ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2019; 15(3):556-567. doi:10.7150/ijbs.29907 This issue Cite
Research Paper
Rosmarinic acid alleviates cardiomyocyte apoptosis via cardiac fibroblast in doxorubicin-induced cardiotoxicity
Xin Zhang*, Jin-Xiu Zhu*, Zhen-Guo Ma, Hai-Ming Wu, Si-Chi Xu, Peng Song, Chun-Yan Kong, Yu-Pei Yuan, Wei Deng ![]() , Qi-Zhu Tang
, Qi-Zhu Tang ![]()
Department of Cardiology, Renmin Hospital of Wuhan University, Wuhan 430060, RP China
Cardiovascular Research Institute of Wuhan University, Wuhan 430060, RP China
Hubei Key Laboratory of Cardiology, Wuhan 430060, RP China
* These authors contributed equally to this work.
Abstract

Cardiomyocyte apoptosis is a key event in the process of doxorubicin (DOX)-induced cardiotoxicity. Our previous study found that rosmarinic acid (RA) could attenuate pressure overload-induced cardiac dysfunction via cardiac fibroblasts (CFs), however its effect in DOX-induced cardiotoxicity remains unknown. In the present study, mice were subjected to a single intraperitoneal injection of DOX (15mg/kg) to generate DOX-induced cardiotoxicity. Histological examination, echocardiography, and molecular markers were used to evaluate the effects of RA. Neonatal rat cardiomyocytes (CMs) and CFs were used to verify the protective effect of RA in vitro. Conditioned medium derived from RA-treated CFs were prepared to illustrate the effect of RA on paracrine interplay between CFs and CMs. We found that RA significantly alleviated DOX-induced cardiomyocyte apoptosis and cardiac dysfunction in vivo, which, however, had almost negligible beneficial effect on DOX directly induced cardiomyocyte apoptosis in vitro. Mechanistically, CFs-derived Fas L was responsible for DOX-induced cardiomyocyte apoptosis, and RA treatment could decrease Fas L expression in CFs and its release to the conditioned medium by suppressing nuclear factor of activated T cells (NFAT) activation and metalloproteinase 7 (MMP7) expression, and exerted the anti-apoptotic effect on CMs via CFs. Ionomycin, and activator of NFAT, abrogated RA-mediated protective effect on cardiomyocyte apoptosis and cardiac dysfunction. In summary, RA alleviated cardiomyocyte apoptosis by inhibiting the expression and release of Fas L in CFs via a paracrine manner, moreover, NFAT as well as MMP7 inhibition were responsible for the suppression of Fas L. RA could be a powerful new therapeutic agent to mitigate cardiomyocyte apoptosis, thereby improving DOX-induced cardiotoxicity.
Keywords: Rosmarinic acid, Apoptosis, Cardiac fibroblast, Fas ligand, Nuclear factor of activated T cells, Metalloproteinase.
Introduction
Doxorubicin (DOX) is a well-established and highly effective antitumor anthracycline antibiotic, which has been commonly used for the treatment of various cancers, including leukemias, lymphomas and solid tumors [1-3]. However, the clinical use of DOX is evidently hampered due to the development of life-threatening cardiotoxicity [4, 5]. Despite the exact mechanism remains largely unknown, multiple factors have been substantiated to be involved in DOX-induced cardiotoxicity. DOX application can increase the production of reactive oxygen species (ROS), facilitate the release of inflammatory mediators and disturb the homeostasis of intracellular calcium cycling in the heart, which eventually evoke apoptotic signaling pathways and lead to cardiomyocyte apoptosis [6-8]. Massive loss of contractile cardiomyocyte contributes to systolic dysfunction, and the replacement with non-contractile and poorly conductive collagen scar further impairs mechanical and electrical functions of the heart and ultimately triggers the occurrence of heart failure and arrhythmogenesis. In addition, our recent study demonstrated that suppression of cardiomyocyte apoptosis significantly attenuated DOX-induced cardiac dysfunction [9]. Therefore, unearthing novel therapeutic agents alleviating cardiomyocyte apoptosis is of paramount importance for treating DOX-induced cardiotoxicity.
The heart is composed of various types of cells, such as cardiomyocytes, cardiac fibroblasts (CFs) and inflammatory cells, etc [10]. It is becoming clear that myocardial cell responses are regulated by direct and indirect communications between cardiomyocytes and non-myocytes [11]. CFs is the most abundant cell type present in the myocardium and is traditionally considered as the main source of extracellular matrix. However, increasing researches proposed an indispensable role of CFs in the regulation of cardiomyocyte function [12]. Previous findings demonstrated that CFs could secrete star miRNA-enriched exosomes and induce cardiomyocyte hypertrophy in a paracrine signaling manner [13]. Results from Dawn et al indicated that CFs-derived paracrine factors altered impulse conduction and ion channel expression, and participated in the regulation of electrical propagation of cardiomyocytes [14]. Besides, a recent study showed that CFs-derived NLRP3 inflammasome and subsequent IL-1β production are responsible for cardiomyocyte apoptosis in sepsis [15]. More importantly, specific deletion of ataxia telangiectasia mutated kinase in CFs alleviated DOX-triggered cardiomyocyte apoptosis via a paracrine regulation between CFs and cardiomyocytes [16]. All these researches defined a pivotal role of CFs in the regulation of cardiomyocyte function and apoptosis.
Rosmarinic acid (α-o-caffeoyl-3, 4-dihydroxyphenyl lactic acid; RA), a natural poly-phenolic compound mainly distributed in rosemary, mint, perilla frutescens and sarcandra glabra, has been reported with a range of biological effects, including anti-inflammatory, anti-oxidative, anti-bacterial and HIV-1 inhibiting properties, etc [17, 18]. Recent studies also identified that RA possessed a powerful anti-apoptotic capacity and could protect cells against hydrogen peroxide-induced DNA damage and apoptosis [19, 20]. Moreover, our recent study found that RA treatment could target CFs and attenuate pressure overload-induced cardiac dysfunction in mice [21]. Based on these findings, we hypothesized that RA might be a promising candidate for the treatment of cardiomyocyte apoptosis and DOX-induced cardiotoxicity. In the current study, we demonstrated that RA treatment alleviated cardiomyocyte apoptosis and cardiac dysfunction in DOX-induced cardiotoxicity, and the cardioprotective effect of RA was achieved via targeting CFs.
