ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2019; 15(5):1010-1019. doi:10.7150/ijbs.29680 This issue Cite
Research Paper
Metformin Inhibits the NLRP3 Inflammasome via AMPK/mTOR-dependent Effects in Diabetic Cardiomyopathy
Fan Yang1, Ying Qin2, Yueqiu Wang1, Songyan Meng3, Huimin Xian4, Hui Che1,2, Jie Lv1, Yang Li1, Yahan Yu5, Yunlong Bai2,5 ![]() , Lihong Wang1,2
, Lihong Wang1,2 ![]()
1. Department of Endocrinology, The Second Affiliated Hospital of Harbin Medical University, Harbin, China;
2. Translational Medicine Research and Cooperation Center of Northern China, Heilongjiang Academy of Medical Sciences, Harbin, China;
3. Department of Geriatrics, The Second Affiliated Hospital of Harbin Medical University, Harbin, China;
4. Department of Cardiology, The Second Affiliated Hospital of Harbin Medical University, Harbin, China;
5. Department of Pharmacology (State-Province Key Laboratories of Biomedicine-Pharmaceutics of China, Key Laboratory of Cardiovascular Medicine Research, Ministry of Education), College of Pharmacy, Harbin Medical University, Harbin, China.
Abstract

Metformin is a widely used antidiabetic drug for type 2 diabetes that can play a cardioprotective role through multiple pathways. It is a recognized agonist of AMP-activated protein kinase (AMPK) that blocks mitochondrial complex I. The NLRP3 inflammasome has been demonstrated to be activated in diabetic cardiomyopathy (DCM). However, the role of metformin in regulating the NLRP3 signaling pathway in DCM remains unclear. It has been reported that AMPK can inhibit NLRP3 by activating autophagy. The aim of this study was to investigate whether metformin can inhibit the NLRP3 inflammasome by activating the AMPK/mTOR pathway in DCM. In this study, streptozotocin-induced C57BL/6 mice and high glucose-treated primary cardiomyocytes from neonatal mice were treated with metformin or an AMPK inhibitor compound C. Echocardiography, hematoxylin-eosin and Masson staining showed that the function and morphology of the diabetic hearts were improved after metformin treatment, whereas these parameters deteriorated after intervention with an AMPK inhibitor. Immunohistochemical staining, immunofluorescence staining and western blot assays indicated that the expression levels of mTOR, NLRP3, caspase-1, IL-1β and GSDMD-N were decreased in the diabetic model treated with metformin and were reversed after the administration of an AMPK inhibitor in vivo and in vitro. Mechanistically, our results demonstrated that metformin can activate AMPK, thus improving autophagy via inhibiting the mTOR pathway and alleviating pyroptosis in DCM. Thus, we provide novel information for the treatment of DCM.
Keywords: metformin, AMPK, autophagy, NLRP3 inflammasome, diabetic cardiomyopathy
Introduction
Diabetes can cause a variety of complications, including diabetic cardiomyopathy (DCM), which is closely related to the increased morbidity of heart failure and arrhythmia [1]. Mitochondrial dysfunction, metabolic abnormalities, autophagy, inflammation and cell death have been indicated to be involved in cardiac hypertrophy and myocardial fibrosis, thus enhancing the risk of heart failure in DCM [2]. Although various medications are used to control hyperglycemia and restore cardiac function, the morbidity and mortality rates of DCM continue to increase. Thus, the mechanisms and treatment of DCM still need to be further elucidated.
Metformin, which can promote the uptake and utilization of glucose in liver, muscle and adipose tissue, is the most widely used drug for treating type 2 diabetes [3]. Recent studies have shown that metformin possesses anti-tumor, anti-aging and cardioprotective effects in addition to its hypoglycemic effect [4-6]. Some important molecular biological mechanisms of metformin are its downregulation of mitochondrial respiratory chain complex I and its upregulation of phosphorylation of AMP-activated protein kinase (AMPK) [7]. AMPK is regarded as a sensor of cellular energy and modulates many cellular statuses [8]. Studies have shown that metformin can protect the heart through inhibiting inflammation, autophagy, apoptosis and other pathways via AMPK [9, 10]. However, studies of the molecular mechanisms underlying the cardioprotective function of metformin remain insufficient.
The nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3) inflammasome is a multiprotein complex consisting of NLRP3, apoptosis-associated speck-like protein containing a caspase-1 recruitment domain (ASC) and caspase-1 [11]. Previous studies have indicated that hyperglycemia activates NLRP3, which promotes the autocatalytic activation of pro-caspase-1 to cleaved caspase-1. Then, cleaved caspase-1 facilitates the maturation of pro-IL-1β [12]. This process causes inflammation-related programmed cell death, which can progress to DCM. The downregulation of the NLRP3 inflammasome restores cardiac function in DCM models [13, 14]. Notably, AMPK activation attenuates NLRP3 inflammasome upregulation in some pathological processes, such as diabetes, pain, ischemic stroke and endoplasmic reticulum stress [15-17]. Furthermore, previous studies indicated that autophagy could downregulate the NLRP3 inflammasome via the mTOR signaling pathway [18]. Therefore, we investigated whether metformin is involved in regulation of NLRP3 inflammasome via the AMPK/mTOR pathway in DCM.
