ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2019; 15(7):1440-1451. doi:10.7150/ijbs.30193 This issue Cite
Research Paper
The Antioxidant MitoQ Protects Against CSE-Induced Endothelial Barrier Injury and Inflammation by Inhibiting ROS and Autophagy in Human Umbilical Vein Endothelial Cells
Sha Chen1*, Yu Wang1*, Hailin Zhang2, Ran Chen1, Fangfang Lv2, Zhengmao Li3, Ting Jiang3, Daopeng Lin2, Hongyu Zhang3, Li Yang4 ![]() , Xiaoxia Kong1
, Xiaoxia Kong1 ![]()
1. School of Basic Medical Sciences, Institute of Hypoxia Medicine, Wenzhou Medical University, Wenzhou, Zhejiang 325035, PR China
2. Department of Children's Respiration, The Second Affiliated Hospital & Yuying Children's Hospital, Wenzhou Medical University, Wenzhou, Zhejiang 325027, PR China
3. School of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou, Zhejiang 325035, PR China
4. Department of Respiratory Medicine, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang 325035, PR China
* These authors contributed equally to this work.
Abstract

Chronic obstructive pulmonary disease (COPD) is a common disease characterized by persistent airflow limitation. Pulmonary vascular endothelial barrier injury and inflammation are increasingly considered to be important pathophysiological processes in cigarette smoke extract (CSE)-induced COPD, but the mechanism remains unclear. To identify the cellular mechanism of endothelial barrier injury and inflammation in CSE-treated human umbilical vein endothelial cells (HUVECs), we investigated the effect of the mitochondrion-targeting antioxidant mitoquinone (MitoQ) on endothelial barrier injury and inflammation. We demonstrated that MitoQ restored endothelial barrier integrity by preventing VE-cadherin disassembly and actin cytoskeleton remodeling, as well as decreased inflammation by the NF-κB and NLRP3 inflammasome pathways in endothelial cells. In addition, MitoQ also maintained mitochondrial function by reducing the production of ROS and excess autophagy. Inhibition of autophagy by 3-MA protected against cytotoxicity that was induced by CSE in HUVECs. Overall, our study indicated that mitochondrial damage is a key promoter in the induction of endothelial barrier dysfunction and inflammation by CSE. The protective effect of MitoQ is related to the inhibition of ROS and excess autophagy in CSE-induced HUVEC injury.
Keywords: Chronic obstructive pulmonary disease, Cigarette smoke extract, Endothelial barrier, Inflammation, ROS, Autophagy
Introduction
Chronic obstructive pulmonary disease (COPD) is a common preventable and treatable disease characterized by persistent airflow limitation. Smoking is considered to be the most important factor leading to COPD [1-3]. However, pulmonary vascular cell injury is increasingly considered to be an important pathophysiological process in cigarette smoke extract (CSE)-induced COPD [4]. The vascular endothelium is formed by a continuous endothelial cell monolayer and plays key roles in the maintenance of vascular integrity [5-7]. CSE causes endothelial dysfunction in the pulmonary circulation in human and animal models [8-10].
Inflammation is also involved in many pathophysiological processes, including COPD [11, 12]. Nuclear factor NF-κB plays a central role in inflammation and apoptosis through diverse signaling cascades [13]. It has been reported that upregulation of cell adhesion molecules by NF-κB in endothelial cells is a critical step that alters the adhesive properties of vasculature and causes uncontrolled infiltration of leukocytes into the inflamed tissue [14]. Pharmacological inhibitors of the NF-κB pathway in endothelial cells have potential therapeutic value in treating inflammatory diseases and cancers [15]. In COPD and other pulmonary inflammatory diseases, the main function of the NLRP3 inflammatory corpuscle and its products is to initiate a primary immune response and prevent pathogen invasion [16]. However, when the NLRP3 inflammasome is excessively activated, leading to the excessive release of IL-1 beta and IL-18, the inflammatory response persists, leading to COPD-related pathophysiological changes, which results in airflow limitation and breathing difficulties [17-20]. Recently, the central role of endothelial dysfunction in inflammation has also been recognized. The maintenance of endothelial function is particularly important for the passage of macromolecules and fluid between the blood and interstitial space. If the barrier is disrupted, excessive leakage may result in tissue inflammation and inflammatory diseases, such as acute respiratory distress syndrome (ARDS) and COPD [21-23]. Therefore, restoring vessel barrier function and controlling inflammation following injury are essential for maintaining tissue fluid balance and reversing lung injury and edema. However, the exact mechanism of CSE-induced vascular barrier dysfunction and inflammation is not yet fully understood.
