ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2019; 15(4):870-881. doi:10.7150/ijbs.30488 This issue Cite
Research Paper
BMI1 Deficiency Results in Female Infertility by Activating p16/p19 Signaling and Increasing Oxidative Stress
Rong Wang1,2*, Xian Xue1,2*, Yang Wang1, Haiyang Zhao1, Yuling Zhang3, Hui Wang1 ![]() , Dengshun Miao1,2
, Dengshun Miao1,2
1. State Key Laboratory of Reproductive Medicine, Department of Anatomy, Histology and Embryology, Nanjing Medical University, Nanjing, Jiangsu, China
2. The Research Center for Bone and Stem Cells, Nanjing Medical University, Nanjing, Jiangsu, China
3. Department of Ultrasound, Taikang Xianlin Drum Tower Hospital, Nanjing, Jiangsu, China
*These authors contributed equally to this work.
Abstract

The polycomb repressor B lymphoma Mo-MLV insertion region 1 (BMI1) is a core composition of polycomb repressive complex 1 (PRC1) and contributes to diverse fundamental cellular processes including cell senescence, apoptosis and proliferation. To investigate the role and mechanism of BMI1 in maintaining normal female reproductive function, we compared the differences in reproductive phenotypes between Bmi1-deficient and wild-type female mice. The Bmi1-deficient female mice were then supplemented with N-acetylcysteine in their drinking water to explore whether antioxidant supplementation could improve reproductive dysfunction caused by BMI1 deficiency. The results revealed that Bmi1 deletion resulted in complete infertility in female mice, estrous cycle disorder, and follicular developmental disorders. The reactive oxygen species levels in the ovarian tissue were increased; the ability of antioxidant enzymes was downregulated; the expression levels of p19 and p53 proteins were significantly upregulated. We also found that oocytes derived from Bmi1-deficient mice could not develop into embryos by in vitro fertilization and in vitro culture of embryos. Furthermore, supplementation with the antioxidant NAC not only improved the reproductive defects caused by Bmi1 deletion, but also largely rescued the ability of Bmi1-deficient oocytes to develop into embryos in vitro. These results indicated that cells lacking Bmi1 resulted in female infertility by activating the p16/p19 signaling pathway, increasing oxidative stress and DNA damage, inhibiting granulosa cell proliferation, and inducing granulosa cell apoptosis. Thus, BMI1 may be a novel potential target for the clinical treatment of female infertility.
Keywords: BMI1, female infertility, oxidative stress, DNA damage, antioxidant
Introduction
B lymphoma Mo-MLV insertion region 1 (BMI1) belongs to the polycomb group (PcG) protein family, which are transcriptional repressors [1]. As a proto-oncogene, Bmi1 is associated with the regulation of cell differentiation, and is expressed highly in stem cells and several types of malignant tumors. BMI1 also plays a role in proliferation and apoptosis of tumor cells, regulation of chromosome stability, and self-renewal capacity [2, 3]. Recent studies have shown that BMI1 is related to cancer stem cells among tissues and organs, including head-and-neck, digestive system, hematopoietic system, respiratory system, mammary gland, genitourinary system, and skin [4-6]. Mechanistic studies revealed that BMI1 deficiency affects premature senescence, which involves oxidative stress and ongoing DNA damage. BMI1 deficiency, through the INK4a/p16 (also known as cyclin dependent kinase inhibitor 2A) and INK4d/p19 (cyclin dependent kinase inhibitor 2D) signaling pathways, inhibits CyclinD1, cell dependent kinase (CDK)4/6, and p53, which causes cell cycle arrest, growth arrest, cell senescence, and apoptosis [7, 8]. Therefore, oxidative stress status and the resulting changes in a series of downstream molecules may be the core mechanism of the negative systemic effect and premature aging caused by BMI1 deficiency.
Oxidative stress plays an essential role in critical biological processes in human reproduction [9]. The phenotype of oxidative damage to the reproductive system is similar to that of reproduction aging, and with age, germ cells are particularly sensitive to oxidative stress. In addition, the imbalance between reactive oxygen species (ROS) and protective antioxidants affects the entire reproductive lifespan in males and females [10]. A previous study suggested that in normal follicle development, there is a certain amount of ROS; however, excessive ROS not only reduces the amount and quality of granulosa cells, but also influences the whole reproductive stage, even causing infertility [11]. This could reduce oocytes numbers and qualities, upregulate aging indicators, and eventually cause ovulated oocyte defects [12]. Oxidative stress could also affect spermatogenesis, sperm function, and the spermatogenic microenvironment, eventually causing infertility [13, 14]. Therefore, researchers are keen to determine the role of BMI1 in the reproductive system and whether it is regulated by oxidative stress.
In our previous studies, we observed that BMI1 is not only expressed in nervous tissue and bone tissue, but also in testes and ovaries. BMI1 deficiency caused infertility in male mice, accompanied by smaller testes, oligospermia, and sperm malformation [15-18]. Studies indicated that BMI1 deficiency reduces testosterone syntheses, increases oxidative stress and DNA damage, activates p16 and p19 signaling pathways, inhibits germ cell proliferation, and inducing germ cell apoptosis and sperm malformation in male fertility [19]. However, it is unclear whether BMI1 deficiency contributes to female infertility, and whether antioxidants could rescue female infertility in mice deficient in BMI1.
Therefore, in the present study, 3-week-old Bmi1-/- mice were randomly treated with or without N-acetylcysteine (NAC) in their drinking water. After 4 weeks of treatment, alterations in DNA damage, cell proliferation, and cell cycle-related parameters were analyzed in the ovaries. This study aimed to clarify the role of BMI1 in sustaining female reproduction, and thus could reveal a potential and effective direction for clinical therapy of female infertility.
Materials and Methods
Animals
The Bmi1 heterozygote (Bmi1+/-) mice (129Ola/ FVB/N hybrid background) provided in this study had been backcrossed in the C57BL/6J background and mated to generate Bmi1 homozygote (Bmi1-/-) and their wild-type (WT) littermates, which were genotyped using PCR, as described previously [19]. In this study, all the mice were housed and bred in the Experimental Animal Center in Nanjing Medical University. Animal use was approved by the Institutional Animal Care and Use Committee of Nanjing Medical University (IACUC-1706001).
Vaginal cytology
To assess the time of pubertal initiation in Bmi1-/- mice, we observed the vaginal opening time at 8:00 am, starting with 3-week-old mice. The estrous cycle also was determined using a sterile stick or swab to wipe the vaginal wall gently, which was then mounted on gelatinized slides. The characterization of the estrous cycle was analyzed according to a previous report [20].
Histological analysis
Ovarian tissues were fixed in 4% formaldehyde solution at 4 ºC overnight. After dehydration, permeabilization, wax immersion, and embedding, 5-μm sections were cut, stained with hematoxylin-eosin, and examined by microscopy (Olympus CX31, Japan).
Electron microscopy analysis
Ovarian specimens were prefixed in glutaraldehyde and post-fixed in osmium acid solution. After dehydration in acetone and embedding in epoxy resin, ultrathin sections were cut and loaded on copper screens. These sections were stained with uranium acetate and lead citrate, and then examined by transmission electron microscopy (TEM; Tecnai G2 Spirit Bio TWIN, USA).