Materials and Methods
Antibodies and reagents
Primary antibodies against the following proteins were purchased from Cell Signaling Technology (Danvers, MA, USA): total-Nuclear factor of activated T cells 3 (T-NFAT3, 2183S), cleaved-caspase3 (C-Caspase3, 9661), Caspase3 (9662P), C-Caspase8 (9429), Caspase8 (4790), BAX (2722) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, 2118). Anti-B-cell lymphoma 2 (BCL-2, ab196495), anti-Fas ligand (Fas L, ab15285), metalloproteinase 7 (MMP7, ab189277) and anti-NFAT3 (ab62613) were obtained from Abcam (Cambridge, UK). RA was obtained from Shanghai Winherb Medical Co. (Shanghai, China, No 160603) and was dissolved in 0.1% DMSO as previously described [21]. DOX (D1515), rh Fas L (S8689) and rat Fas L Elisa Kit (RAB0179) were purchased from Sigma-Aldrich (St. Louis, MO, USA), whereas ionomycin (9995) was obtained from Cell Signaling Technology (Danvers, MA, USA). ApopTag® Plus In Situ Apoptosis Fluorescein Detection Kit (S7111) was purchased from Millipore (Billerica, MA, USA), and Alexa Fluor 568-goat anti-rabbit secondary antibodies were purchased from LICOR Biosciences (Lincoln, USA). PE Annexin V Apoptosis Detection Kit I (Cat: 559763) was purchased from BD Pharmingen (San Diego, CA, USA). The BCA protein assay kit was from Pierce (Rockford, IL, USA).
Animals and treatments
All animal care and experimental procedures were in compliance with the Guidelines for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH Publication, revised 2011) and approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University. C57/B6 male mice (8-10 weeks old, 23.5-27.5 g) were purchased from the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences (Beijing, China) and were subjected to an adaptive feeding for 1 week before the study commenced. All mice were maintained under specific pathogen-free, environmentally controlled (Temperature: 20-25°C; Humidity: 50±5%) barrier conditions in individual ventilated cages and were fed with sterile food and water ad libitum. DOX-induced cardiotoxicity was generated with a single intraperitoneal injection of DOX (15mg/kg) as our previously described and the control group received equal volume of normal saline (NS)12[9]. On the second day of the injection, mice were orally administered with RA (100mg/kg/d) or vehicle (Veh) for consecutive 7 days and then were sacrificed with heart samples collected for further study.
Echocardiography and hemodynamics
Transthoracic echocardiography was performed according to our previous studies[21-23]. Briefly, mice were anesthetized with 1.5% isoflurane and were placed in a supine position on a pre-warming pad. Two-dimensional (2D) guided M-mode echocardiography was conducted using MyLab 30CV ultrasound (Esaote SpA, Genoa, Italy) equipped with a 10 MHz phased array transducer. Caution was taken to avoid excessive pressure over the chest wall, which could induce bradycardia and deformation of the heart. Images of the heart at the level of papillary muscle were recorded in 2D mode in the parasternal short-axis view with a depth setting of 2 cm. Echocardiographic parameters were averaged from three to five cardiac cycles, whereas heart rate was averaged over 10 consecutive cycles.
Invasive hemodynamic monitoring was performed by PowerLab system (AD Instruments Ltd., Oxford, UK) using a 1.4-French Millar pressure-volume catheter (SPR-839; Millar Instruments, Houston, TX) as our previously described [21-23]. Data collection and analysis were performed in a blinded manner.
Western blot and quantitative real-time PCR
Western blot was performed referring to our previous articles [24-26]. In brief, proteins from cultured cells or homogenized left ventricles were separated by 10% SDS-PAGE and were electrophoretically transferred onto PVDF membranes (EMD Millipore, Billerica, MA, USA; No. IPFL00010). Membranes were blocked with 5% nonfat milk for 60min at room temperature, and were subsequently probed with indicating primary antibodies overnight at 4°C, followed with the secondary antibodies at 37°C for 1 hour. The bands were scanned and quantified by Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE, USA), and protein expressions were normalized to total proteins or GAPDH. Nuclear and cytosolic protein fractions were separated by a commercial kit as our previously described [9, 21]. Proteins from cytosolic lysates were normalized to GAPDH, whereas proteins from nuclear lysates were normalized to PCNA.
Total RNA was isolated using TRIzol according to our previous studies and reverse transcribed with Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, MA, USA)[27]. The expression level of each individual transcript was normalized to Gapdh.
Cell culture and treatments
Neonatal rat cardiomyocytes (CMs) and CFs were separated and cultured by the methods according to previous literatures [24]. Bromodeoxyuridine (0.1mM) was added to inhibit the proliferation of CFs in CMs. CMs were seeded onto 6-well plates or glass slides and were cultured in Dulbecco's modified Eagle's medium (DMEM)/F12 (GIBCO, C11995) with 15% fetal bovine serum (FBS, GIBCO, 10099) for 48 hours to achieve 70-80% confluency. After synchronization, CMs were pretreated with RA (20μg/ml) or equal volume of vehicle for 16 hours followed with phosphate buffer solution (PBS), DOX (1μM) or recombinant human Fas L (rh Fas L, 5μg/ml) for another 8 hours. To notify the paracrine interplay between CMs and CFs, CMs were incubated with the indicating conditioned medium (ConM) from CFs for 24 hours. rh Fas L was replenished to CFs-derived ConM before incubated with CMs to further corroborate its role in mediating the paracrine apoptosis.
To further verify the effect of RA, we isolated adult mouse heart CFs from different groups as previously described [25]. Briefly, left ventricles were harvested and digested with 0.125% trypsin and collagenase at 37 °C. The adult CFs were then suspended in DMEM/F12 medium with 10% FBS and cultured for 90 min. Then the adult CFs were harvested for further analysis.
Conditioned medium produced from cardiac fibroblast
ConM from CFs were prepared according to previous studies with minor modifications [16, 28]. Briefly, confluent CFs were pretreated with RA (30μg/ml) or equal volume of vehicle for 16 hours followed with PBS or DOX (1μM) stimulation for another 8 hours. Then the cells were replaced with fresh medium and cultured for 24 hours, after that the medium were collected to obtain indicating ConM as ConM-PV (PBS+Veh), ConM-PR (PBS+RA), ConM-DV (DOX+Veh) and ConM-DR (DOX+RA). To block the effect of Fas L in vitro, CFs were pretreated with Fas-Fc (a neutralizing agent of Fas L, 10μg/ml) for 16 hours followed by incubation with DOX for another 8 hours. Ionomycin (100nM) was added to CFs with RA to induce NFAT activation and nuclear translocation.
Immunofluorescence staining
Immunofluorescence staining was performed as previously described [21, 29]. In brief, cell coverslips were fixed with 4% formaldehyde, permeabilized in 0.2% Triton X-100 and blocked with 10% goat serum for 60 min at 37°C. Then cells were stained with T-NFAT3 (1:100) overnight at 4°C, followed with Alexa Fluor 568-goat anti-rabbit secondary antibody (1:200) for 60 minutes at 37°C, and DAPI was used for nuclei observation. Images were observed using a special OLYMPUS DX51 fluorescence microscope (Tokyo, Japan) in a blind manner.