Some studies have shown that ROS may play an important role in the pathogenesis of COPD. Elevated ROS levels have been shown to have important roles in regulating a number of physiological and pathophysiological events, including cell apoptosis, inflammation, and disruption of the endothelial barrier [24]. CSE increases mitochondrion-derived ROS (mtROS), further promotes the activation of endothelial cells and increases proinflammatory cytokines in synergy with cytosolic ROS [25]. Antioxidants targeted to the mitochondria for the maintenance of function reduce endothelial inflammation in hypertensive animal models [26]. The administration of antioxidants reduces CSE- and/or inflammatory-dependent BBB damage. Previous studies have demonstrated that exposure of ECs to ROS induces actin stress fibers and redistribution of VE-cadherin to the cell periphery, which results in EC barrier dysfunction [27-30]. Therefore, it is often important to improve oxidative stress and the quantity of mitochondria for the maintenance of EC function and to decrease inflammation.
In this study, the mitochondrion-targeted antioxidant mitoquinone (MitoQ) was used to suppress mitochondrial ROS to evaluate the protective effect of MitoQ on CSE-induced endothelial dysfunction and inflammation in HUVECs. We also used a nonspecific inhibitor of PI3-kinase, 3-MA, to evaluate the role of autophagy in MitoQ-protected HUVECs treated with CSE and further evaluated whether the protective effect of MitoQ is closely related to the autophagy response. Our data demonstrated that MitoQ clearly restored endothelial barrier function and further decreased NF-κB/NLRP3-mediated inflammatory activation by maintaining mitochondrial function. Moreover, the effect of 3-MA suggested that CSE-mediated autophagy likely has a destructive role in endothelial injury. Therefore, antioxidants and restored autophagy may contribute to the rescue of CSE-induced COPD.
Materials and Methods
Cell culture
HUVECs were obtained from American Type Culture Collection (ATCC). HUVECs were cultured in Endothelial Cell Medium (ECM, SclenCell) containing 10% fetal bovine serum (FBS, SclenCell) and 1% Endothelial Cell Growth Supplement (ECGS, SclenCell) and 1% penicillin/streptomycin solution (P/S, SclenCell) and were maintained at 37°C and 5% CO2. HUVECs were utilized for experimentation at 70-80% confluency.
Preparation of CSE
The extraction of cigarette smoke is prepared according to previous reports [31] with some modifications. Briefly, one cigarette (Double Happiness, Shanghai Tobacco Corporation, Shanghai, China) was burned and the smoke passed through a filter for removing particles and bacteria into a vessel containing phosphate-buffered saline (ECM, 10 mL per one cigarette) using a vacuum pump. This 100% CSE was adjusted to a pH of 7.4 and was diluted to the concentrations of 0, 1.25, 2.5 and 5% and used for the following experiment within 30 min of preparation.
Drug treatment
At the cell level, the experiment was divided into four groups: Control group, CSE group, MitoQ+CSE group and MitoQ group. HUVECs were treated by 5% CSE for 24 hours with (or not) MitoQ of 100nmol/L.