Immunohistochemical staining
Briefly, ovarian sections were deparaffinized, hydrated, and immersed in H2O2 in methanol (1:10) to inactivate endogenous peroxidase. Non-specific binding of antibodies was blocked by incubating tissues with 10% goat serum for 1 hour at room temperature. Tissues were then incubated with antibodies recognizing BMI1 (Abjent, USA), proliferating cell nuclear antigen (PCNA) (Abcam, USA), Caspase-3 (BD, USA), 8-Oxo-2'-deoxyguanosine (8-OHdG) (Abcam, USA), phosphorylated H2A histone family member X (γ.H2AX) (CST, USA), or bromodeoxyuridine (BrdU) (CST, USA) overnight at 4 ºC. The sections were then rinsed in phosphate-buffered saline (PBS) and incubated with biotin-conjugated secondary antibodies for 1 hour at room temperature. After washing, the sections were incubated with Elite ABC (Vector, USA) for 1 hour at room temperature, and diaminobenzidine (DAB) (Vector, USA) until the desired stain intensity developed. Sections were counter-stained with hematoxylin, and analyzed under microscopy (Axioskop 2 plus; Zeiss, Germany).
Western blotting
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer containing 10% phenylmethanesulfonylfluoride (PMSF). The solution was precipitated for 1 hour on ice, and then centrifuged for 15 min at 13000 × g. The pellet was dissolved in loading buffer. The protein samples were then resolved through sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and the separated proteins were transferred to polyvinylidene difluoride (PVDF) membranes. After washing with PBS, the PVDF membranes were blocked with 5% skim milk solution, and then incubated with various primary antibodies at 4 ºC overnight. The antibodies used were those recognizing caspase-3 (BD, USA), B-cell CLL/ lymphoma 2 (BCL2) (Santa Cruz Biotechnology, USA), p16 (Santa Cruz Biotechnology, USA), p19 (Cell Signaling Technology, USA), p53 (Santa Cruz Biotechnology, USA), superoxide dismutase 1 (SOD1) (Abcam, USA), superoxide dismutase 2 (SOD2) (Navus, USA), and β-actin (Abcam, USA). The membranes were then incubated with horseradish peroxidase (HRP)-conjugated goat anti-mouse, rabbit anti-goat, or goat-anti rabbit antibodies (KPL, USA). Immunoreactive protein bands were visualized using the enhanced chemiluminescence (ECL) solution (Nanjing Jiancheng Bioengineering Institute, China) and quantified using Scion Image Beta 4.02 (Scion Corporation, NIH).
Real-time reverse transcription-PCR
Total RNA was isolated from cell pellets or cartilage using the TRIzol reagent (Sigma, Japan). For reverse transcription (RT), mRNA was used as the template for single stranded DNA synthesis. A commercial RT-kit (Vazyme, China) was used. The primer sequences (forward and reverse, respectively) were: mouse Sod1 (5′-GGTGAACCAGTTGTGTTGTC-3′, 5′-CCGTCCTTTCCAGCAGTC-3′), mouse Sod2 (5′-GACCTGCCTTACGACTATG-3′, 5′-GAAGAGCGACCTGAGTTG-3′), mouse Gpx1 (glutathione peroxidase 1) (5′-CAATCAGTTCGGACACCAGGAG-3′, 5′ -TCTCACCATTCACTTCGCACTTC-3′), mouse Gsr (glutathione-disulfide reductase) (5′-GGATTGGCTGTGATGAGATG-3′, 5′-CTGAAGAGGTAGGATGAATGG-3′), mouse Cat (catalase) (5′-CAGGTGCGGACATTCTAC-3′, 5′-TTGCGTTCTTAGGCTTCTC-3′), and mouse Txnrd1 (thioredoxin reductase 1) (5′-TCCCTCTCATCAGTTCTATGG-3′, 5′-ACTTGGTGGTTTGCTACGAC-3′). For real-time PCR, the single stranded DNA was used as template with specific primers for the different genes. A commercial kit (Vazyme, China) was used to detect mRNA expression.
Flow cytometry analysis
ROS production in ovaries was tested using diacetyldichlorofluorescein staining (DCFDA, Invitrogen, USA). Fresh ovaries were placed in PBS at 4 ºC, and then the single cell suspension was collected by squeezing the ovaries through gauze. DCFDA (5 mM) was added to the cells. After 30 min of incubation at 37 ºC, the cells were centrifuged at 2000 × g for 5 min and the pellet was washed with PBS twice. Finally, the samples were analyzed using flow cytometry with a FACS-calibur flow cytometer from Becton Dickinson (Germany).
N-acetylcysteine Supplementation
For in vivo administration, Bmi1-/- female mice were randomized to receive drinking water with or without NAC (1 mg×ml-1) for 4 weeks until they were 7 weeks old, which was based on previously published reports [7, 12] and our preliminary research.
Oocyte collection
Mice were stimulated via intraperitoneal injection of 5 IU of pregnant mare serum gonadotropin (PMSG, Ningbo Sansheng, China). To retrieve ovulated oocytes, 46 hours later, mice were injected with 5 IU human chorionic gonadotropin (hCG, NingboSansheng, China). Fourteen hours later, cumulus-oocyte complexes were released by ampulla puncture. The cumulus masses were removed by incubation in hyaluronidase medium (Sigma, Aldrich).
In vitro fertilization and embryo culture
Sperm was separated from the epididymis of 10-week-old male mice. After maintaining in human tubal fluid (HTF) (Shanghai, China) fertilization medium containing bovine serum albumin (BSA; Nanjing, China) for 1.5 hours, dispersed spermatozoa were added to HTF drops containing ovulated oocytes at 37 °C for 4 hours. The embryos were then transferred into potassium simplex optimization medium (KSOM) medium (Millipore, MR-020P-D, USA) and cultured to the blastocyst stage in 5% CO2 in air, and 95% relative humidity at 37 °C. Micrographs were then acquired under a stereoscope (Nikon, Burlingame, California, USA).
Statistical analysis
All data from the image analyses are presented as the mean ± the standard error of the mean (SEM). Statistical analyses were performed using SPSS 16.0 (Version 16.0, SPSS Inc., Chicago, IL, USA). Statistical comparisons were evaluated using Student's test and two-way analysis of variance (ANOVA) when appropriate. Statistical significance was confirmed at P < 0.05.