TdT-mediated dUTP nick end-labelling (TUNEL) staining
TUNEL staining was performed to detect the apoptosis index both in vivo and in vitro according to the manufacturer's instructions using a commercially available kit. To evaluate the cell types in the apoptotic cell populations, double fluorescent staining of TUNEL with cardiomyocyte-specific marker (α-actin) was done referring to previous studies [30]. Briefly, cardiac sections or cell coverslips were first underwent standard TUNEL staining, and then were incubated with α-actin (1:100) overnight at 4°C after carefully rinsed with PBS for 4 times. On the second day, they were stained with Alexa Fluor 568-goat anti-rabbit secondary antibody (1:200) and DAPI was used for nuclei observation. Images were captured by a special OLYMPUS DX51 fluorescence microscope (Tokyo, Japan) in a blind manner. For TUNEL detection in vivo, only apoptotic nuclei in the center of the cardiomyocyte were regarded as apoptotic cardiomyocytes [30].
Flow cytometry
Cell apoptosis was further detected by flow cytometry (BD, FACS AriaIII) using PE Annexin V Apoptosis Detection Kit I according to manufacturer's recommendation. Briefly, cells were trypsinized and collected via centrifugation at 1,000g for 5 minutes. After washed and calculated, cells were stained with 7-aminoactinomycin D (7-AAD) and Annexin V for 15 minutes in the dark, and then 400μl of 1×binding buffer was added to each tube before analysis.
Enzyme linked immunosorbent assay
Medium from treated CFs was collected and the concentration of soluble Fas L in cell supernatant was detected by enzyme linked immunosorbent assay (Elisa) according to the manufacturer's protocol using a commercial kit from Sigma-Aldrich (RAB0179).
Statistical analysis
All data in this research were reported as mean ± standard error of the mean (SEM) and analyzed by SPSS22.0 software. Statistical analyses among three or more groups were done by one way ANOVA followed by Tukey post hoc test. A P value less than 0.05 was considered statistically significant.
Results
RA attenuated DOX-induced cardiotoxicity in mice
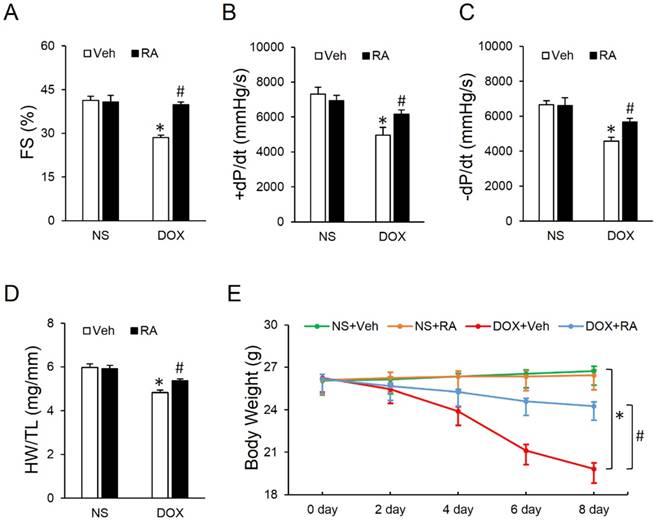
Mice were challenged with a single intraperitoneal injection of DOX (15mg/kg) for 8 days to generate DOX-induced cardiotoxicity according to our previous study [9]. As shown in Figure 1A-C, DOX exposure led to markedly decreased fractional shortening (FS) and ±dp/dt, which were significantly attenuated by RA administration. Studies from our lab and the others have addressed that DOX injection could result in massive cell death in the heart, which contributes to the progression of cardiac dysfunction [9, 31]. Consistent with the loss of cardiac cells, we observed that heart weight (HW) was decreased after DOX injection, whereas RA treatment could minimize the magnitude of HW reduction, as evidenced by the ratio of HW and tibia length (HW/TL) (Figure 1D). Besides, the ultrasound structure of ventricular wall became thinner in response to DOX treatment, as determined by the decreased interventricular septal thickness at systole (IVSs) and left ventricular posterior wall thickness at end-systole (LVPWs), which were also improved after RA supplementation (Table S1). Meanwhile, our data showed a decline of heart rate and blood pressure in DOX-treated mice compared with that in control group, however, mice with RA protection had unchanged heart rate and blood pressure whatever in the presence or absence of DOX (Table S1), which was in line with our previous data [21]. Body weight loss due to chemotherapeutic agents and cachexia syndrome predicts bad prognosis in cancer patients via compromising functions of skeletal muscle, adipose tissue and also internal organs including the liver, kidneys, lungs, especially the heart [32]. Previous studies indicated that DOX application significantly decreased the body weight in cancer patients [33], but intriguingly, we found that systematic administration of RA could attenuate DOX-induced body weight loss in mice, which raises the possibility for its clinical use (Figure 1E). Collectively, we concluded that RA could attenuate DOX-induced cardiotoxicity in mice.
Rosmarinic acid (RA) attenuated doxorubicin (DOX)-induced cardiotoxicity in mice. (A) Fractional shortening (FS) of mice with or without RA protection after DOX injection (n=8). (B-C) Hemodynamic analysis (n=8). (D) Statistical results of the heart weight (HW)/ tibia length (TL) (n=8). (E) Alteration of body weight (BW) among the four groups (n=8). Values represent the mean±SEM. *P<0.05 versus normal saline (NS)+Vehicle (Veh), #P<0.05 versus DOX+Veh.
RA significantly mitigated DOX-induced cardiomyocyte apoptosis in vivo but less in vitro
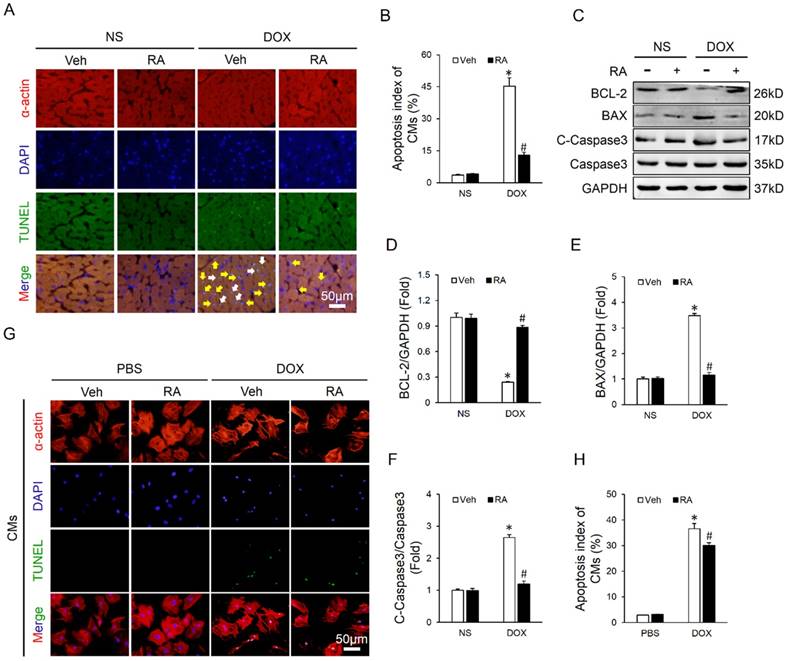
Cardiomyocyte apoptosis is an important event in the process of DOX-induced cardiotoxicity and contributes to the initiation and progression of cardiac dysfunction [9, 31]. As shown in Figure 2A-B, DOX injection resulted in distinct cardiomyocyte apoptosis in vivo, and RA lavage mitigated this pathological alteration. Concomitantly, there was a marked attenuation of apoptosis-related proteins, as confirmed by the upregulated BCL-2 and downregulated BAX and C-Caspase3 (Figure 2C-F). To verify the protective effect of RA on DOX-triggered cardiomyocyte apoptosis in vitro, neonatal rat CMs were prepared and treated. Surprisingly, we found that RA treatment had almost negligible beneficial effect on DOX directly induced cardiomyocyte apoptosis in vitro (Figure 2G-H). The paradoxical results revealed that cardiomyocytes might not be the direct target of RA in the regulation of cardiomyocyte apoptosis, or in other words, RA might regulate DOX-induced cardiomyocyte apoptosis via other cell types.