Cell viability assay
The Cell Counting Kit-8 (CCK-8) assay kit (Dojindo, Kumamoto, Japan) was performed as per the manufacturer's instructions. Cells were plated in 96-well plates at a density of 5× 104/well in sextuplicate. At indicated drug concentration points, 10 μl CCK-8 solutions were added to the cells for 2 hours at 37 °C, and the absorbance of the cells was measured at 450nm using an enzyme standard instrument according to the manufacturer's instructions.
Western blotting analysis
HUVECs were lysed in RadioImmunoprecipitation Assay (RIPA) buffer (Beyotime, P0013B) with PMSF lysis buffer. The protein concentration was measured using a BCA assay kit (Beyotime). Equal protein amounts (usually 30μg) were separated by 8%-15% SDS-PAGE. Proteins were then transferred to PVDF membranes (Immobilon, IPVH00010). Blots were blocked and immunoblotted with NLRP3 (Cell Signal, 1:1000, 15101), Caspase-1 (Cell Signal, 1:1000, 2225), IL-1β (Cell Signal, 1:1000, 12426), GAPDH (Bioworld, 1:50000, AP0063), Cleaved-Caspase-3 (Cell Signal, 1:500, 9664), F-actin (Abcam, 1:500, ab205), VE-Cadherin(Abcam, 1:1000, ab33168), NF-κB p65 (Sigma, 1:500, 510038), P-IκBα (Cell Signal, 1:1000, 2859), IκBα (Cell Signal, 1:1000, 4814), Histone (Abcam, 1:1000, ab1791), Beclin1 (Santa Cruz, 1:1000, sc-48341), LC3A/B (Abcam, 1:500, ab128025), TOMM20 (Santa Cruz, 1:100, sc-17764), p62 (Abcam, 1:500, ab56416) at 4 °C overnight, followed by HRP-conjugated secondary antibodies (typically 1:2000 dilution) for 1h at room temperature. The results were analyzed with Image J Analysis Software (Image J 1.44p, Wayne Rasband, National Institutes of Health, USA).
Intracellular ROS determination
The level of intracellular ROS was examined by using cell-permeable fluorescent probe 2',7'-dichlorofluorescin-diacetate (DCFH-DA). Intracellular detection of ROS in different groups was achieved by incubating cells with 10μM DCFH-DA for 30 min at 37°C in the dark. Cells were then detached by gentle scraping with a cell scraper and analyzed by fluorescence microplate. The fluorescence of DCFH-DA was excited at 488 nm, and the emission was collected at 525 nm.
Immunofluorescence staining
HUVECs were cultured on glass slides, washed in PBS and fixed with 4% paraformaldehyde for 30 min at room temperature. Cells were then permeabilized, blocked with 5% BSA, and incubated with primary and secondary antibodies. Cells were co-stained with DAPI for the nuclei. Images were captured from six or more randomly chosen fields at 400× magnification using a Nikon ECLIPSE Ti microscope (Nikon, Tokyo, Japan). Data from repeated experiments were used for statistical analysis.
Statistical analysis
All data are expressed as means ± SEM (n=5) from at least three separate experiments. Student's t-test was used for statistical analysis for comparison of two groups. For more than two groups, statistical evaluation of the data was performed using a one-way analysis-of-variance (ANOVA) test, followed by Tukey's post hoc test. Values of *P < 0.05 and **P < 0.01 were considered statistically significant.