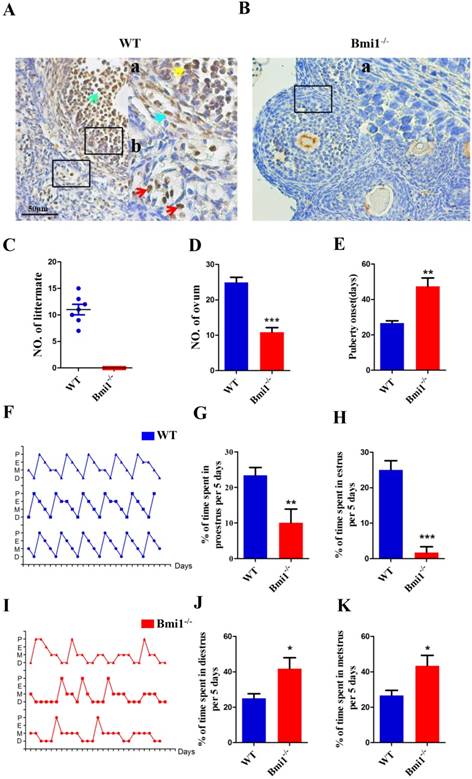
The localization of Bmi1 in ovarian and the effects of BMI1 deficiency on the fertility and estrous cycle. (A) and (B) Bmi1 immunohistochemical staining from 7-week old WT and Bmi1-/- female mice. (A) Green arrow: cumulus cell. (Aa) Yellow arrow: granulosa cell. (Aa) Blue arrow: theca cell. (Ab) Red arrows: mesenchymal cells. Scale bar: 50 µm. (C) Five-week old Bmi1-/- and WT female mice were mated with WT male mice and checked for the number of littermates after 5 weeks. (D) The result of multiple ovulation from 3-week old WT and Bmi1-/- female mice. (E) Statistical result for vaginal opening time. (F) and (I) are estrous cycles of WT and Bmi1-/- female mice. (G) The percentage of time at proestrus per 5 days. (H) The percentage of time at estrus per 5 days. (J) The percentage of time at diestrus per 5 days. (K) The percentage of time at metestrus per 5 days. Each value is the mean ± SEM of determinations in five mice of each group. *: p < 0.05; **: p < 0.01; ***: p < 0.001 compared with WT mice. Bmi1, B lymphoma Mo-MLV insertion region 1; WT, wild-type; SEM; standard error of the mean.
Results
The localization of BMI1 in ovaries and the effects of BMI1 deficiency on the fertility and estrous cycle
To identify the localization of BMI1 in ovaries, we performed immunohistochemical staining on ovarian sections of WT female mice using anti-BMI1 antibodies (Fig. 1A-B). The results revealed that in female mice, BMI1 was expressed in granulosa cells, including both mural granulosa cells (GCs) and cumulus cells (CCs), and in theca cells and interstitial cells. To identify whether BMI1 was required to sustain female fertility, we crossed WT or Bmi1-/- female mice with WT male mice in adulthood for 1 month. Compared with the WT female mice, Bmi1-/- female mice were completely infertile (Fig. 1C). To evaluate the influence of BMI1 deficiency on ovulation, the WT and Bmi1-/- mice received an injection with PMSG, and then hCG after 44 hours. The egg masses were taken out and counted from the ampulla portion after 16 hours. The results showed that BMI1 deficiency attenuated ovulation (Fig. 1D). Compared with that in WT female mice, the Bmi1-/- mice vaginal opening time was markedly delayed (Fig. 1E). Estrous cycle dysfunction was also observed (Fig. 1F, 1I). In addition, the proestrus and estrus times were reduced significantly (Fig. 1G-H). In contrast, the diestrus and metestrus times were prolonged significantly (Fig. 1J-K).
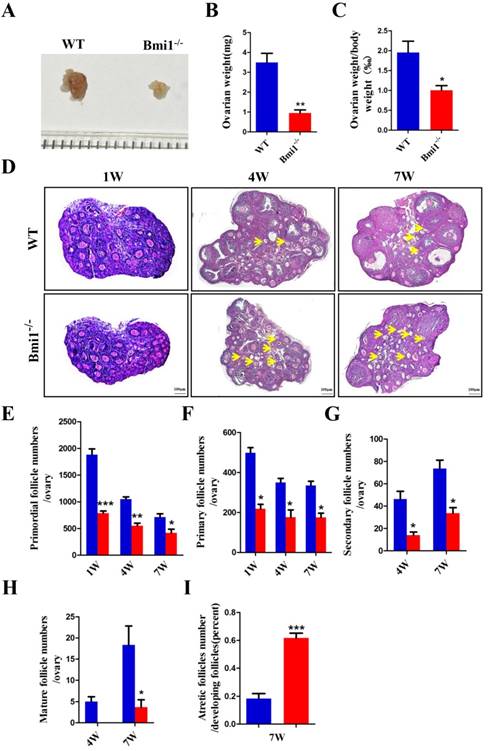
BMI1 deficiency results in ovarian and follicular development defects in mice
To explore whether the infertility in Bmi1-/- mice contributed to defective ovarian and follicular development, we compared the ovarian weight, ovarian weight/body weight ratio, and each stage follicles between Bmi1-/- and WT mice. We noted that the ovarian size, weight, and the total body weight ratio decreased dramatically in Bmi1-/- mice compared with those in WT mice (Fig. 2A-C). To further clarify the changes in the amounts of follicles and their proportion among different stages, the numbers of primordial, primary, secondary, mature, and atretic follicles were assessed in serial ovarian H&E stained sections from 1-week, 4-week, and 7-week-old WT mice and Bmi1-/- mice (Fig. 2D). The results showed that a decrease in primordial and primary follicles from 1 to 7-week-old WT mice was accompanied by a dramatic decline in Bmi1-/- mice at the same age (Figs. 2E-F). We also found the numbers of secondary and mature follicles increased from 4 to 7-week-old WT mice; however, their numbers were dramatically reduced in Bmi1-/-mice at the same age (Figs. 2G-H). In addition, the number of atretic follicles increased notably in 7-week-old Bmi1-/-mice compared with that in age-matched WT mice (Figs. 2I). Collectively, the data suggested that BMI1 deficiency led to defective ovarian and follicle development.
BMI1 deficiency results in ovarian and follicular development defects in mice. (A) Ovarian size and (B) weight from 7-week old mice, (C) ovarian weight/body weight ratio. (D) Representative images of ovarian sections from 1-week, 4-week, and 7-week-old WT and Bmi1-/- mice. Scale bar: 100 µm. Yellow arrow: atretic follicle. (E-I) The proportion of each stage follicles in WT mice and Bmi1-/- mice. Each value is the mean ± SEM of determinations in five mice of each group. *: p < 0.05; **: p < 0.01; ***: p < 0.001 compared with WT mice. Bmi1, B lymphoma Mo-MLV insertion region 1; WT, wild-type; SEM; standard error of the mean.