RA alleviated cardiomyocyte apoptosis via cardiac fibroblast in vitro
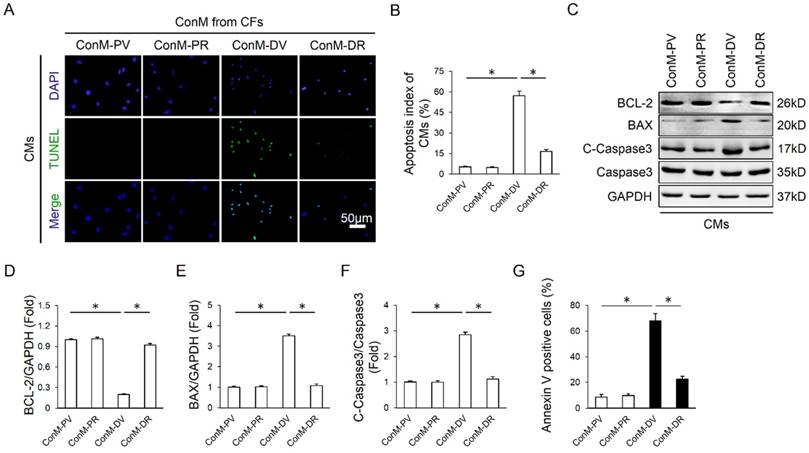
Emerging researches showed that multiple cell types were involved in the regulation of cardiomyocyte apoptosis in a direct or indirect manner, including endothelial cells, inflammatory cells and CFs, etc [16, 34]. Considering the fact that CFs are the most abundant cell type in the heart and our previous data also disclosed that RA could target CFs, we therefore laid our emphasis on CFs. Because earlier studies supposed the existence of a paracrine interaction between CFs and CMs, we hypothesized that RA might directly control the expression of paracrine factors in CFs and alleviate cardiomyocyte apoptosis in a paracrine-regulating manner. As expected, ConM from DOX-treated CFs (ConM-DV) elicited a significant increase of cardiomyocyte apoptosis in vitro, even more obvious than DOX directly induced cardiomyocyte apoptosis, which is coincident with a recent study demonstrating that CFs are the key lineage responsible for DOX-induced cardiotoxicity (Figure 3A-B, Figure 2G-H) [16]. However, ConM from DOX-treated CFs with RA protection (ConM-DR) caused less cardiomyocyte apoptosis compared with that treated with ConM-DV, further corroborated by the attenuation of apoptosis-related proteins (Figure 3A-F). And the protective effect of RA was further verified by flow cytometry data in vitro (Figure 3G). Therefore, we concluded that inhibition of the paracrine interplay between CMs and CFs might be responsible for RA-mediated protective effects on cardiomyocyte apoptosis.
RA significantly mitigated DOX-induced cardiomyocyte apoptosis in vivo but less in vitro. (A-B) Co-staining of α-actin with TUNEL in hearts and the statistical results (n=6). White arrows indicate TUNEL-positive cardiomyocytes, whereas yellow arrows indicate TUNEL-positive non-myocytes. (C-F) Western blot and statistical results (n=6). (G-H) Co-staining of α-actin with TUNEL in neonatal rat cardiomyocytes and the statistical results (n=4). Values represent the mean±SEM. *P<0.05 versus NS+Veh, #P<0.05 versus DOX+Veh. In figure 2h, *P<0.05 versus phosphate buffered saline (PBS)+Veh, #P<0.05 versus DOX+Veh.
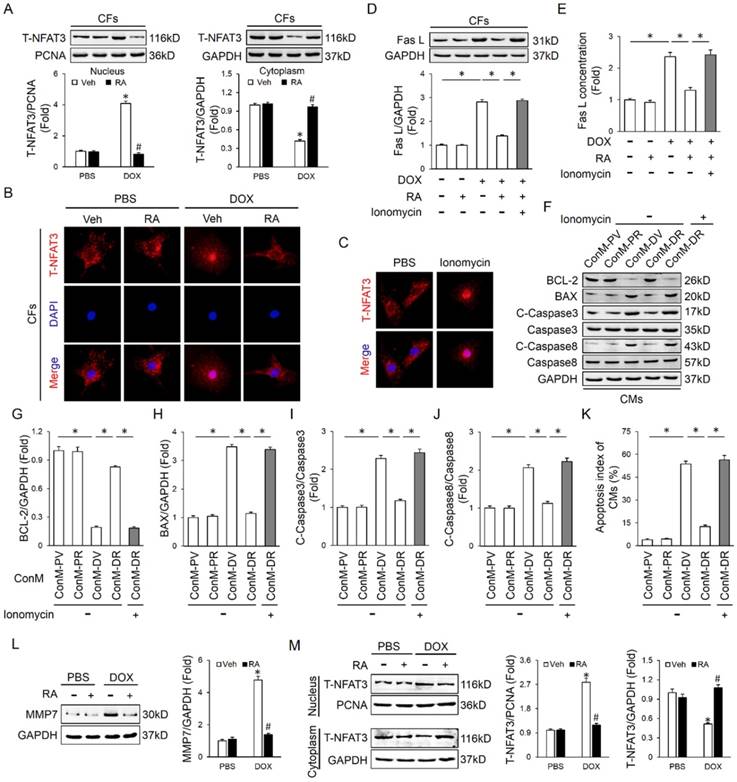
RA suppressed Fas L expression and release in cardiac fibroblasts that mediate DOX-induced cardiotoxicity
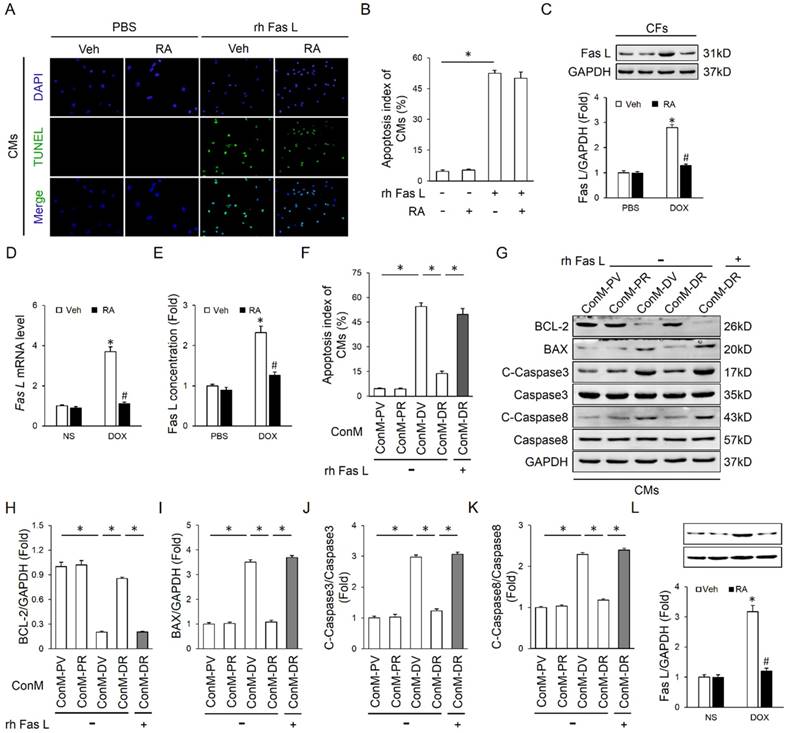
Fas/Fas L system plays a crucial role in delivering apoptotic signals in organisms and has been implicated in DOX-induced cardiomyocyte apoptosis and cardiotoxicity [6, 16]. More importantly, Fas L could be cleaved and released out of the cell to induce adjacent cell apoptosis via a paracrine manner [16]. As shown in Figure S1A-B, Fas L expression in CFs was notably induced after DOX treatment, which was less in CMs whatever in the baseline or after induction. In addition, DOX treatment could also increase the level of Fas L in ConM from CFs (Figure 4D). To confirm whether the apoptosis of cardiomyocytes was induced by Fas L secreted from CFs, Fas-Fc, which can neutralize Fas L, was added to the medium before stimulation by DOX. Significantly, CMs cultured in medium conditioned by CFs treated with DOX and Fas-Fc exhibited less apoptosis when compared with cells cultured in medium conditioned by CFs treated with DOX only (Figure S1C-D), thus indicating that Fas L produced by CFs could facilitate cardiomyocyte apoptosis.
We first detected whether RA performed as a neutralizing agent of the existing Fas L and thus blocked its biological effect. As shown in Figure 4A-B, RA treatment could not offset rh Fas L-induced cardiomyocyte apoptosis, which suggested that RA treatment was unable to block the apoptosis-inducing effect of the existing Fas L. Nevertheless, the mRNA and protein level of Fas L in DOX-treated CFs was decreased after RA treatment with no alteration in cardiomyocytes, and meanwhile DOX-triggered release of Fas L to ConM was also suppressed (Figure 4C-E, Figure S1E). Therefore, RA-mediated protective effect on cardiomyocyte apoptosis might be ascribed to its