Results
CSE induced the internalization of VE-cadherin and the rearrangement of the actin cytoskeleton of human umbilical vein endothelial cells
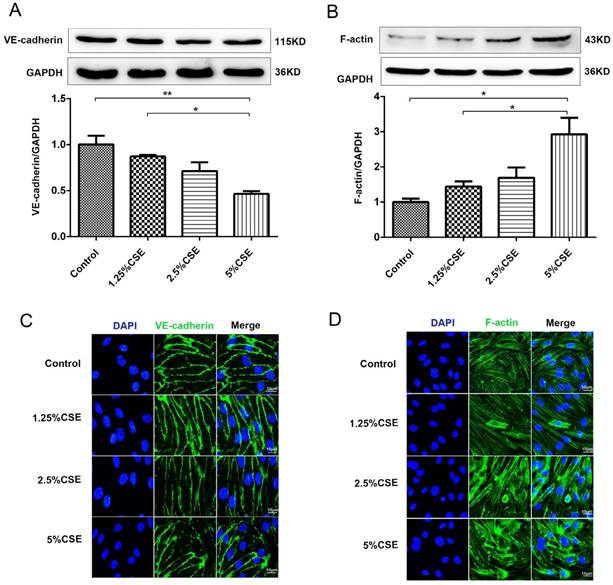
We examined the effect of CSE stimulation on endothelial barrier integrity in cultured HUVECs. First, the expression of the membrane proteins VE-cadherin and F-actin in HUVECs was analyzed by Western blotting. As shown in Fig. 1A, the expression level of VE-cadherin clearly decreased after 2.5 and 5% CSE stimulation for 24 hours. There was no significant difference in the expression level of VE-cadherin after 1.25% CSE treatment compared with the control group. In contrast, the expression of F-actin increased significantly in the 5% CSE-treated group (Fig. 1B). Furthermore, CSE-induced morphological changes in HUVECs were analyzed by immunofluorescence microscopy. HUVEC cell monolayers seeded in 6-well plates were stimulated with different concentrations of CSE for 24 hours. Under the control conditions, VE-cadherin was observed as a continuous line along the interendothelial cell border. Immunostaining for F-actin with rhodamine-phalloidin revealed a predominance of circumferential actin bundles and a few stress fibers. After treatment with different concentrations of CSE for 24 hours, the immunostaining results showed that the expression of VE-cadherin clearly decreased in HUVECs treated with 5% CSE, leading to the formation of intercellular gaps (Fig. 1C). Furthermore, CSE induced F-actin reorganization and intercellular gap formation and increased stress fiber formation (Fig. 1D). These results demonstrated that CSE induced VE-cadherin disassembly and actin cytoskeleton remodeling in endothelial cells, thereby disrupting endothelial barrier integrity.
CSE induced NF-κB and NLRP3 inflammasome activation in human umbilical vein endothelial cells
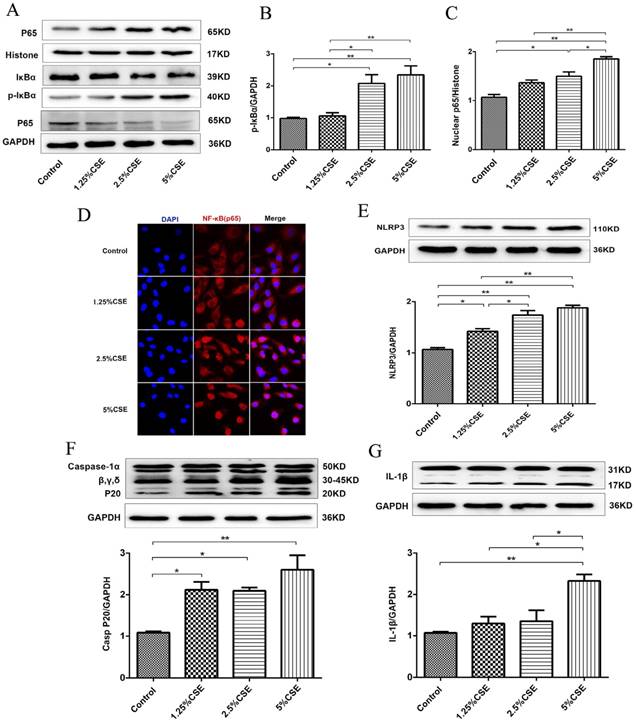
To further examine the effect of CSE on endothelial cells, we analyzed the classical inflammatory signaling pathways of NF-κB and NLRP3 inflammasome signaling. As shown in Fig. 2, the expression levels of p-IκBα and nuclear NF-κB p65 protein significantly increased after 5% CSE treatment (Fig. 2A-C), and CSE markedly induced NF-κB p65 nuclear translocation, which was determined by immunofluorescence (Fig. 2D). Then, we analyzed the NLRP3 signaling pathway in HUVECs treated with CSE and found that CSE significantly increased the levels of NLRP3 (P < 0.01) (Fig. 2E) and activated caspase-1 (P < 0.01) (Fig. 2F), as well as contributed to IL-1β maturation (Fig. 2G). These results indicated that CSE activated the NF-κB and NLRP3 inflammasome signaling pathways in HUVECs.