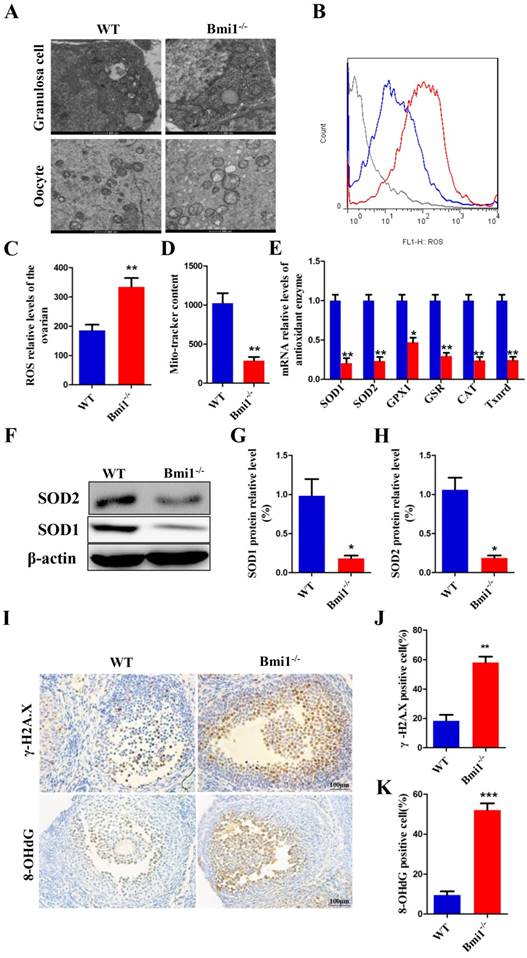
BMI1 deficiency causes oxidative stress status and antioxidant capacity decline in ovaries
To evaluate whether mice lacking BMI1 undergo oxidative stress in their ovaries, first, we detected the mitochondrial contents and structure in oocytes and granulosa cells using TEM and Mito- tracker. In the WT mice, the mitochondrial outer membrane was smooth and clear, with the mitochondria cristae being clearly visible and in good order. In the Bmi1-/- mice, the mitochondrial outer membrane was indistinct and imperfect, with some crest mitochondria blurring or even swelling, and the number of mitochondria was reduced (Figs. 3A, 3D). We then detected the changes in ROS levels in 7-week-old Bmi1-/- mice and WT mice using flow cytometry analysis. The results revealed increased ROS levels in the ovaries of Bmi1-/- mice compared with those in WT mice (Figs. 3B-C). To explore whether the levels of various antioxidant proteins was related to BMI1 deficiency, we preformed real-time RT-PCR and western blotting. These results indicated that the mRNA levels of Sod1, Sod2, Gpx1, Cat, and Gsr and the protein levels of SOD1 and SOD2 in ovaries were downregulated markedly in Bmi1-/- mice compared with those in WT mice (Figs. 3E-H). As shown in Fig. 3I, we sought to assess whether oxidative stress in ovaries increased DNA damage using immunohistochemistry for 8-OHdG and γ.H2A.X. We observed a significant increase in cells positive for 8-OHdG and γ.H2A.X in Bmi1-/- mice compared with that in WT mice (Fig. 3J-K).
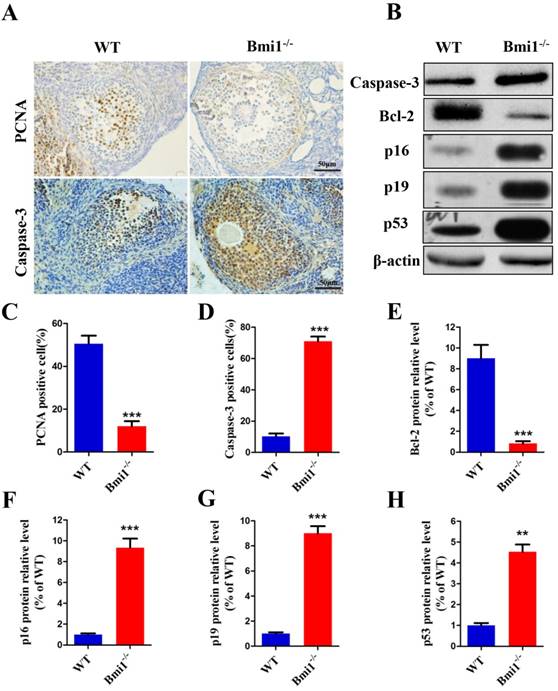
BMI1 deficiency reduces proliferation and induces apoptosis of granulosa cells by activating the p16/p19 signaling pathway in ovaries
Granulosa cells are essential to regulate follicular development, so the large atretic follicles caused by deletion of Bmi1 were assumed to be related to granulosa cells. The proliferation and apoptosis of granulosa cells were examined in 7-week-old mice (Fig. 4A). As expected, the percentage of PCNA-positive cells was significantly reduced in Bmi1-/- mice (Fig. 4C) and the percentage of Caspase-3-positive cells increased (Fig. 4D). To identify whether the changes in granulosa cells caused by BMI1 deficiency were related to activation of the p16 and p19 pathways in ovaries, the expression levels of p16 and p19 pathway-associated molecules were measured (Fig. 4B). We observed that the relative protein expression levels were upregulated markedly in the Bmi1-/-mice (Fig. 4F-H). Coincident with this upregulation, there was a marked decrease in the level of anti-apoptotic protein BCL-2 in the Bmi1-/-mice (Fig. 4E).
NAC supplementation improves ovarian and follicular developmental defects caused by BMI1 deficiency
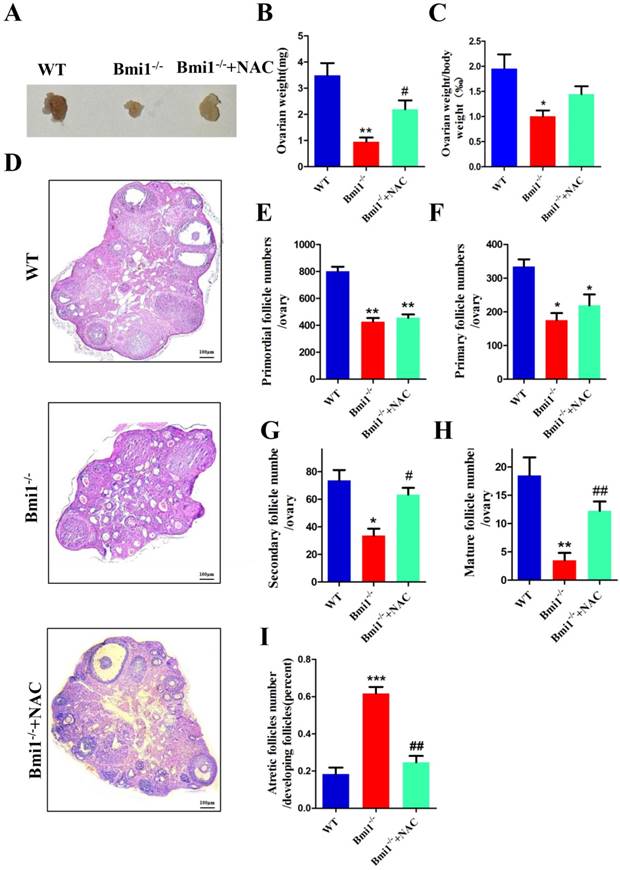
To determine whether NAC supplementation could improve defective ovarian and follicle development in Bmi1-/- mice, 3-week-old Bmi1-/- mice were given NAC in their drinking water until they were 7 weeks old and the phenotypes of the ovaries were examined. The results showed that the ovarian size, weight, and body weight ratio increased markedly in NAC-treated Bmi1-/- mice compared with that in vehicle-treated Bmi1-/- mice (Fig. 5A-C). In contrast to that in untreated Bmi1-/- mice, the number of primordial and primary follicles observed in the antioxidant-treated Bmi1-/- mice increased insignificantly (Fig. 5E-F). In addition, a significant increase in the number of secondary and mature follicles was observed (Fig. 5G-H). The number of atretic follicles decreased dramatically in the NAC-treated Bmi1-/- mice (Fig. 5I).