suppression of Fas L in CFs. To address this question, we added rh Fas L to ConM-DR to replenish the decreased Fas L, and observed that Fas L replenishment could abolish the protective effect of RA on cardiomyocyte apoptosis (Figure 4F-J). Specially, Caspase8 is the classical downstream effector of Fas/Fas L pathway that can be cleaved to C-Caspase8 to initiate Fas L-induced cell apoptosis [35]. And we found that RA treatment overtly downregulated the expression of C-Caspase8, which was reversed with the supplementation of rh Fas L (Figure 4G, K). Taken together, we suggested that RA protected cardiomyocyte apoptosis via suppressing Fas L expression in CFs and its release to the conditioned medium, which could trigger cardiomyocyte apoptosis via a paracrine manner.
RA alleviated cardiomyocyte apoptosis via cardiac fibroblast in vitro. (A-B) Representative TUNEL images and statistical results of conditioned medium (ConM) from cardiac fibroblasts (CFs) on cardiomyocyte apoptosis. Cardiomyocytes (CMs) were incubated for another 24h in ConM prepared from CFs pretreated with RA (20μg/ml) 16 hours followed by DOX (1μM)) for 8h, and then immunostained for TUNEL assay in CMs (n=4). (C-F) Western blot and statistical analysis of cultured CMs treated with ConM (n=4). (G) Flow cytometry results (n=4). Values represent the mean±SEM. *P<0.05 versus the matched control.
NFAT inactivation and MMP7 suppression was responsible for RA-mediated inhibition on Fas L
Finally, we tried to clarify the possible mechanism that is responsible for RA-mediated inhibition on Fas L in CFs. A previous study demonstrated that RA inhibited NFAT activation in T cells, and NFAT is intimately related to cell apoptosis [36]. Therefore, we speculated whether NFAT was responsible for RA-mediated inhibition on Fas L. Due to the fact that NFAT3 (NFATc4) in CFs is predominantly located in cytoplasm under control conditions and markedly accumulated in the nucleus after activation, we thus detected NFAT3 nuclear translocation as a reflection of NFAT activation [37]. In line with the previous finding, we also found that RA treatment could inhibit the activation of NFAT, as reflected by NFAT3 nuclear translocation (Figure 5A-B). To evaluate the necessity of NFAT inactivation in RA-mediated inhibition on Fas L, we treated CFs with ionomycin (an activator of NFAT) and found that ionomycin treatment could abrogate the inhibitory effect of RA on Fas L expression and its release to the conditioned medium (Figure 5C-E). Accordingly, attenuated molecular alterations together with the improved cardiomyocyte apoptosis by RA were also retarded (Figure 5F-K). Previous studies indicated that MMPs were responsible for Fas L cleavage and RA has been proved to suppressed MMPs expression in tumor cells, we thus detected whether RA treatment decreased MMPs in CFs. In line with the decreased release of Fas L by CFs, we also observed that RA incubation inhibited MMP7 expression, the one was involved in Fas L cleavage and release. These data defined a crucial role of NFAT inactivation and MMPs suppression in RA-mediated suppression on Fas L and protective effect on cardiomyocyte apoptosis.
RA suppressed Fas L expression and release in cardiac fibroblasts that mediate DOX-induced cardiotoxicity. (A-B) Effect of RA on recombinant human Fas L (rh Fas L)-induced cardiomyocyte apoptosis. CMs were pretreated with RA (20μg/ml) or equal volume of vehicle for 16 hours followed with PBS or rh Fas L (5μg/ml) for another 8 hours, and then were performed with TUNEL staining (n=4). (C-D) Protein and mRNA level of Fas L in neonatal rat heart CFs (n=4). (E) Fas L concentration in ConM (n=5). (F) Replenishment of rh Fas L abolished the anti-apoptotic effect of RA on CMs. rh Fas L was replenished (5μg/ml) to neonatal rat heart CFs-derived ConM before incubated with CMs (n=4). (G-K) Western blot and statistical results in CMs (n=4). (L) Fas L expression in adult mouse heart CFs (n=4). Values represent the mean±SEM. *P<0.05 versus the matched control. In figure 4C-E, *P<0.05 versus PBS+Veh, #P<0.05 versus DOX+Veh.
