced its activation. Furthermore, inhibition of NF-κB markedly impaired the ING4 knockdown-induced upregulation of miR-155 and downregulation of FOXO3a. Our data implied that ING4 suppresses miR-155 expression and upregulates FOXO3a expression in HCC cells via inactivating NF-κB pathway.
Taken together, our study provided the first compelling evidence that ING4 can suppress human HCC growth and metastasis to a great extent via a NF-κB/miR-155/FOXO3a pathway. Downregulation of ING4 in human HCC may facilitate the activation of NF-κB, upregulation of miR-155 and downregulation of FOXO3a, leading to HCC progression.
Abbreviations
HCC: hepatocellular carcinoma; ING4: inhibitor of growth 4; ING: inhibitor of growth; FOXO3a: forkhead box 3a; FOXO: Forkhead box class O; NF-κB: nuclear factor-κB; miRNA/miR: microRNA; VEGF: vascular endothelial growth factor; eNOS: endothelial nitric oxide synthase; EMT: epithelial-mesenchymal transition; EMT-TF: EMT-inducing transcription factor; MMP: matrix metalloproteinase; IL: interlukin; HIF-1α: hypoxia inducible factor-1α; AJCC: American Joint Committee's Cancer; DMEM: Dulbecco's modified Eagle's medium; FBS: fetal bovine serum; PBS: Phosphate-Buffered Saline; GFP: green fluorescent protein; BSD: Blasticidin S; CCK-8: Cell Counting Kit-8; PI: propidium iodide; PE: phycoerythrin; 7-AAD: 7-aminoactinomycin D; PVDF: polyvinylidene difluoride; BCA: bicinchoninic acid; DAB: 3,3'-diaminobenzidine; BCIP/NBT: 5-Bromo-4-Chloro-3-Indolyl Phosphate/Nitro-Blue-Tetrazolium; FasL: Fas ligand; TRAIL: tumor necrosis factor-related apoptosis-inducing ligand; DIG: digoxigenin; HRP: horseradish peroxidase; AP: alkaline phosphatase; NC: negative control; TU: transducing unit; MOI: multiplicity of infection; OD: optical density; HE: hematoxylin & eosin; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis; qRT-PCR: real-time quantitative RT (Reverse Transcription)-PCR (Polymerase Chain Reaction); siRNA: small interfering RNA; IHC: immunohistochemistry; 3'-UTR: 3'-untranslational region; AKT: protein kinase B; ERK: extracellular signal-regulated kinase; IKKβ: IκB kinase β.
Acknowledgements
This research work was supported by grants from the National Natural Science Foundation of China (NNSFC) (Nos. 81372443, 81572992 and 81772645), the Science and Technology Department of Jiangsu Province (Nos. BY2015039-03 and BL2014039), the Health and Family Planning Commission of Jiangsu Province (No. LGY2016030), the Science and Technology Development Bureau of Xiangcheng District of Suzhou City (No. XJ201534), the Pushin HK Jiangsu Medical Technology Ltd., Inc. (No. P112200315) and the Wu Jieping Medical Foundation (No. P112200914).