In the present study, we investigated the effects of metformin on the NLRP3 pathway in high glucose-treated cardiomyocytes and diabetic mice and further explored the underlying mechanisms of metformin involved in the AMPK/mTOR signaling pathway. Our results provide novel insight into the mechanism of metformin in the regulation of DCM.
Materials and Methods
Mouse model establishment
Male mice (C57BL/6, 8 weeks old) were bred at the Second Affiliated Hospital of Harbin Medical University. All mice were housed in standard conditions (temperature 22±1 °C; 12 h light and dark cycles) and fed conventional diets. The mice were divided randomly into four groups (n=5). The control group was administered normal saline intragastrically daily. The diabetes mellitus (DM) group received intraperitoneal injections of 50 mg/kg freshly prepared streptozotocin (STZ, Sigma) dissolved in citrate buffer (10 mM, pH=4.5) daily for five days. One week after the injections, tail vein blood glucose values≥16.7 mmol/L measured by glucometer (Accu-Chek, Roche Diagnostics) proved successful model establishment. Some mice with DM were administered metformin (Sigma, 200 mg/kg/day) [9] with or without intraperitoneal injection of the AMPK inhibitor compound C (Selleckchem Chemicals, Houston, TX, USA, 20 mg/kg/day) for eight weeks as previously described [16]. Then, the mice were used for the following experiments. The experiments were approved by the Ethics Committee of Harbin Medical University. The use of animals in the experiments was in accordance with the Interdisciplinary Principles and Guidelines for the Use of Animals in Research, Testing, and Education by the New York Academy of Sciences, Ad Hoc Animal Research Committee.
Cell culture and transfection
Primary cardiomyocytes were isolated from 1- to 3-day-old neonatal C57BL/6 mice according to previous methods [19]. Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Hyclone, Logan, USA) were used to culture the cardiomyocytes, which were divided into four groups and treated with low glucose (5.5 mM glucose, control) or high glucose (30 mM glucose, HG) with or without metformin (2 mM, HG+Met) or the AMPK inhibitor compound C (10 μM, HG+Met+compound C) for 24 h according to a previous study [20]. The control group was also treated with a corresponding amount of mannitol for osmolarity. All cells were incubated at 37 °C with 5% CO2.
Echocardiography
Mice were anesthetized via intraperitoneal injection of avertin for echocardiography. Ejection fraction (EF) and fractional shortening (FS) of the left ventricle were derived by two-dimensional M-mode echocardiography with a Vevo1100 high-resolution imaging system (VisualSonics, Toronto, ON, Canada).
Hematoxylin and eosin (HE) and Masson's trichrome staining
Left ventricle samples were fixed in 4% paraformaldehyde at room temperature, embedded in paraffin and cut into 5-μm-thick sections. Subsequently, the sections were separately stained with HE and Masson's trichrome [21]. Finally, the morphological changes and collagen content of the cardiac tissues were observed by fluorescence microscopy (Nikon 80i, Otawara, Tochigi, Japan).
Immunohistochemical analyses
Left ventricle samples were also stained with primary antibodies against caspase-1 (Cell Signaling Technology, MA, USA, 1:200), NLRP3 (Boster Biological Technology, Wuhan, China, 1:200) and IL-1β (R&D Systems, Minneapolis, MN, 1:200) at 4 °C overnight, followed by secondary antibodies. Next, the sections were stained with diaminobenzidine. Finally, a fluorescence microscope (Nikon 80i) was used to obtain images of the tissues.
Immunofluorescence staining
Primary cardiomyocytes were treated with 4% buffered paraformaldehyde for 20 min and then blocked with 1% BSA and 0.1% Triton-X for 2 h at room temperature. Next, the cells were treated with a primary antibody against the N terminal of gasdermin D (GSDMD-N) (Bioss, Beijing, China, 1:200) at 4 °C overnight, followed by treatment with a secondary antibody for 1 h at room temperature [22]. The nuclei were stained with DAPI (Beyotime, Shanghai, China), and the results were captured using a fluorescence microscope.
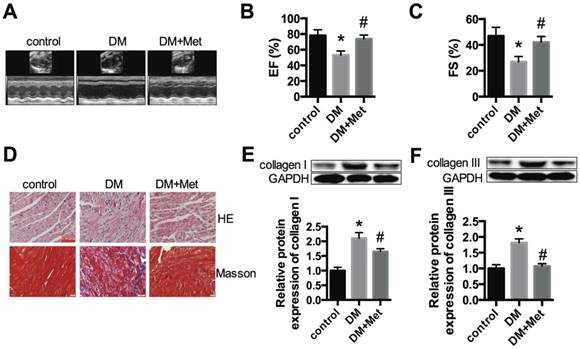
Cardiac function and morphology in diabetic mice treated with or without metformin. (A) M-mode echocardiograms of the left ventricles are shown. EF (B) and FS values (C) are shown. (D) The results of HE and Masson's trichrome staining are shown. Scale bar, 60 μm. (E and F) Western blot analysis of collagen I and collagen III expression in the different groups. *P<0.05 compared to the control group, #P<0.05 compared to the DM group. n=5.