NAC supplementation inhibits oxidative stress, DNA damage, and cellular apoptosis, and stimulates cellular proliferation
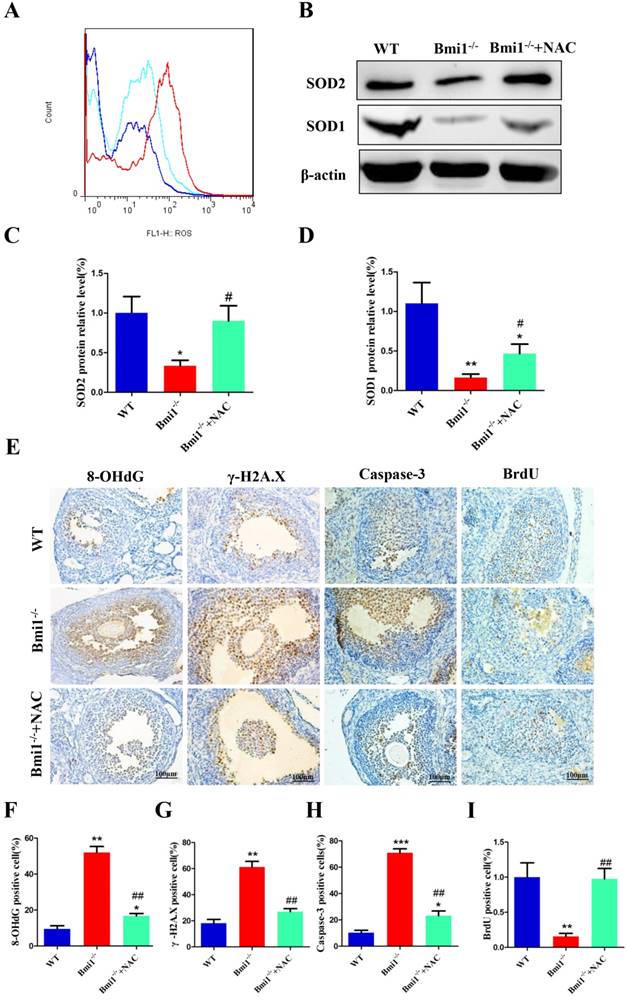
To evaluate whether the improvements in ovarian and follicular development induced by NAC supplementation were associated with inhibiting oxidative stress, DNA damage, and cellular apoptosis, and stimulating cellular proliferation, in Bmi1-/- mice, we assessed any alterations in ROS levels, protein expression levels of anti-oxidative enzymes in ovaries, DNA damage, cellular apoptosis, and proliferation in 7-week-old mice. The ROS levels were notably decreased in NAC-treated Bmi1-/- mice compared with those in Bmi1-/- mice (Fig. 6A), and the protein levels of SOD1 and SOD2 were also upregulated significantly. Overall, the parameters increased notably in NAC-treated Bmi1-/- mice compared with untreated Bmi1-/- mice (Figs. 6B-D). A similar result was noted for the increased level of 8-OHdG, γ.H2A.X and Caspase-3, positive cells in Bmi1-/- mice compared with WT mice, and fewer BrdU-positive cells were noted in mice lacking BMI1 (Figs. 6E-I). Most, but not all, of these parameters could be rescued by NAC administration (Figs. 6A-I).
NAC supplementation ameliorates the developmental potential of early embryos from Bmi1-/- mice
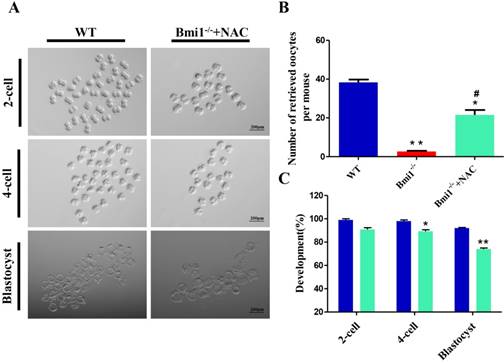
Next, we investigated whether NAC supplementation could promote the developmental competence of subsequent embryos. To address this question, we performed in vitro fertilization (IVF) of oocytes from WT mice, vehicle-treated Bmi1-/- mice, and NAC-treated Bmi1-/- mice. The embryos were then cultured to check their development. We noticed that fewer oocytes derived from ovaries in the vehicle-treated Bmi1-/- mice compared with that in WT mice; however, the number of oocytes increased significantly in the NAC-treated Bmi1-/- mice, although they did not reach the WT mouse levels (Fig. 7B). Oocytes derived from WT mice with IVF could develop into early embryos, including 2-cells, 4-cells, and blastocysts in in vitro cultures (Figs. 7A, C); however, oocytes derived from Bmi1-/- mice treated with IVF did not developed into early embryos, even 2-cell stage embryos. Surprisingly, oocytes derived from NAC-treated Bmi1-/- mice with IVF developed into early embryos, including 2-cells, 4-cells, and blastocysts in in vitro culture, which approached the level of the WT mice (Figs. 7A, C). These observations demonstrated that the compromised developmental potential of early embryos from Bmi1-/- mice could be partially rescued by the antioxidant NAC.
Discussion
In the present study, we found that BMI1 is expressed in follicular granulosa cells, follicular cells, and mesenchymal cells, which suggested that mice lacking BMI1 might have female reproduction defects. The effect of BMI1 was studied using a mouse model with the homozygous deletion of Bmi1. Bmi1 knockout female mice exhibited complete infertility, with ovarian and follicular developmental defects and estrous cycle dysfunction. Oocytes derived from Bmi1-/- mice could not develop into embryos in vitro, as demonstrated through IVF and embryo culture. Mechanistically, cells lacking BMI1 showed inhibited granulosa cell proliferation, induction of granulosa cell apoptosis, and female infertility by increasing oxidative stress and ongoing DNA damage, triggering the p16/p19 signaling pathway. Furthermore, supplementation with the antioxidant NAC not only improved the reproductive defects caused by Bmi1 deletion in vivo, but also rescued the potential of BMI1-deficient oocytes to develop into embryos.