Author Contributions
QFL, QL, SBR and XYF conceived and designed the experiments. QFL, HQQ, TYL, WJ and LDP performed the experiments and collected data. QFL, WJ, TM and XYF analyzed and interpreted data. QFL, QL, SBR and XYF wrote the paper. All the authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Torre LA, Bray F, Siegel RL. et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87-108
2. El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118-1127
3. El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142:1264-1273.e1
4. Aravalli RN, Steer CJ, Cressman EN. Molecular mechanisms of hepatocellular carcinoma. Hepatology. 2008;48:2047-2063
5. Lam EW, Brosens JJ, Gomes AR. et al. Forkhead box proteins: tuning forks for transcriptional harmony. Nat Rev Cancer. 2013;13:482-495
6. Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013;14:83-97
7. Myatt SS, Lam EW. The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer. 2007;7:847-859
8. Yang JY, Hung MC. A new fork for clinical application: targeting forkhead transcription factors in cancer. Clin Cancer Res. 2009;15:752-757
9. Yang JY, Hung MC. Deciphering the role of forkhead transcription factors in cancer therapy. Curr Drug Targets. 2011;12:1284-1290
10. Karadedou CT, Gomes AR, Chen J. et al. FOXO3a represses VEGF expression through FOXM1-dependent and -independent mechanisms in breast cancer. Oncogene. 2012;31:1845-1858
11. Wang J, Zheng X, Zeng G. et al. Purified vitexin compound 1 inhibits growth and angiogenesis through activation of FOXO3a by inactivation of Akt in hepatocellular carcinoma. Int J Mol Med. 2014;33:441-448
12. Potente M, Urbich C, Sasaki K. et al. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J Clin Invest. 2005;115:2382-2392
13. Chou CC, Lee KH, Lai IL. et al. AMPK reverses the mesenchymal phenotype of cancer cells by targeting the Akt-MDM2-Foxo3a signaling axis. Cancer Res. 2014;74:4783-4795
14. Ni D, Ma X, Li HZ. et al. Downregulation of FOXO3a promotes tumor metastasis and is associated with metastasis-free survival of patients with clear cell renal cell carcinoma. Clin Cancer Res. 2014;20:1779-1790
15. Shiota M, Song Y, Yokomizo A. et al. Foxo3a suppression of urothelial cancer invasiveness through Twist1, Y-box-binding protein 1, and E-cadherin regulation. Clin Cancer Res. 2010;16:5654-5663
16. Liu H, Yin J, Wang H. et al. FOXO3a modulates WNT/beta-catenin signaling and suppresses epithelial-to-mesenchymal transition in prostate cancer cells. Cell Signal. 2015;27:510-518
17. Coomans de Brachene A, Demoulin JB. FOXO transcription factors in cancer development and therapy. Cell Mol Life Sci. 2016;73:1159-1172
18. Zhang R, Jin J, Shi J. et al. INGs are potential drug targets for cancer. J Cancer Res Clin Oncol. 2017;143:189-197
19. Guerillon C, Bigot N, Pedeux R. The ING tumor suppressor genes: status in human tumors. Cancer Lett. 2014;345:1-16
20. Yuan S, Jin J, Shi J. et al. Inhibitor of growth-4 is a potential target for cancer therapy. Tumour Biol. 2016;37:4275-4279
21. Cui S, Gao Y, Zhang K. et al. The emerging role of inhibitor of growth 4 as a tumor suppressor in multiple human cancers. Cell Physiol Biochem. 2015;36:409-422
22. Yan R, He L, Li Z. et al. SCF(JFK) is a bona fide E3 ligase for ING4 and a potent promoter of the angiogenesis and metastasis of breast cancer. Genes Dev. 2015;29:672-685
23. Shiseki M, Nagashima M, Pedeux RM. et al. p29ING4 and p28ING5 bind to p53 and p300, and enhance p53 activity. Cancer Res. 2003;63:2373-2378