Discussion
DOX is widely used for cancer therapy with usage limitation due to the cumulative dose-dependent cardiotoxicity that leads to irreversible degenerative cardiomyopathy and heart failure in cancer patients [1, 7]. Our group and others have verified the crucial role of cardiomyocyte apoptosis in the process of DOX-induced cardiotoxicity, and pharmacologic inhibition of cardiomyocyte apoptosis evidently improved DOX-induced cardiac dysfunction [9, 31]. In the present study, we found that cardiomyocyte apoptosis in response to DOX was caused by Fas L secreted from CFs, which is in line with a recent study [16], and RA supplementation could inhibit the expression of Fas L in CFs and its release to ConM, and subsequently alleviate cardiomyocyte apoptosis and protect against DOX-induced cardiac dysfunction. However, RA pretreatment was incapable of blocking the apoptosis-inducing effect of the pre-existing Fas L. Previous studies revealed a fact that the antitumor activity of DOX was likely to be distinct from the mechanism of its cardiotoxicity [38, 39], consistently, most currently available data suggested that the chemotherapeutic effect of DOX on malignancies was not Fas/Fas L-dependent but rather was executed through a P53-mediated pathway [39, 40], which was diametrically opposed to that in DOX-induced cardiotoxicity [41]. More inspiring, in addition to the efficiency in alleviating cardiomyocyte apoptosis, RA has been also reported to induce apoptosis and necrosis of cancer cells [17, 42]. Moreover, a recent study from Han et al indicated that RA possessed the effect of inhibiting metastasis of colorectal cancer [43]. All the results proved that RA could be an effective and safe therapeutic agent against cardiomyocyte apoptosis and DOX-induced cardiotoxicity.
NFAT inactivation was responsible for RA-mediated inhibition on Fas L. (A-B) Western blot and representative immunofluorescence images of NFAT3 nuclear translocation of CFs (n=4). (C) Ionomycin (100nM) was added to CFs with RA to induce NFAT activation and nuclear translocation (n=5). (D-E) Expression of Fas L in CFs and its release to ConM (n=4-5). (F-K) Western blot and statistical results of CMs (n=4). (L) MMP7 protein levels in neonatal rat heart CFs with or without RA protection (n=4). (M) NFAT3 nuclear translocation data in adult mouse heart CFs by western blot (n=4). Values represent the mean±SEM. *P<0.05 versus the matched control. In figure 5A, L-M, *P<0.05 versus PBS+Veh, #P<0.05 versus DOX+Veh.
Fas/Fas L system is ubiquitously expressed in various cell types and delivers apoptotic signals in organisms via either paracrine or autocrine manner. This death signaling is initiated through the interaction of Fas L and Fas, and propagated via the downstream caspase cascade [44, 45]. Previous studies indicated that the expression of Fas L in cardiac cells was upregulated in response to DOX [6, 16], and DOX treatment could also increase the protein level of Fas in cardiac tissues [46]. Meanwhile, Kalivendi et al reported that DOX treatment led to the upregulation of Fas L expression and caspase activation in H9C2 cells and elicited the apoptotic cascade [6]. More importantly, supplementation with Fas L neutralizing antibody notably alleviated DOX-induced cardiomyocyte apoptosis both in vivo and in vitro [41, 47]. Despite all these findings suggested the contribution of Fas/Fas L to cardiomyocyte apoptosis and DOX-induced cardiotoxicity, most of them focused only on CMs with few studies on CFs [41]. However, in the present study, we found that DOX treatment significantly impelled the expression of Fas L in CFs that was less in CMs, which is consistent with the findings reported by Zhan et al [16]. In addition to the transmembrane form, Fas L could also be processed to soluble Fas L by MMPs and released to the extracellular milieu, which may serve to induce cell apoptosis via a paracrine manner [48]. Herein, we found that DOX stimulation enhanced the release of soluble Fas L from CFs, which in turn facilitated cardiomyocyte apoptosis. RA pretreatment inhibited the induction and release of Fas L and blocked cardiomyocyte apoptosis, whereas Fas L replenishment abrogated the protective effect of RA. In addition, we also found that RA treatment had almost negligible beneficial effect on DOX directly induced cardiomyocyte apoptosis. Based on these findings, we concluded that RA might alleviate DOX-induced cardiomyocyte apoptosis via CFs in a paracrine dependent manner.
NFAT family was initially identified in T cells and has been shown to regulate the expression of a wide range of genes involved in immune system responses [49]. However, increasing studies implied that NFAT family extended well beyond its immune-regulating function in T cells and played a crucial role in orchestrating cell apoptosis [50]. Previous studies showed that NFAT1 knockout or NFAT1/NFAT4 double knockout mice were linked to resistance to cell death [51, 52]. Besides, as a transcriptional factor, NFAT family was also implicated in the regulation of various apoptosis-related genes and triggered cell apoptosis in different cell types [53-56], the most important of which was Fas L that can mediate either paracrine or autocrine apoptosis [57]. In the present study, we found that RA treatment significantly alleviated DOX-induced cardiomyocyte apoptosis and cardiac dysfunction via suppressing Fas L expression and release in a NFAT-dependent manner in CFs. Since NFAT is a transcription factor that controls the expression of Fas L, therefore the decreased release of Fas L might be secondary to the downregulation of the total expression levels. In addition to the total expression levels, the release of soluble Fas L is also dependent on the cleaved process by MMPs [48]. Considering numerous investigations have verified the inhibitory activity of RA on MMPs [43], we therefore detected the expression of MMP7 in CFs, for its role in Fas L cleavage and found that RA treatment also suppressed MMP7 expression in CFs.
Taken together, we found that RA administration alleviated cardiomyocyte apoptosis via cardiac fibroblast in a paracrine manner. The beneficial effect of RA on cardiomyocyte apoptosis might be ascribed to its inhibition on NFAT activation- and MMP7 cleavage-dependent Fas L expression in CFs and release to the extracellular milieu. Our research collectively provided evidence for the application of RA in the treatment of DOX-induced cardiomyocyte apoptosis and cardiotoxicity.
Supplementary Material
Supplementary figure and table.