The effect of CSE on the expression of VE-cadherin and the actin cytoskeleton in HUVECs. (A) The expression levels of the VE-cadherin and (B) F-actin in cell lysates from HUVECs were detected by Western blotting. Expression of VE-cadherin (C) and F-actin (D) in HUVECs after CSE treatment was analyzed by immunofluorescent staining. Scale bar is 10μm. Green indicates cells labeled with F-actin and VE-cadherin; blue, DAPI. All data were compared with control group and presented as mean ±SEM (n=5 in each group), *P < 0.05, **P < 0.01.
Cigarette smoke extract induced mitochondrial dysfunction in human umbilical vein endothelial cells
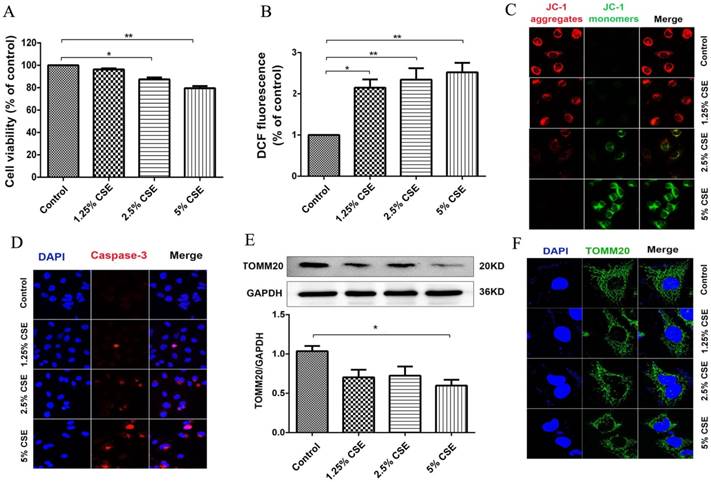
To investigate the effect of CSE on mitochondrial integrity, HUVECs were treated with different concentrations of CSE in model of CSE exposure in vitro. The HUVECs were treated with CSE at 0, 1.25, 2.5 and 5% concentrations for 24 hours, and cell viability was analyzed using a CCK-8 assay. Noticeable decreases in cell viability were observed after the cells were treated with CSE concentrations of 2.5 and 5% for 24 hours (Fig. 3A). ROS analysis showed that CSE significantly increased ROS production in a dose-dependent manner (Fig. 3B). Furthermore, the mitochondrial membrane potential was analyzed by immunofluorescence staining. The results showed that the mitochondrial membrane potential significantly decreased after CSE treatment (Fig. 3C). Furthermore, the protein levels of Caspase-3 were identified by immunofluorescence. Our results showed that CSE significantly increased expression of protein Caspase-3 (Fig. 3D), Finally, we evaluated mitochondrial integrity by analyzing the expression of TOMM20, and the results showed that CSE induced markedly decreased expression levels of TOMM20, which was determined by Western blotting and immunofluorescence staining (Fig. 3E and F). These data suggested that CSE exposure induced endothelial barrier dysfunction and inflammation, which is related to ROS production and mitochondrial damage in HUVECs.