24. Unoki M, Shen JC, Zheng ZM. et al. Novel splice variants of ING4 and their possible roles in the regulation of cell growth and motility. J Biol Chem. 2006;281:34677-34686
25. Xie Y, Zhang H, Sheng W. et al. Adenovirus-mediated ING4 expression suppresses lung carcinoma cell growth via induction of cell cycle alteration and apoptosis and inhibition of tumor invasion and angiogenesis. Cancer Lett. 2008;271:105-116
26. Gong A, Ye S, Xiong E. et al. Autophagy contributes to ING4-induced glioma cell death. Exp Cell Res. 2013;319:1714-1723
27. Zhang X, Xu LS, Wang ZQ. et al. ING4 induces G2/M cell cycle arrest and enhances the chemosensitivity to DNA-damage agents in HepG2 cells. FEBS Lett. 2004;570:7-12
28. Xie Y, Sheng W, Miao J. et al. Enhanced antitumor activity by combining an adenovirus harboring ING4 with cisplatin for hepatocarcinoma cells. Cancer Gene Ther. 2011;18:176-188
29. Wang R, Huang J, Feng B. et al. Identification of ING4 (inhibitor of growth 4) as a modulator of docetaxel sensitivity in human lung adenocarcinoma. Mol Med. 2012;18:874-886
30. Ling C, Xie Y, Zhao D. et al. Enhanced radiosensitivity of non-small-cell lung cancer (NSCLC) by adenovirus-mediated ING4 gene therapy. Cancer Gene Ther. 2012;19:697-706
31. Zhao Y, Su C, Zhai H. et al. Synergistic antitumor effect of adenovirus-mediated hING4 gene therapy and (125)I radiation therapy on pancreatic cancer. Cancer Lett. 2012;316:211-218
32. Shen JC, Unoki M, Ythier D. et al. Inhibitor of growth 4 suppresses cell spreading and cell migration by interacting with a novel binding partner, liprin alpha1. Cancer Res. 2007;67:2552-2558
33. Li J, Martinka M, Li G. Role of ING4 in human melanoma cell migration, invasion and patient survival. Carcinogenesis. 2008;29:1373-1379
34. Qu H, Yin H, Yan S. et al. Inhibitor of growth 4 suppresses colorectal cancer growth and invasion by inducing G1 arrest, inhibiting tumor angiogenesis and reversing epithelial-mesenchymal transition. Oncol Rep. 2016;35:2927-2935
35. Garkavtsev I, Kozin SV, Chernova O. et al. The candidate tumour suppressor protein ING4 regulates brain tumour growth and angiogenesis. Nature. 2004;428:328-332
36. Colla S, Tagliaferri S, Morandi F. et al. The new tumor-suppressor gene inhibitor of growth family member 4 (ING4) regulates the production of proangiogenic molecules by myeloma cells and suppresses hypoxia-inducible factor-1 alpha (HIF-1alpha) activity: involvement in myeloma-induced angiogenesis. Blood. 2007;110:4464-4475
37. Lu M, Ma J, Xue W. et al. The expression and prognosis of FOXO3a and Skp2 in human hepatocellular carcinoma. Pathol Oncol Res. 2009;15:679-687
38. Fang F, Luo LB, Tao YM. et al. Decreased expression of inhibitor of growth 4 correlated with poor prognosis of hepatocellular carcinoma. Cancer Epidemiol Biomarkers Prev. 2009;18:409-416
39. Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010;17:1471-1474
40. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101-1108
41. Ding SJ, Li Y, Shao XX. et al. Proteome analysis of hepatocellular carcinoma cell strains, MHCC97-H and MHCC97-L, with different metastasis potentials. Proteomics. 2004;4:982-994
42. Carbajo-Pescador S, Mauriz JL, Garcia-Palomo A. et al. FoxO proteins: regulation and molecular targets in liver cancer. Curr Med Chem. 2014;21:1231-1246
43. Xu D, He X, Chang Y. et al. Inhibition of miR-96 expression reduces cell proliferation and clonogenicity of HepG2 hepatoma cells. Oncol Rep. 2013;29:653-661
44. Chiu CF, Chang YW, Kuo KT. et al. NF-kappaB-driven suppression of FOXO3a contributes to EGFR mutation-independent gefitinib resistance. Proc Natl Acad Sci U S A. 2016;113:E2526-2535