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (No: 81270303, 81470516, 81470402, 81500184, 81700254), the Key Project of the National Natural Science Foundation (No. 81530012), Scientific Action Plans for the Prevention and Treatment of Major Diseases-Cardiovascular Diseases (2016ZX-008-01), the Fundamental Research Funds for the Central Universities (No. 2042017kf0085, 2042015kf0073), China Postdoctoral Science Foundation (No. 2014M562068), Natural Science Foundation of Hubei Province of China.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Carvalho C, Santos RX, Cardoso S. et al. Doxorubicin: the good, the bad and the ugly effect. Curr Med Chem. 2009;16:3267-3285
2. Moliterni A, Menard S, Valagussa P. et al. HER2 overexpression and doxorubicin in adjuvant chemotherapy for resectable breast cancer. J Clin Oncol. 2003;21:458-462
3. Petrioli R, Fiaschi AI, Francini E. et al. The role of doxorubicin and epirubicin in the treatment of patients with metastatic hormone-refractory prostate cancer. Cancer Treat Rev. 2008;34:710-718
4. Takemura G, Fujiwara H. Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog Cardiovasc Dis. 2007;49:330-352
5. Silber JH, Barber G. Doxorubicin-induced cardiotoxicity. N Engl J Med. 1995;333:1359-1360
6. Kalivendi SV, Konorev EA, Cunningham S. et al. Doxorubicin activates nuclear factor of activated T-lymphocytes and Fas ligand transcription: role of mitochondrial reactive oxygen species and calcium. Biochem J. 2005;389:527-539
7. Octavia Y, Tocchetti CG, Gabrielson KL. et al. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol. 2012;52:1213-1225
8. Zhang S, Liu X, Bawa-Khalfe T. et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18:1639-1642
9. Yuan YP, Ma ZG, Zhang X. et al. CTRP3 protected against doxorubicin-induced cardiac dysfunction, inflammation and cell death via activation of Sirt1. J Mol Cell Cardiol. 2018;114:38-47
10. Kamo T, Akazawa H, Komuro I. Cardiac nonmyocytes in the hub of cardiac hypertrophy. Circ Res. 2015;117:89-98
11. Tirziu D, Giordano FJ, Simons M. Cell communications in the heart. Circulation. 2010;122:928-937
12. LaFramboise WA, Scalise D, Stoodley P. et al. Cardiac fibroblasts influence cardiomyocyte phenotype in vitro. Am J Physiol Cell Physiol. 2007;292:C1799-C1808
13. Bang C, Batkai S, Dangwal S. et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. 2014;124:2136-2146
14. Pedrotty DM, Klinger RY, Kirkton RD. et al. Cardiac fibroblast paracrine factors alter impulse conduction and ion channel expression of neonatal rat cardiomyocytes. Cardiovasc Res. 2009;83:688-697
15. Zhang W, Tao A, Lan T. et al. Carbon monoxide releasing molecule-3 improves myocardial function in mice with sepsis by inhibiting NLRP3 inflammasome activation in cardiac fibroblasts. Basic Res Cardiol. 2017;112:16
16. Zhan H, Aizawa K, Sun J. et al. Ataxia telangiectasia mutated in cardiac fibroblasts regulates doxorubicin-induced cardiotoxicity. Cardiovasc Res. 2016;110:85-95
17. Moon DO, Kim MO, Lee JD. et al. Rosmarinic acid sensitizes cell death through suppression of TNF-alpha-induced NF-kappaB activation and ROS generation in human leukemia U937 cells. Cancer Lett. 2010;288:183-191
18. Han J, Wang D, Ye L. et al. Rosmarinic Acid Protects against Inflammation and Cardiomyocyte Apoptosis during Myocardial Ischemia/Reperfusion Injury by Activating Peroxisome Proliferator-Activated Receptor Gamma. Front Pharmacol. 2017;8:456
19. Gerogianni PS, Chatziathanasiadou MV, Diamantis DA. et al. Lipophilic ester and amide derivatives of rosmarinic acid protect cells against H2O2-induced DNA damage and apoptosis: The potential role of intracellular accumulation and labile iron chelation. Redox Biol. 2018;15:548-556
20. Lee HJ, Cho HS, Park E. et al. Rosmarinic acid protects human dopaminergic neuronal cells against hydrogen peroxide-induced apoptosis. Toxicology. 2008;250:109-115
21. Zhang X, Ma ZG, Yuan YP. et al. Rosmarinic acid attenuates cardiac fibrosis following long-term pressure overload via AMPKalpha/Smad3 signaling. Cell Death Dis. 2018;9:102
22. Xu SC, Ma ZG, Wei WY. et al. Bezafibrate Attenuates Pressure Overload-Induced Cardiac Hypertrophy and Fibrosis. Ppar Res. 2017;2017:5789714
23. Ma ZG, Dai J, Wei WY. et al. Asiatic Acid Protects against Cardiac Hypertrophy through Activating AMPKalpha Signalling Pathway. Int J Biol Sci. 2016;12:861-871
24. Ma ZG, Dai J, Zhang WB. et al. Protection against cardiac hypertrophy by geniposide involves the GLP-1 receptor / AMPKalpha signalling pathway. Br J Pharmacol. 2016;173:1502-1516
25. Ma ZG, Yuan YP, Zhang X. et al. Piperine Attenuates Pathological Cardiac Fibrosis Via PPAR-gamma/AKT Pathways. EBioMedicine. 2017;18:179-187
26. Wei WY, Ma ZG, Xu SC. et al. Pioglitazone Protected against Cardiac Hypertrophy via Inhibiting AKT/GSK3beta and MAPK Signaling Pathways. Ppar Res. 2016;2016:9174190
27. Ma ZG, Zhang X, Yuan YP. et al. A77 1726 (leflunomide) blocks and reverses cardiac hypertrophy and fibrosis in mice. Clin Sci (Lond). 2018;132:685-699
28. Ma ZG, Dai J, Yuan YP. et al. T-bet deficiency attenuates cardiac remodelling in rats. Basic Res Cardiol. 2018;113:19
29. Ma ZG, Yuan YP, Xu SC. et al. CTRP3 attenuates cardiac dysfunction, inflammation, oxidative stress and cell death in diabetic cardiomyopathy in rats. Diabetologia. 2017;60:1126-1137
30. Yoon YS, Uchida S, Masuo O. et al. Progressive attenuation of myocardial vascular endothelial growth factor expression is a seminal event in diabetic cardiomyopathy: restoration of microvascular homeostasis and recovery of cardiac function in diabetic cardiomyopathy after replenishment of local vascular endothelial growth factor. Circulation. 2005;111:2073-2085
31. Mukhopadhyay P, Batkai S, Rajesh M. et al. Pharmacological inhibition of CB1 cannabinoid receptor protects against doxorubicin-induced cardiotoxicity. J Am Coll Cardiol. 2007;50:528-536
32. Simons JP, Schols AM, Buurman WA. et al. Weight loss and low body cell mass in males with lung cancer: relationship with systemic inflammation, acute-phase response, resting energy expenditure, and catabolic and anabolic hormones. Clin Sci (Lond). 1999;97:215-223