BMI1 deficiency causes oxidative stress status and antioxidant capacity decline in ovaries. (A) TEM images of granulosa cells and oocytes in WT mice and Bmi1-/- mice. Scale bar: 500 nm. (B) Representative graphs of flow cytometry analysis of granulosa cells for DCFDA and (C) the statistical result. (D) The mitochondrial morphology in ovarian sections was detected using Mito-tracker probes with FCM. (E) Real-time reverse transcription (RT)-PCR analysis of ovarian extracts from 7-week old mice for expressions of Sod1, Sod2, Gpx1, Gsr, Cat, and Txnrd1. The mRNA expression levels were determined using real-time reverse transcription (RT)-PCR is calculated as a ratio relative to Gapdh expression, and expressed relative to that in WT mice. (F) Western blotting showing the expression of SOD1 and SOD2 in 7-week old mice. (G) SOD1 and (H) SOD2 protein relative levels assessed by densitometry analysis calculated as a ratio relative to β-actin protein levels and expressed relative to their levels in WT mice. (I) Representative micrographs of ovarian sections from 7-week old mice stained immunohistochemically for γ-H2A.X and 8-OHdG. Scale bar: 100 µm. Percentages of (J) γ-H2A.X and (K) 8-OHdG positive cells in mouse ovaries. Each value is the mean ± SEM of determinations in 5 mice of each group. *: p < 0.05; **: p < 0.01 compared with WT mice. Bmi1, B lymphoma Mo-MLV insertion region 1; WT, wild-type; SEM; standard error of the mean; TEM, transmission electron microscopy; DCFDA, diacetyldichlorofluorescein; FCM, flow cytometry; Sod1, superoxide dismutase 1; Sod2, superoxide dismutase 2; Gpx1, glutathione peroxidase 1; Gsr, glutathione-disulfide reductase; Cat, catalase; Txnrd1, thioredoxin reductase 1; Gapdh, glyceraldehyde-3-phosphate dehydrogenase; γ-H2A.X, phosphorylated H2A histone family member X; 8-OHdG, 8-Oxo-2'-deoxyguanosine.
BMI1 deficiency reduces the proliferation and induces apoptosis of granulosa cells by activating p16/p19 signaling pathways in ovaries. (A) Representative micrographs of ovarian sections from 7-week old mice stained immunohistochemically for PCNA and Caspase-3. Percentages of (C) PCNA and (D) Caspase-3 positive cells. Scale bar: 100 µm. (B) Western blotting showing the levels of Caspase-3, BCL-2, p16, p19, and p53. (E) BCL-2, (F) p16, (G) p19 and (H) p53 relative protein levels were assessed by densitometry analysis, calculated as relative to β-actin protein levels and expressed relative to levels in the WT mice. Each value is the mean ± SEM of determinations in five mice of each group. **: p < 0.01; ***: p < 0.001 compared with WT mice. Bmi1, B lymphoma Mo-MLV insertion region 1; WT, wild-type; SEM; standard error of the mean; PCNA, proliferating cell nuclear antigen; Bcl-2, B-cell CLL/lymphoma 2; cyclin dependent kinase inhibitor 2A, p16; cyclin dependent kinase inhibitor 2D, p19; tumor protein p53, p53.
BMI1 deficiency causes a generalized failure-to- thrive and a significant shortening of lifespan [21, 22]. In the present study, we found that compared with WT mice, the ovaries were smaller, the numbers of follicles at all stags decreased dramatically, and a large number of atresia appeared in Bmi1-/- female mice (Fig. 2). Previous studies demonstrated that BMI1 deficiency increased the level of testicular oxidative stress in male mice, and resulted in DNA damage in germ cells [19]. In this study, we found the relative ROS relative levels in the ovaries increased significantly in Bmi1-/- female mice. Meanwhile, levels antioxidant enzymes, including SOD1, SOD2, GPX1, CAT, GSR, and TXNRD were downregulated markedly. These data revealed that BMI1 deficiency led to oxidative stress. The decreased content of mito-tracker suggested damage to ovarian mitochondria in the Bmi1-/- female mice; indeed, TEM revealed an indistinct outer mitochondrial membrane. Mitochondria are the cellular energy factory, and except for the nucleus, they are the only organelle carrying DNA [23, 24]. The reduced number of mitochondria could result in a lower ATP level, which would activate the mammalian target of rapamycin (mTOR) signaling pathway, and result in abnormal activation, the loss of more follicles, prematurely depleted follicles, and a decline in the ovarian reserve (DOR) [25]. Moreover, the available evidence indicates that oxidative stress can cause DNA damage [11], which could lead to several cell responses, including inhibiting intracellular signal transduction, gene transcription, and DNA repair; delay or blockage of the cell cycle; induction of cell apoptosis; and the development of tumors [26-28]. DNA double-strand breaks are one type of DNA damage, and phosphorylation of H2A.X is a timely response of eukaryotic cells to DNA double-strand breaks [29, 30]. 8-OHdG is a good target for oxidative stress-induced DNA damage [31]. In this study, we found that there were large increases in γ-H2AX and 8-OHdG positive cells in Bmi1-/- female mice (Fig. 3). Consequently, increased oxidative stress induced by BMI1 deficiency may be related to mitochondria dysfunction and DNA damage, which could further damage female reproductive function.
NAC supplementation improves ovarian and follicular developmental defects caused by BMI1 deficiency. (A) Ovarian size and (B) weight from 7-week old WT, Bmi1-/-, Bmi1-/-+NAC mice, (C) ovarian weight/body weight ratio. (D) Representative micrographs of ovarian sections from 7-week old mice. Scale bar: 100 µm. (E-I) The proportion of each stage follicles in mice. Each value is the mean ± SEM of determinations in five mice of each group. *: p < 0.05; **: p < 0.01 compared with WT mice. #: p < 0.05; ##: p < 0.01 compared with Bmi1-/- mice. Bmi1, B lymphoma Mo-MLV insertion region 1; WT, wild-type; SEM; standard error of the mean; NAC, N-acetylcysteine.
NAC supplementation inhibits oxidative stress, DNA damage and cellular apoptosis, stimulates cellular proliferation. (A) Representative graphs of flow cytometry analysis of granulosa cells from 7-week-old mice stained for DCFDA. (B) Western blotting showing the expression of SOD1 and SOD2 in WT, vehicle Bmi1-/-, and Bmi1-/-+NAC mice. (C) SOD2 and (D) SOD1 relative protein levels assessed by densitometry analysis, calculated as a ratio relative to β-actin protein levels and expressed relative to the levels in WT mice. (E) Representative micrographs of ovarian sections from 7-week old mice stained immunohistochemically for 8-OHdG, γ-H2A.X, Caspase-3, and BrdU. Percentages of (F) 8-OHdG, (G) γ-H2A.X, (H) Caspase-3 and (I) BrdU positive cells in the ovaries. Scale bar: 100 µm. Each value is the mean ± SEM of determinations in five mice of each group. *: p < 0.05; **: p < 0.01; ***: p < 0.001 compared with WT mice. ##: p < 0.01 compared with Bmi1-/- mice. Bmi1, B lymphoma Mo-MLV insertion region 1; WT, wild-type; SEM; standard error of the mean; NAC, N-acetylcysteine; DCFDA, diacetyldichlorofluorescein; Sod1, superoxide dismutase 1; Sod2, superoxide dismutase 2; γ-H2A.X, phosphorylated H2A histone family member X; 8-OHdG, 8-Oxo-2'-deoxyguanosine; BrdU, bromodeoxyuridine.