45. Yang WB, Chen PH, Hsu Ts. et al. Sp1-mediated microRNA-182 expression regulates lung cancer progression. Oncotarget. 2014;5:740-753
46. Wei Z, Liu Y, Wang Y. et al. Downregulation of Foxo3 and TRIM31 by miR-551b in side population promotes cell proliferation, invasion, and drug resistance of ovarian cancer. Med Oncol. 2016;33:126
47. Fu Q, Du Y, Yang C. et al. An oncogenic role of miR-592 in tumorigenesis of human colorectal cancer by targeting Forkhead Box O3A (FoxO3A). Expert Opin Ther Targets. 2016;20:771-782
48. Nozell S, Laver T, Moseley D. et al. The ING4 tumor suppressor attenuates NF-kappaB activity at the promoters of target genes. Mol Cell Biol. 2008;28:6632-6645
49. Hou Y, Zhang Z, Xu Q. et al. Inhibitor of growth 4 induces NFkappaB/p65 ubiquitin-dependent degradation. Oncogene. 2014;33:1997-2003
50. Cox J, Weinman S. Mechanisms of doxorubicin resistance in hepatocellular carcinoma. Hepat Oncol. 2016;3:57-59
51. Yang Z, Liu S, Zhu M. et al. PS341 inhibits hepatocellular and colorectal cancer cells through the FOXO3/CTNNB1 signaling pathway. Sci Rep. 2016;6:22090
52. Zhou Y, Liang C, Xue F. et al. Salinomycin decreases doxorubicin resistance in hepatocellular carcinoma cells by inhibiting the beta-catenin/TCF complex association via FOXO3a activation. Oncotarget. 2015;6:10350-10365
53. Brunet A, Bonni A, Zigmond MJ. et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857-868
54. Plas DR, Thompson CB. Akt activation promotes degradation of tuberin and FOXO3a via the proteasome. J Biol Chem. 2003;278:12361-12366
55. Yang JY, Zong CS, Xia W. et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008;10:138-148
56. Hu MC, Lee DF, Xia W. et al. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117:225-237
57. Fu X, Wen H, Jing L. et al. MicroRNA-155-5p promotes hepatocellular carcinoma progression by suppressing PTEN through the PI3K/Akt pathway. Cancer Sci. 2017;108:620-631
58. Xue X, Liu Y, Wang Y. et al. MiR-21 and MiR-155 promote non-small cell lung cancer progression by downregulating SOCS1, SOCS6, and PTEN. Oncotarget. 2016;7:84508-84519
59. Thompson RC, Vardinogiannis I, Gilmore TD. Identification of an NF-kappaB p50/p65-responsive site in the human MIR155HG promoter. BMC Mol Biol. 2013;14:24
60. Thompson RC, Herscovitch M, Zhao I. et al. NF-kappaB down-regulates expression of the B-lymphoma marker CD10 through a miR-155/PU.1 pathway. J Biol Chem. 2011;286:1675-1682
Author contact
![]() Corresponding authors: Prof. Yufeng Xie (Email: sdxyfcom, Tel: +86-0512-67780645), Department of Oncology and Dr. Lei Qin (Email: qinleiedu.cn, Tel: +86-0512-67780178), Department of General Surgery, the First Affiliated Hospital of Soochow University, No. 188 Shizi Street, Suzhou 215006, Jiangsu Province, China; and Prof. Bairong Shen (Email: bairong.shenedu.cn, Tel: +86-0512-65110951), Center for Systems Biology, Soochow University, No. 1 Shizi Street, Suzhou 215006, Jiangsu Province, China
Corresponding authors: Prof. Yufeng Xie (Email: sdxyfcom, Tel: +86-0512-67780645), Department of Oncology and Dr. Lei Qin (Email: qinleiedu.cn, Tel: +86-0512-67780178), Department of General Surgery, the First Affiliated Hospital of Soochow University, No. 188 Shizi Street, Suzhou 215006, Jiangsu Province, China; and Prof. Bairong Shen (Email: bairong.shenedu.cn, Tel: +86-0512-65110951), Center for Systems Biology, Soochow University, No. 1 Shizi Street, Suzhou 215006, Jiangsu Province, China
Received 2018-7-10
Accepted 2018-11-29
Published 2019-1-1