Protein extraction and western blot analyses
Briefly, total protein was extracted, and the samples were separated via 10% SDS-PAGE and then transferred onto nitrocellulose membranes. Subsequently, the membranes were blocked with 5% nonfat milk dissolved in PBS for 2 h at room temperature. Next, the membranes were probed with primary antibodies against collagen I (Abcam, Cambridge, UK, 1:800), collagen III (Abcam, 1:800), NLRP3 (1:800), caspase-1 (1:1000), IL-1β (1:1000), GSDMD-N (1:1000), p-AMPK (Cell Signaling Technology, 1:800), AMPK (Cell Signaling Technology, 1:800), LC3 (Cell Signaling Technology, 1:1000), mTOR (Cell Signaling Technology, 1:1000) and GAPDH (ZSGB-BIO, Beijing, China, 1:1000) at 4°C overnight. The membranes were then washed with PBST three times and incubated with the corresponding secondary antibodies for 1 h at room temperature. GAPDH was used as an internal control. The western blot bands were imaged and quantified using the GelDox XR System (Bio-Rad, CA, USA) and Quantity One software.
Mitochondrial complex I activity assay
Mitochondrial complex I activity was detected using a Complex I Activity Assay kit (MitoSciences, Eugene, OR) according to the manufacturer's instructions. Cell proteins were extracted and loaded on 96-well plates and incubated with a mitochondrial complex I antibody for 3 h at room temperature. A Nano Drop spectrophotometer was used to measure the optical density (OD 450 nm).
Data analyses
The values in this study were analyzed with GraphPad Prism 6 and are shown as the mean ± SD. Unpaired Student's t-tests were used for comparisons between two groups. Differences among different groups were analyzed using one-way ANOVA. P<0.05 was considered statistically significant.
Results
Metformin restores the function and morphology of cardiac tissue in diabetic mice
We established a STZ-induced diabetic model with C57BL/6 mice. Echocardiography indicated that the function of the left ventricle deteriorated in the DM group, as shown by the EF and FS values. Metformin significantly improved cardiac function in diabetic mice (Fig. 1A-C). HE and Masson's trichrome staining indicated myocardial hypertrophy and collagen deposition in the DM group, which were improved after metformin treatment (Fig. 1D). The protein expression levels of collagen I and collagen III were significantly increased in the DM group, and these changes were alleviated after treatment with metformin (Fig. 1E and F).
NLRP3 pathway changes are ameliorated by metformin in diabetic hearts
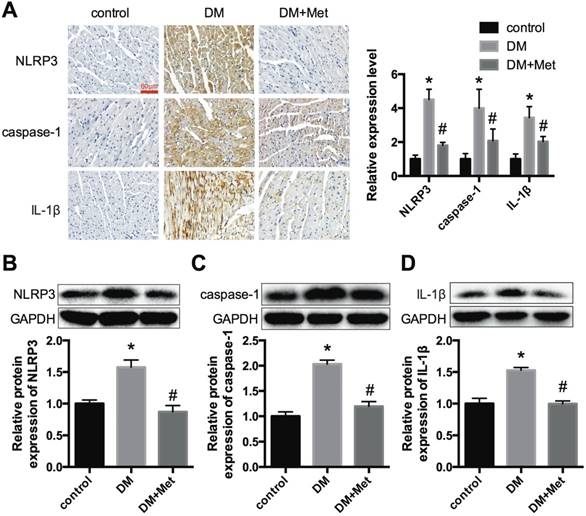
Immunohistochemistry and western blot analyses indicated that NLRP3, caspase-1 and IL-1β were significantly overexpressed in the cardiac tissues of diabetic mice and were decreased after the administration of metformin (Fig. 2A-D). These data demonstrated that metformin could exert a cardiac protective effect and inhibit the NLRP3 pathway in diabetic hearts.
The effects of metformin on the NLRP3 pathway in high glucose-treated cardiomyocytes
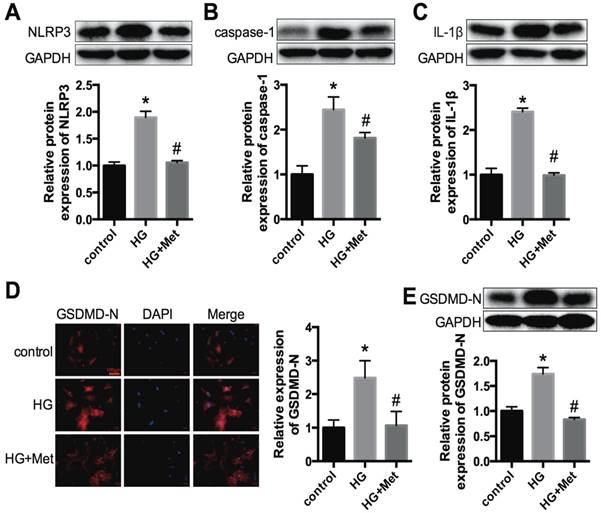
To confirm the effects of metformin on the NLRP3 pathway in vitro, primary cardiomyocytes from C57BL/6 mice were cultured in 5.5 mmol/L (control) and 30 mmol/L glucose (high glucose, HG) and treated with or without metformin for 24 h. Western blotting was conducted to detect the protein expression levels of NLRP3, caspase-1 and IL-1β. The results showed that high glucose remarkably enhanced the expression levels of NLRP3, caspase-1 and IL-1β in cardiomyocytes. After metformin treatment, these increases were alleviated significantly (Fig. 3A-C). We also detected the expression level of GSDMD-N, a key enzyme in the process of pyroptosis, using immunofluorescence staining and western blotting. Metformin inhibited the expression of GSDMD-N (Fig. 3D and E). These results were consistent with the animal experiments described above.