PCNA is a proliferative nuclear antigen, and caspase-3 is a critical downstream factor that causes apoptosis directly [32, 33]. Therefore, we measured these two factors. The number of PCNA positive cells in ovaries decreased, while the number of caspase-3 positive granulosa cells increased dramatically. In addition, the amount of apoptosis inhibitory protein BCL-2 also decreased significantly. The results showed that BMI1 deficiency could reduce proliferation and increase apoptosis, thus accelerating ovarian aging. The above molecular alterations were consistent with the results of previous studies. Moreover, we examined the levels of the anti-oncogenes p16, p19, and p53 by western blotting, and all of them were upregulated significantly in Bmi1-/- female mice (Fig. 4). As previously reported, BMI1 mainly works by inhibiting Ink4a/Arf transcription, which encodes the cell cycle regulators p16 (Ink4a) and p19 (Arf) [1]. Therefore, BMI1 deficiency may result in high levels of p16Ink4a transcription, which would suppress cell cycle processes and break the p16Ink4a balance [34]. Besides, the p19Arf/p53 pathway contributes to the progression of cell apoptosis, and is an essential signaling pathway [35]. There is evidence that p19Arf and p53 expression increased in neurons from BMI1 deficient mice [36]. That study also suggested that p53 is involved in the majority of apoptotic phenotypes [36]. In addition, a study showed that inhibiting the expression of p53 could attenuate apoptosis of granulosa cells and the number of atretic follicles [37], which correspond with the results of the present study. Thus, BMI1 deficiency can induce apoptosis of oocytes and granulosa cells by activating the p16-Rb and p19-p53 pathways.
NAC supplementation ameliorates the developmental potential of early embryos from Bmi1-/- mice. (A) Representative phase contrast images of 2-cell, 4-cell, and blastocyst embryos derived from WT and Bmi1-/- oocytes. Scale bar: 200 µm. (B) Number of retrieved oocytes from the three groups. (n = 150 for WT, n = 8 for Bmi1-/-, n = 111 for Bmi1-/-+NAC). (C) The percentage of embryos that successfully progressed to the blastocyst stage during in vitro culture. Each value is the mean ± SEM of the determinations in three independent experiments. *: p < 0.05; **: p < 0.01 compared with WT mice. #: p < 0.05 compared with Bmi1-/- mice. Bmi1, B lymphoma Mo-MLV insertion region 1; WT, wild-type; SEM; standard error of the mean; NAC, N-acetylcysteine.
NAC, as an endogenous antioxidant, is involved in the decomposition of peroxides by balancing oxidative stress via replenishing intracellular glutathione stores [38]. NAC treatment downregulated conjunctival epithelial cell dysfunction by inhibiting ROS production [39]. Clinically, NAC has been used to treat lung injury patients with chronic obstructive pulmonary disease, and has the advantage of clearing ROS [40, 41]. Previous data demonstrated that oxidative stress causes a reduction in the number and quality of oocytes, and upregulation of indicators of cell ageing. The administration of NAC improved the quantity and quality of oocytes, and significantly promoted follicular development [12]. In addition, NAC supplementation slowed organ aging and extended lifespan after drinking water containing NAC was administered to Bmi1-/- mice [42].
To determine whether oxidative stress plays a crucial role on female infertility induced by BMI1 deficiency, the Bmi1-/- mice were treated with drinking water containing NAC for 4 weeks. In contrast to untreated Bmi1-/- mice, NAC treatment reduced the ROS levels in the ovary, enhanced the antioxidative capacity, decreased DNA damage, and alleviated the obstacle to follicle growth (Fig. 5-6). Furthermore, we demonstrated that female infertility induced by BMI1 deficiency could be largely rescued by antioxidant supplement (Fig. 7). Bmi1-/- female mice are totally infertile; however, the developmental defects of early embryos in Bmi1-/- mice were markedly rescued by NAC administration. In addition the average member of retrieved oocytes increased significantly. Nevertheless, there is a limitation: The results for embryo development may be discrepant because of the low fertility of Bmi1-/- mice (n = 150 for the WT, n = 8 for Bmi1-/-, n = 111 for Bmi1-/-+NAC). Also, the exact mechanism by which antioxidants regulate these molecules remains to be investigated.
Our results implied that BMI1 deficiency results in female infertility with increased ROS production and ongoing DNA damage, whereas these alterations were largely rescued by NAC administration. These results indicated that BMI1 has a crucial role in promoting follicle growth and early embryo development by scavenging ROS, controlling the balance between cell proliferation and apoptosis, preventing DNA damage, and restraining p16Ink4a/ Rb and p19Afr/p53 signaling pathways, which are involved in cell cycle regulation.
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (81501326 and 81774075).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Biehs B, Hu JK, Strauli NB, Sangiorgi E, Jung H, Heber RP. et al. BMI1 represses Ink4a/Arf and Hox genes to regulate stem cells in the rodent incisor. Nat Cell Biol. 2013;15:846
2. Bognar Z, Fekete K, Antus C, Hocsak E, Bognar R, Tapodi A. et al. Desethylamiodarone—A metabolite of amiodarone—Induces apoptosis on T24 human bladder cancer cells via multiple pathways. PLoS ONE. 2017;12:e0189470
3. Luo H, Yang R, Li C, Tong Y, Fan L, Liu X. et al. MicroRNA-139-5p inhibits bladder cancer proliferation and self-renewal by targeting the Bmi1 oncogene. Tumour Biol. 2017;39:1010428317718414
4. Molofsky AV, Pardal R, Iwashita T, Park IK, Clarke MF, Morrison SJ. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 2003;425:962
5. Park Ik, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL. et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302
6. Zhang HW, Ding J, Jin JL, Guo J, Liu JN, Karaplis A. et al. Defects in mesenchymal stem cell self-renewal and cell fate determination lead to an osteopenic phenotype in Bmi-1 null mice. J Bone Miner Res. 2010;25:640-52
7. Jin J, Lv X, Chen L, Zhang W, Li J, Wang Q. et al. Bmi-1 plays a critical role in protection from renal tubulointerstitial injury by maintaining redox balance. Aging Cell. 2014;13:797-809
8. Jin J, Tao J, Gu X, Yu Z, Wang R, Zuo G. et al. P16 INK4a Deletion Ameliorated Renal Tubulointerstitial Injury in a Stress-induced Premature Senescence Model of Bmi-1 Deficiency. Sci Rep. 2017;7:7502
9. Becatti M, Fucci R, Mannucci A, Barygina V, Mugnaini M, Criscuoli L. et al. A biochemical approach to detect oxidative stress in infertile women undergoing assisted reproductive technology procedures. Int J Mol Sci. 2018;19:592