33. Kratz F, Roth T, Fichiner I. et al. In vitro and in vivo efficacy of acid-sensitive transferrin and albumin doxorubicin conjugates in a human xenograft panel and in the MDA-MB-435 mamma carcinoma model. J Drug Target. 2000;8:305-318
34. Kuramochi Y, Cote GM, Guo X. et al. Cardiac endothelial cells regulate reactive oxygen species-induced cardiomyocyte apoptosis through neuregulin-1beta/erbB4 signaling. J Biol Chem. 2004;279:51141-51147
35. Wieder T, Essmann F, Prokop A. et al. Activation of caspase-8 in drug-induced apoptosis of B-lymphoid cells is independent of CD95/Fas receptor-ligand interaction and occurs downstream of caspase-3. Blood. 2001;97:1378-1387
36. Kang MA, Yun SY, Won J. Rosmarinic acid inhibits Ca2+-dependent pathways of T-cell antigen receptor-mediated signaling by inhibiting the PLC-gamma 1 and Itk activity. Blood. 2003;101:3534-3542
37. Herum KM, Lunde IG, Skrbic B. et al. Syndecan-4 signaling via NFAT regulates extracellular matrix production and cardiac myofibroblast differentiation in response to mechanical stress. J Mol Cell Cardiol. 2013;54:73-81
38. Kluza J, Marchetti P, Gallego MA. et al. Mitochondrial proliferation during apoptosis induced by anticancer agents: effects of doxorubicin and mitoxantrone on cancer and cardiac cells. Oncogene. 2004;23:7018-7030
39. Wang S, Konorev EA, Kotamraju S. et al. Doxorubicin induces apoptosis in normal and tumor cells via distinctly different mechanisms. intermediacy of H(2)O(2)- and p53-dependent pathways. J Biol Chem. 2004;279:25535-25543
40. Eischen CM, Kottke TJ, Martins LM. et al. Comparison of apoptosis in wild-type and Fas-resistant cells: chemotherapy-induced apoptosis is not dependent on Fas/Fas ligand interactions. Blood. 1997;90:935-943
41. Nakamura T, Ueda Y, Juan Y. et al. Fas-mediated apoptosis in adriamycin-induced cardiomyopathy in rats: In vivo study. Circulation. 2000;102:572-578
42. Wu CF, Hong C, Klauck SM. et al. Molecular mechanisms of rosmarinic acid from Salvia miltiorrhiza in acute lymphoblastic leukemia cells. J Ethnopharmacol. 2015;176:55-68
43. Han YH, Kee JY, Hong SH. Rosmarinic Acid Activates AMPK to Inhibit Metastasis of Colorectal Cancer. Front Pharmacol. 2018;9:68
44. Nakamura T, Imai Y, Matsumoto T. et al. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell. 2007;130:811-823
45. Esmailzadeh S, Huang Y, Su MW. et al. BIN1 tumor suppressor regulates Fas/Fas ligand-mediated apoptosis through c-FLIP in cutaneous T-cell lymphoma. Leukemia. 2015;29:1402-1413
46. Lien YC, Daosukho C, St CD. TNF receptor deficiency reveals a translational control mechanism for adriamycin-induced Fas expression in cardiac tissues. Cytokine. 2006;33:226-230
47. Yamaoka M, Yamaguchi S, Suzuki T. et al. Apoptosis in rat cardiac myocytes induced by Fas ligand: priming for Fas-mediated apoptosis with doxorubicin. J Mol Cell Cardiol. 2000;32:881-889
48. Vargo-Gogola T, Crawford HC, Fingleton B. et al. Identification of novel matrix metalloproteinase-7 (matrilysin) cleavage sites in murine and human Fas ligand. Arch Biochem Biophys. 2002;408:155-161
49. Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472-484
50. Mognol GP, Carneiro FR, Robbs BK. et al. Cell cycle and apoptosis regulation by NFAT transcription factors: new roles for an old player. Cell Death Dis. 2016;7:e2199
51. Caetano MS, Vieira-de-Abreu A, Teixeira LK. et al. NFATC2 transcription factor regulates cell cycle progression during lymphocyte activation: evidence of its involvement in the control of cyclin gene expression. Faseb J. 2002;16:1940-1942
52. Ranger AM, Oukka M, Rengarajan J. et al. Inhibitory function of two NFAT family members in lymphoid homeostasis and Th2 development. Immunity. 1998;9:627-635
53. Robbs BK, Lucena PI, Viola JP. The transcription factor NFAT1 induces apoptosis through cooperation with Ras/Raf/MEK/ERK pathway and upregulation of TNF-alpha expression. Biochim Biophys Acta. 2013;1833:2016-2028
54. Ueffing N, Schuster M, Keil E. et al. Up-regulation of c-FLIP short by NFAT contributes to apoptosis resistance of short-term activated T cells. Blood. 2008;112:690-698
55. Gomez-Sintes R, Lucas JJ. NFAT/Fas signaling mediates the neuronal apoptosis and motor side effects of GSK-3 inhibition in a mouse model of lithium therapy. J Clin Invest. 2010;120:2432-2445
56. Jayanthi S, Deng X, Ladenheim B. et al. Calcineurin/NFAT-induced up-regulation of the Fas ligand/Fas death pathway is involved in methamphetamine-induced neuronal apoptosis. Proc Natl Acad Sci U S A. 2005;102:868-873
57. Su X, Cheng J, Liu W. et al. Autocrine and paracrine apoptosis are mediated by differential regulation of Fas ligand activity in two distinct Jurkat T cell populations. J Immunol. 1998;160:5288-5293
Author contact
![]() Corresponding authors: Qi-Zhu Tang, Department of Cardiology, Renmin Hospital of Wuhan University, Cardiovascular Research Institute, Hubei Key Laboratory of Cardiology, Wuhan University at Jiefang Road 238, Wuhan 430060, RP China. Tel.: +86 27 88073385; Fax: +86 27 88042292. E-mail: qztangedu.cn or Wei Deng, Department of Cardiology, Renmin Hospital of Wuhan University, Cardiovascular Research Institute, Hubei Key Laboratory of Cardiology, Wuhan University at Jiefang Road 238, Wuhan 430060, RP China. Tel.: +86 27 88073385; Fax: +86 27 88042292. E-mail: vivideng1982edu.cn
Corresponding authors: Qi-Zhu Tang, Department of Cardiology, Renmin Hospital of Wuhan University, Cardiovascular Research Institute, Hubei Key Laboratory of Cardiology, Wuhan University at Jiefang Road 238, Wuhan 430060, RP China. Tel.: +86 27 88073385; Fax: +86 27 88042292. E-mail: qztangedu.cn or Wei Deng, Department of Cardiology, Renmin Hospital of Wuhan University, Cardiovascular Research Institute, Hubei Key Laboratory of Cardiology, Wuhan University at Jiefang Road 238, Wuhan 430060, RP China. Tel.: +86 27 88073385; Fax: +86 27 88042292. E-mail: vivideng1982edu.cn
Received 2018-9-13
Accepted 2018-11-9
Published 2019-1-1