Metformin activates mitochondrial complex I/p-AMPK expression
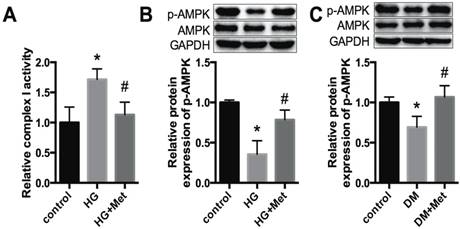
A previous study proposed that mitochondrial complex I is a the direct target of metformin, and thus regulates the phosphorylation of AMPK [23]. In our study, the activity of mitochondrial complex I was increased in HG-treated cardiomyocytes, whereas it was significantly downregulated after metformin treatment (Fig. 4A). In addition, p-AMPK was reduced dramatically in high glucose-treated cardiomyocytes, whereas it was improved after metformin treatment (Fig. 4B). Moreover, a similar effect was observed in the cardiac tissues of diabetic mice (Fig. 4C).
NLRP3, caspase-1 and IL-1β expression levels in vivo. (A) Immunohistochemistry analysis was performed to detect the expression of NLRP3, caspase-1 and IL-1β. Scale bar, 60 μm. The results were quantified and statistically analyzed. Western blotting analyses were conducted to detect the expression levels of NLRP3 (B), caspase-1 (C) and IL-1β (D). *P<0.05 compared to the control group, #P<0.05 compared to the DM group. n=5.
NLRP3, caspase-1, IL-1β and GSDMD-N expression levels in vitro. Primary cardiomyocytes were treated with 5.5 mmol/L (control) and 30 mmol/L glucose (HG). The cardiomyocytes in the HG group were treated with or without 2 mM metformin. Western blotting was used to detect the expression levels of NLRP3 (A), caspase-1 (B) and IL-1β (C). The expression levels of GSDMD-N were detected by an immunofluorescence assay (D) and western blotting (E). Scale bar, 100 μm. *P<0.05 compared with the control group, #P<0.05 compared to the HG group. n=3.
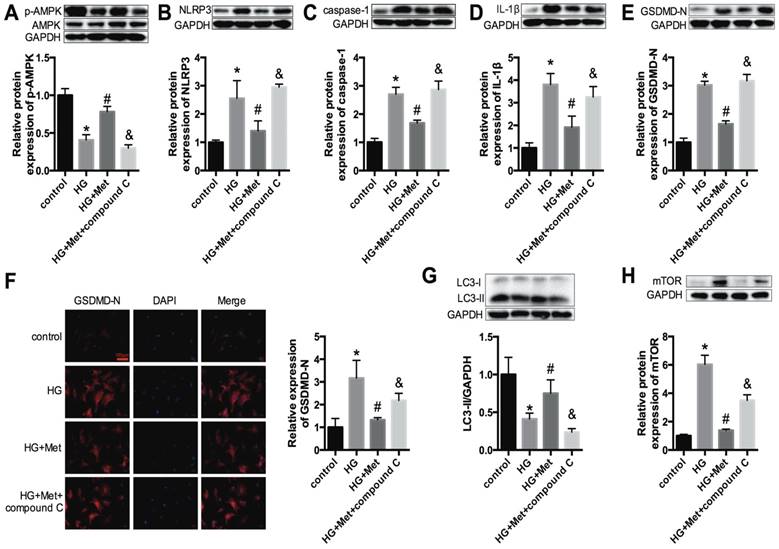
An AMPK inhibitor blocked the suppressive effect of metformin on the NLRP3 pathway via regulating autophagy in vitro
Next, to gain insight into the relationships among metformin, AMPK and the NLRP3 pathway, the AMPK inhibitor compound C (10 μM) was administered with metformin to high glucose-treated cardiomyocytes (HG+Met+compound C). Figure 5A shows that compound C effectively inhibited p-AMPK expression. Upon combination with the AMPK inhibitor, the protective effect of metformin on the NLRP3 pathway was abrogated, as shown by the increased expression levels of NLRP3, caspase-1, IL-1β and GSDMD-N (Fig. 5B-F).