10. Adeoye O, Olawumi J, Opeyemi A, Christiania O. Review on the role of glutathione on oxidative stress and infertility. JBRA Assist Reprod. 2018;22:61
11. Agarwal A, Gupta S, Sharma R. Role of oxidative stress in female reproduction. Reprod Biol Endocrinol. 2005;3:28
12. Liu J, Liu M, Ye X, Liu K, Huang J, Wang L. et al. Delay in oocyte aging in mice by the antioxidant N-acetyl-L-cysteine (NAC). Hum Reprod. 2012;27:1411-20
13. Yu B, Lin H, Yang L, Chen K, Luo H, Liu J. et al. Genetic variation in the Nrf2 promoter associates with defective spermatogenesis in humans. J Mol Med (Berl). 2012;90:1333-42
14. O'Flaherty C, Matsushita-Fournier D. Reactive oxygen species and protein modifications in spermatozoa. Biol Reprod. 2017;97:577-85
15. EL-Hadaad HA. Immunohistochemical expression and prognostic relevance of Bmi-1, a stem cell factor, in epithelial ovarian cancer. Ann Diagn Pathol. 2014;18:58-62
16. Zhang F, Sui L, Xin T. Correlations of BMI-1 expression and telomerase activity in ovarian cancer tissues. Exp Oncol. 2008. 1812 -9269
17. Gjerstorff MF, Relster MM, Greve KB, Moeller JB, Elias D, Lindgreen JN. et al. SSX2 is a novel DNA-binding protein that antagonizes polycomb group body formation and gene repression. Nucleic Acids Res. 2014;42:11433-46
18. Jiang L, Li J, Song L. Bmi-1, stem cells and cancer. Acta Biochim Biophys Sin. 2009;41:527-34
19. Dai X, Zhang Q, Yu Z, Sun W, Wang R, Miao DS. Bmi1 Deficient Mice Exhibit Male Infertility. Int J Biol Sci. 2018;14:358
20. Caligioni CS. Assessing reproductive status/stages in mice. Curr Protoc Neurosci. 2009;48:A-4I
21. Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164
22. Bruggeman SW, Valk-Lingbeek ME, van der Stoop PP, Jacobs JJ, Kieboom K, Tanger E. et al. Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Gene Dev. 2005;19:1438-43
23. Yonutas H, Vekaria H, Sullivan P. Mitochondrial specific therapeutic targets following brain injury. Brain Res. 2016;1640:77-93
24. Treulen Fn, Uribe P, Boguen R, Villegas JV. Mitochondrial permeability transition increases reactive oxygen species production and induces DNA fragmentation in human spermatozoa. Hum Reprod. 2015;30:767-76
25. Slotkin EK, Patwardhan PP, Vasudeva SD, de Stanchina E, Tap WD, Schwartz GK. MLN0128, an ATP-competitive mTOR kinase inhibitor with potent in vitro and in vivo antitumor activity, as potential therapy for bone and soft-tissue sarcoma. Cancer Ther. 2015;14:395-406
26. Pan MR, Li K, Lin SY, Hung WC. Connecting the dots: from DNA damage and repair to aging. Int J Mol Sci. 2016;17:685
27. Al-Oqail MM, Siddiqui MA, Al-Sheddi ES, Saquib Q, Musarrat J, Al-Khedhairy AA. et al. Verbesina encelioides: cytotoxicity, cell cycle arrest, and oxidative DNA damage in human liver cancer (HepG2) cell line. BMC Complement Altern Med. 2016;16:126
28. Giromini C, Rebucci R, Fusi E, Rossi L, Saccone F, Baldi A. et al. Cytotoxicity, apoptosis, DNA damage and methylation in mammary and kidney epithelial cell lines exposed to ochratoxin A. Cell Biol Toxicol. 2016;32:249-58
29. Liang S, Jin YX, Yuan B, Zhang JB, Kim NH. Melatonin enhances the developmental competence of porcine somatic cell nuclear transfer embryos by preventing DNA damage induced by oxidative stress. Sci Rep. 2017;7:11114
30. Lorković ZJ, Park C, Goiser M, Jiang D, Kurzbauer MT, Schlögelhofer P. et al. Compartmentalization of DNA damage response between heterochromatin and euchromatin is mediated by distinct H2A histone variants. Curr Biol. 2017;27:1192-9
31. Risom L, Moller P, Loft S, Mutagenesis M. Oxidative stress-induced DNA damage by particulate air pollution. Mut Res. 2005;592:119-37
32. Vital-Reyes V, Rodíguez-Burford C, Chhieng DC, Alvarado-Cabrero I, Reyes-Fuentes A, Grizzle WE. et al. Ovarian expression of markers associated with proliferation or apoptosis in women with diminished ovarian reserve. Fertil Steril. 2006;86:176-85
33. Barberino RS, Menezes VG, Ribeiro AE, Palheta RC, Jiang X, Smitz JE. et al. Melatonin protects against cisplatin-induced ovarian damage in mice via the MT1 receptor and antioxidant activity. Biol Reprod. 2017;96:1244-55
34. De Jonge HJ, Woolthuis CM, de Bont ES, Huls G. Paradoxical down-regulation of p16Ink4a mRNA with advancing age in acute myeloid leukemia. Aging (Albany NY). 2009;1:949
35. Mudhasani R, Zhu Z, Hutvagner G, Eischen CM, Lyle S, Hall L. et al. Loss of miRNA biogenesis induces p19Arf-p53 signaling and senescence in primary cells. J Cell Biol. 2008;181:1055-63
36. Chatoo W, Abdouh M, David J, Champagne M-P, Ferreira J, Rodier F. et al. The polycomb group gene Bmi1 regulates antioxidant defenses in neurons by repressing p53 pro-oxidant activity. J Neurosci. 2009;29:529-42
37. Takeuchi A, Mishina Y, Miyaishi O, Kojima E, Hasegawa T, Isobe K. Heterozygosity with respect to Zfp148 causes complete loss of fetal germ cells during mouse embryogenesis. Nat Genet. 2003;33:172
38. Small DM, Coombes JS, Bennett N, Johnson DW, Gobe GC. Oxidative stress, antioxidant therapies and chronic kidney disease. Nephrology. 2012;17:311-21
39. Park JH, Kang SS, Kim JY, Tchah H. The antioxidant N-acetylcysteine inhibits inflammatory and apoptotic processes in human conjunctival epithelial cells in a high-glucose environment. Invest Ophthalmol. 2015;56:5614-21
40. Bulacio RP, Anzai N, Ouchi M, Torres AM. Organic anion transporter 5 (Oat5) urinary excretion is a specific biomarker of kidney injury: evaluation of urinary excretion of exosomal Oat5 after N-Acetylcysteine prevention of cisplatin induced nephrotoxicity. Chem Res Toxicol. 2015;28:1595-602
41. Cazzola M, Calzetta L, Page C, Jardim J, Chuchalin AG, Rogliani P. et al. Influence of N-acetylcysteine on chronic bronchitis or COPD exacerbations: a meta-analysis. Eur Respir Rev. 2015;24:451-61
42. Sato A, Okada M, Shibuya K, Watanabe E, Seino S, Narita Y. et al. Pivotal role for ROS activation of p38 MAPK in the control of differentiation and tumor-initiating capacity of glioma-initiating cells. Stem Cell Res. 2014;12:119-31
Author contact
![]() Corresponding author: Dr. Hui Wang, The State Key Laboratory of Reproductive Medicine, Department of Anatomy, Histology and Embryology, Nanjing Medical University, Nanjing, Jiangsu, 211166, The People's Republic of China. Tel: 86-25-86869380; E-mail: anniewangedu.cn
Corresponding author: Dr. Hui Wang, The State Key Laboratory of Reproductive Medicine, Department of Anatomy, Histology and Embryology, Nanjing Medical University, Nanjing, Jiangsu, 211166, The People's Republic of China. Tel: 86-25-86869380; E-mail: anniewangedu.cn
Received 2018-10-8
Accepted 2019-1-12
Published 2019-3-1