Metformin activates mitochondrial complex I/p-AMPK expression. (A) The activity of mitochondrial complex I was detected in cardiomyocytes. (B) The protein expression levels of p-AMPK and AMPK in cardiomyocytes were detected by western blotting. GAPDH was used as internal control. n=3. (C) The expression levels of p-AMPK and AMPK in mice were detected. n=5. *P<0.05 compared to the control group. #P<0.05 compared to the HG or the DM group.
Furthermore, previous studies revealed that autophagy could be activated by AMPK and that the NLRP3 inflammasome was negatively regulated by autophagy [18]. Thus, the protein expression levels of LC3 and mTOR were measured. The results demonstrated that the levels of LC3 II were significantly reduced in HG-treated cardiomyocytes and were improved after metformin treatment. However, this amelioration was abolished after treatment with compound C (Fig. 5G). The expression levels of mTOR were increased in the HG group, and metformin suppressed the expression of mTOR. Nevertheless, compound C increased the expression level of mTOR (Fig. 5H). These results confirmed that metformin activated p-AMPK and autophagy, thus inhibiting the NLRP3 inflammasome.
An AMPK inhibitor blocked the suppressive effect of metformin on the NLRP3 pathway in cardiomyocytes. In HG-induced cardiomyocytes treated with 2 mM metformin or 2 mM metformin and 10 μM compound C, the expression levels of p-AMPK and AMPK (A), NLRP3 (B), caspase-1 (C) and IL-1β (D) were detected by western blotting. The expression levels of GSDMD-N were determined by western blotting (E) and immunofluorescence staining (F). Scale bar, 100 μm. LC3 (G) and mTOR (H) were detected by western blotting. *P<0.05 compared to the control group, #P<0.05 compared to the HG group. &P<0.05 compared to the HG+Met group. n=3.
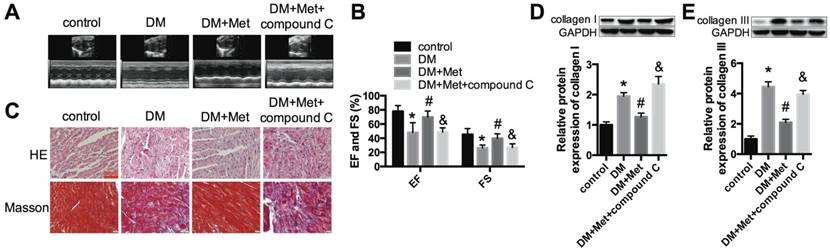
An AMPK inhibitor blocked the protective effect of metformin on cardiac function. (A) M-mode echocardiograms of the left ventricles are shown. (B) EF and FS values are shown. (C) HE and Masson's trichrome staining are shown. Scale bar, 60 μm. (D and E) Western blot analysis of collagen I and collagen III expression in the different groups. *P<0.05 compared to the control group, #P<0.05 compared to the DM group, &P<0.05 compared to the DM+Met group. n=5.
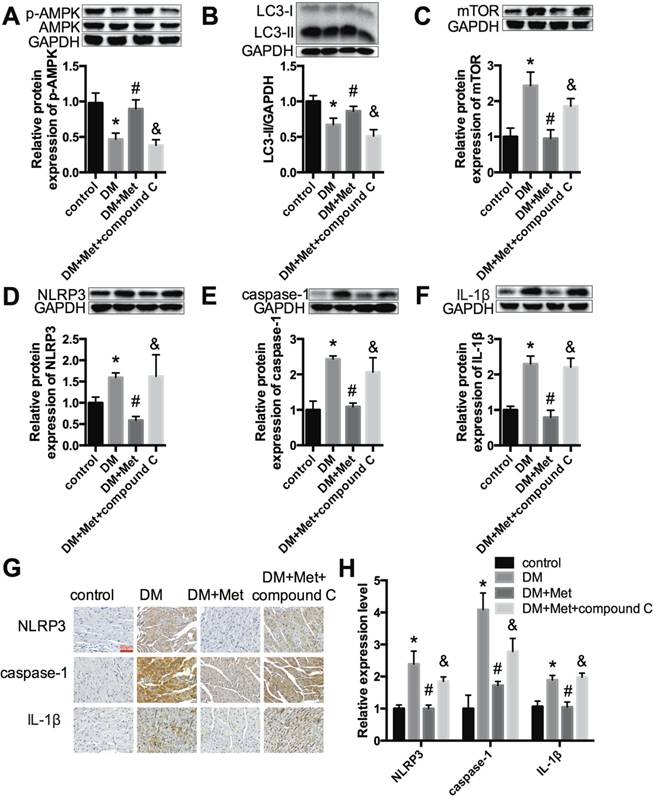
Metformin inhibits the NLRP3 pathway via AMPK/mTOR-dependent effects in diabetic mice
Echocardiography indicated that the effects of metformin on improving the cardiac function of diabetic mice were abrogated by compound C (Fig. 6A and B). Myocardial hypertrophy and collagen deposition were increased after treatment with metformin and compound C, as shown by HE and Masson's trichrome staining (Fig. 6C) as well as the increased expression levels of collagen I and collagen III (Fig. 6D and E). In addition, the results indicated that metformin increased p-AMPK and LC3 II levels and decreased mTOR levels. In contrast, these changes were reversed by compound C (Fig. 7A-C). Furthermore, western blotting and immunohistochemical staining indicated that compound C abolished the effects of metformin on decreasing NLRP3, caspase-1 and IL-1β (Fig. 7D-H).
Discussion
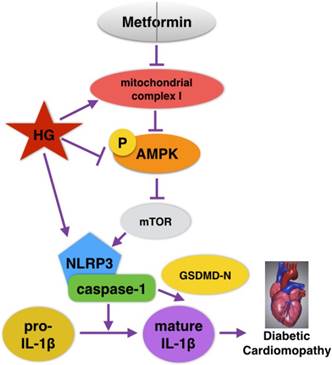
Recently, extensive studies on the cardioprotective effects of metformin in DCM have been conducted, but whether metformin could alleviate NLRP3 inflammasome changes in DCM is unknown. In the present study, NLRP3 inflammasome levels in both cardiac tissues of diabetic mice and HG-induced cardiomyocytes were significantly reduced after the administration of metformin. Moreover, p-AMPK and autophagy were activated by metformin. However, the inhibition of AMPK by compound C blocked the effects of metformin on autophagy and the NLRP3 pathway. Our study verified that metformin could inhibit the NLRP3 inflammasome via AMPK/mTOR signaling in DCM (Fig. 8). We thus proposed a new mechanism for metformin's cardioprotective function.
Metformin inhibits the NLRP3 pathway via AMPK/mTOR-dependent effects in diabetic mice. The expression levels of p-AMPK and AMPK (A), LC3 (B), mTOR (C), NLRP3 (D), caspase-1 (E) and IL-1β (F) were detected by western blotting. (G & H) Immunohistochemistry analysis was performed to detect NLRP3, caspase-1 and IL-1β expression. Scale bar, 60 μm. *P<0.05 compared to the control group, #P<0.05 compared to the DM group, &P<0.05 compared to the DM+Met group. n=5.
Graphical abstract of how metformin inhibits the NLRP3 pathway via AMPK/mTOR-dependent effects in DCM.
Previous studies showed that in DCM, the NLRP3 inflammasome could regulate cell death and fibrosis, which are vital to the structure and function of diabetic hearts [24-26]. Inhibiting the NLRP3 inflammasome and inflammatory factors significantly alleviated DCM. For example, Luo et al. [27] found that NLRP3 gene silencing exerted a protective effect in DCM. In addition, a variety of drugs have been shown to inhibit the NLRP3 pathway in DCM. Luo et al. [28] indicated that rosuvastatin, a type of anti-hyperlipidemic agent, inhibited the NLRP3 inflammasome via the MAPK pathway in DCM. Ye et al. [29] found that dapagliflozin, a sodium-glucose cotransporter 2 (SGLT2) inhibitor, could suppress the expression of the NLRP3 inflammasome by increasing AMPK phosphorylation and alleviating DCM in mice. These authors also demonstrated that better inhibition effects were gained after combination with saxagliptin, a dipeptidyl peptidase-4 inhibitor (DPP4I). However, research on the effects of the classic oral hypoglycemic drug metformin on the NLRP3 inflammasome in DCM is lacking. In our study, NLRP3, caspase-1 and IL-1β were downregulated by metformin both in vivo and in vitro. Our findings highlight the mechanism of metformin and the need for further study.
Rather than relying on its indirect hypoglycemic effects, metformin has multiple mechanisms of protecting cardiac function. The results of the UK Prospective Diabetes Study (UKPDS), a large study of cardiovascular events in diabetes, verified that although there was no significant difference in the reduction of glycosylated hemoglobin among metformin, sulfonylurea and insulin treatments, metformin could reduce all-cause mortality and diabetes-related endpoint events more effectively [30, 31]. In addition, other studies have shown that metformin can alleviate the cardiac dysfunction caused by global ischemia via activating AMPK in nondiabetic mice without affecting blood glucose [32-35]. This evidence suggests that metformin exerts cardiovascular protective effects independent of its hypoglycemic effects [36]. Our results illustrated that metformin protects cardiac function by directly suppressing the NLRP3 inflammasome.
A recent study proposed that mitochondrial complex I is a direct target of metformin and thus regulates the phosphorylation of AMPK [23]. AMPK functions as a cell energy sensor and controls a variety of pathophysiological mechanisms, such as autophagy, apoptosis and protein synthesis [37]. AMPK is considered to be a key factor regulating cardiac metabolism and function. Autophagy, the process of organism self-clearance and self-purification that facilitates the turnover of damaged organelles and proteins, is inhibited in diabetic hearts and is manifested by decreased LC3 and activation of the mTOR signaling pathway [38]. Accumulating evidence indicates that autophagy can decrease NLRP3 inflammasome levels via the mTOR signaling pathway [18, 39, 40]. Mechanistically, autophagy can downregulate the NLRP3 inflammasome through multiple mechanisms, including directly inhibiting NLRP3 inflammasome activation by clearing endogenous NLRP3 agonists (e.g., damaged mitochondria), degrading lysosomes to inhibit the maturation and secretion of IL-1β and destroying the components of the NLRP3 inflammasome (e.g., NLRP3 and ASC) [40, 41]. In this study, we showed that metformin increased p-AMPK levels and decreased mTOR and NLRP3 inflammasome levels. However, the AMPK inhibitor compound C abolished the effects of metformin on autophagy and pyroptosis. Therefore, we demonstrated that metformin inhibits the NLRP3 pathway via mitochondrial complex I/AMPK/mTOR-dependent effects in diabetic mice.
In conclusion, our findings strongly demonstrate that metformin possesses cardioprotective and anti-inflammatory effects by activating AMPK/autophagy and subsequently inhibiting the NLRP3 inflammasome in DCM. Collectively, we provide a comprehensive understanding of the molecular mechanisms of metformin treatment in DCM.
Abbreviations
AMPK: AMP-activated protein kinase; ASC: apoptosis-associated speck-like protein containing a caspase-1 recruitment domain; DCM: diabetic cardiomyopathy; DM: diabetes mellitus; DMEM: Dulbecco's modified Eagle's medium; DPP4I: dipeptidyl peptidase-4 inhibitor; EF: ejection fraction; FBS: fetal bovine serum; FS: fractional shortening; GSDMD-N: the N terminal of gasdermin D; HE: hematoxylin and eosin; HG: high glucose; IL: interleukin; Met: metformin; NLRP3: nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3; SGLT2: sodium-glucose cotransporter; STZ: streptozotocin; UKPDS: UK Prospective Diabetes Study.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81770809 and 81673426), Bethune-Merck Diabetes Research Foundation (G2017044) and the Graduate Innovation Fund of Harbin Medical University (YJSCX2017-59HYD).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Boudina S, Abel ED. Diabetic Cardiomyopathy Revisited. Circulation. 2007;115:3213-23
2. Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Reviews in Endocrine & Metabolic Disorders. 2010;11:31-9
3. Forslund K, Hildebrand F, Nielsen T, Falony G, Chatelier EL, Sunagawa S. et al. Corrigendum: Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature. 2017;545:116
4. Iliopoulos D, Hirsch HA, Struhl K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. 2011;71:3196-201
5. Belcher G, Lambert C, Goh KL, Edwards G, Valbuena M. Cardiovascular effects of treatment of type 2 diabetes with pioglitazone, metformin and gliclazide. Int J Clin Pract. 2004;58:833-7
6. Cabreiro F, Au C, Leung KY, Vergarairigaray N, Cochemé HM, Noori T. et al. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell. 2013;153:228
7. Choi YK, Park KG. Metabolic roles of AMPK and metformin in cancer cells. Molecules & Cells. 2013;36:279
8. Meijer AJ, Codogno P. AMP-Activated Protein Kinase and Autophagy. Autophagy. 2007;3:238-40
9. Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B. et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770-8
10. Xiao H, Ma X, Feng W, Fu Y, Lu Z, Xu M. et al. Metformin attenuates cardiac fibrosis by inhibiting the TGFbeta1-Smad3 signalling pathway. Cardiovasc Res. 2010;87:504-13
11. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221
12. Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger?. Science. 2010;327:296-300
13. Li X, Du N, Zhang Q, Li J, Chen X, Liu X. et al. MicroRNA-30d regulates cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic cardiomyopathy. Cell Death & Disease. 2014;5:e1479
14. Yang F, Qin Y, Lv J, Wang Y, Che H, Chen X. et al. Silencing long non-coding RNA Kcnq1ot1 alleviates pyroptosis and fibrosis in diabetic cardiomyopathy. Cell Death Dis. 2018;9:1000
15. Jing Q, Min W, Zhang J, Cai Q, Dan L, Li Y. et al. The neuroprotection of Sinomenine against ischemic stroke in mice by suppressing NLRP3 inflammasome via AMPK signaling. International Immunopharmacology. 2016;40:492-500
16. Bullon P, Alcocergómez E, Carrión AM, Garridomaraver J, Marinaguilar F, Románmalo L. et al. AMPK phosphorylation modulates pain by activation of NLRP3-inflammasome. Antioxidants & Redox Signaling. 2016;24:157
17. Li Y, Li J, Li S, Li Y, Wang X, Liu B. et al. Curcumin attenuates glutamate neurotoxicity in the hippocampus by suppression of ER stress-associated TXNIP/NLRP3 inflammasome activation in a manner dependent on AMPK. Toxicology & Applied Pharmacology. 2015;286:53
18. Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, NLRP3 inflammasome and auto-inflammatory/immune diseases. Clinical & Experimental Rheumatology. 2016;34:12-6
19. Jentzsch C, Leierseder S, Loyer X, Flohrschütz I, Sassi Y, Hartmann D. et al. A phenotypic screen to identify hypertrophy-modulating microRNAs in primary cardiomyocytes. Journal of Molecular & Cellular Cardiology. 2012;52:13-20
20. Guo S, Meng XW, Yang XS, Liu XF, Ouyang CH, Liu C. Curcumin administration suppresses collagen synthesis in the hearts of rats with experimental diabetes. Acta Pharmacologica Sinica. 2018;39:195
21. Yong Z, Xiaoguang L, Qingwei Z, Jiamin L, Jiaming J, Ning D. et al. Berberine hydrochloride prevents postsurgery intestinal adhesion and inflammation in rats. Journal of Pharmacology & Experimental Therapeutics. 2014;349:417-26
22. Zhang Y, Li X, Li J, Zhang Q, Chen X, Liu X. et al. The anti-hyperglycemic efficacy of a lipid-lowering drug Daming capsule and the underlying signaling mechanisms in a rat model of diabetes mellitus. Scientific Reports. 2016;6:34284
23. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochemical Journal. 2000;348:607-14
24. Li C, Gan S, Cai F. GW26-e2392 NADPH Oxidase-Dependent NLRP3 Inflammasome Activation and its Important Role in Diabetic cardiomyopathy. Journal of the American College of Cardiology. 2015;66:C12-C3
25. Luo B, Huang F, Liu Y, Liang Y, Wei Z, Ke H. et al. NLRP3 Inflammasome as a Molecular Marker in Diabetic Cardiomyopathy. Frontiers in Physiology. 2017;8:519
26. Sharma A, Tate M, Mathew G, Vince JE, Ritchie RH, Haan JBD. Oxidative Stress and NLRP3-Inflammasome Activity as Significant Drivers of Diabetic Cardiovascular Complications: Therapeutic Implications. Frontiers in Physiology. 2018:9
27. Luo B, Li B, Wang W, Liu X, Xia Y, Zhang C. et al. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. Plos One. 2014;9:e104771
28. Luo B, Li B, Wang W, Liu X, Liu X, Xia Y. et al. Rosuvastatin Alleviates Diabetic Cardiomyopathy by Inhibiting NLRP3 Inflammasome and MAPK Pathways in a Type 2 Diabetes Rat Model. Cardiovascular Drugs & Therapy. 2014;28:33-43
29. Ye Y, Bajaj M, Yang HC, Perez-Polo JR, Birnbaum Y. SGLT-2 Inhibition with Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor. Cardiovasc Drugs Ther. 2017;31:119-32
30. Listed N. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:854
31. I M S, A I A, H A N, D R M, S E M, C A C. et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. Bmj. 2000;321:405
32. Verma S, Mcneill JH. Metformin improves cardiac function in isolated streptozotocin-diabetic rat hearts. Am J Physiol. 1994;266:H714
33. Legtenberg RJ, Houston RJ, Oeseburg B, Smits P. Metformin improves cardiac functional recovery after ischemia in rats. Hormone and Metabolic Research. 2002;34:182-5
34. Xie Z, Dong Y, R, Neumann D, Zou M. Phosphorylation of LKB1 at serine 428 by protein kinase C-zeta is required for metformin-enhanced activation of the AMP-activated protein kinase in endothelial cells. Circulation. 2008;117:952-62
35. Zhonglin X, Yunzhou D, Junhua Z, Roland S, Dietbert N, Ming-Hui Z. Identification of the serine 307 of LKB1 as a novel phosphorylation site essential for its nucleocytoplasmic transport and endothelial cell angiogenesis. Molecular & Cellular Biology. 2009;29:3582-96
36. Kirpichnikov D, Mcfarlane SI, Sowers JR. Metformin: an update. Annals of Internal Medicine. 2002;137:25-33
37. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251-62
38. Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B. et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770-8
39. Harris J, Lang T, Thomas JP, Sukkar MB, Nabar NR, Kehrl JH. Autophagy and inflammasomes. Molecular Immunology. 2017
40. K N, JA H, VA R, SJ L, T D, HC L. et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nature immunology. 2011;12:222-30
41. Li X, Edwards M, Swaney KF, Singh N, Bhattacharya S, Borleis J. et al. Mutually inhibitory Ras-PI(3,4)P(2) feedback loops mediate cell migration. Proceedings of the National Academy of Sciences of the United States of America. 2018;115:E9125-e34
Author contact
![]() Corresponding authors: Dr. Lihong Wang, Department of Endocrinology, The Second affiliated Hospital of Harbin Medical University, Harbin 150001, Heilongjiang Province, China. Tel. +86-0451-86605163; E-Mail. nd6688com and Dr. Yunlong Bai, Department of Pharmacology, Harbin Medical University, Harbin 150081, Heilongjiang Province, China. Tel. +86-0451-86671354; E-Mail. baiyunlonghrbmu.edu.cn
Corresponding authors: Dr. Lihong Wang, Department of Endocrinology, The Second affiliated Hospital of Harbin Medical University, Harbin 150001, Heilongjiang Province, China. Tel. +86-0451-86605163; E-Mail. nd6688com and Dr. Yunlong Bai, Department of Pharmacology, Harbin Medical University, Harbin 150081, Heilongjiang Province, China. Tel. +86-0451-86671354; E-Mail. baiyunlonghrbmu.edu.cn
Received 2018-9-3
Accepted 2019-1-15
Published 2019-3-10