MitoQ alleviated CSE-induced damage of endothelial barrier function
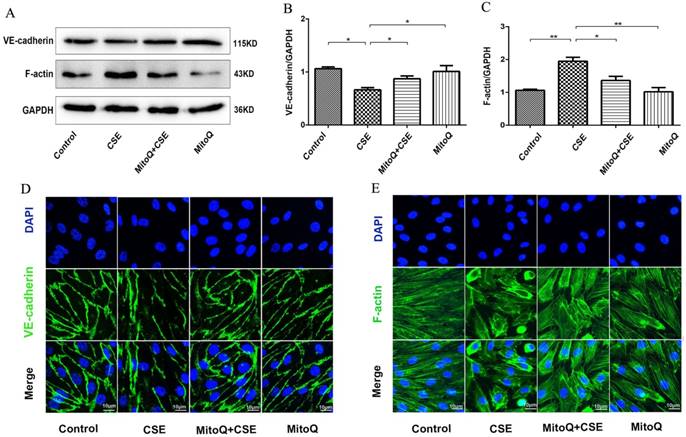
To confirm the effect of ROS on endothelial barrier damage induced by CSE, the mitochondrion-targeted antioxidant mitoquinone (MitoQ) was added to HUVECs. The protein expression levels of VE-cadherin and F-actin were analyzed by Western blotting (Fig. 4). The results showed that MitoQ significantly inhibited the decrease in the VE-cadherin expression level and the increase in the F-actin expression level induced by CSE stimulation. There was no significant difference between the MitoQ group and the control group (Fig. 4A-C). Furthermore, immunofluorescence staining showed that MitoQ significantly decreased CSE-induced VE-cadherin internalization (Fig. 4D) and F-actin reorganization (Fig. 4E). These results demonstrated that MitoQ decreased CSE-induced VE-cadherin disassembly and actin cytoskeleton remodeling and improved endothelial barrier integrity in endothelial cells.
CSE induced NF-κB and NLRP3 inflammasome activation in HUVECs. (A) Expression of protein IκBα and p-IκBα in cell lysates from HUVECs were detected by Western blotting. GAPDH served as the standards. Expression of protein nuclear p65 was detected by cytoplasmic separation Kit. Histone was used as loading control. Quantification analysis of protein p-IκBα (B) and nuclear p65 (C) levels. (D) Nuclear translocation of p65 (red, 400×) was detected by immunofluorescent staining. Expression of protein NLRP3 (E), Caspase-1 (F) and IL-1β (G) was detected by Western blotting. All data were compared with control group and presented as mean ±SEM (n=5 in each group), *P < 0.05, **P < 0.01.
CSE caused mitochondrial dysfunction in HUVECs. (A) HUVECs were incubated with CSE at different concentration for 24h. The cell viability was measured by CCK8 kit. (B) Reactive oxygen species levels were analyzed by DCFH-DA kit. (C) After treatment the cells were subjected JC-1 staining (400×) for mitochondrial membrane potential assessment. Red staining indicates polarized mitochondria in JC-1 staining. Green staining indicates depolarized mitochondria in JC-1 staining. (D) Immunofluorescence staining for caspase-3 (red) and nuclei (blue) (400×). (E) Expression of protein TOMM20 in HUVECs was shown by Western blotting. GAPDH served as the standards. (F) Representative mitochondrial morphology (1000×) in HUVECs incubated for 24h with CSE. Blue: DAPI; green: TOMM20 (mitochondria). All data were compared with control group and presented as mean ±SEM (n=5 in each group), *P < 0.05, **P < 0.01.
The effect of MitoQ on endothelial barrier dysfunction induced by CSE. (A) Western blotting was used to detect expression of protein VE-cadherin and F-actin. Quantification analysis of protein VE-cadherin (B) and F-actin (C) levels. Expression of protein VE-cadherin (green) (D) and F-actin (green) (E) were detected by immunofluorescence. The HUVECs were grown to about 90% confluence. Scale bar is 10μm. Data are expressed as mean ± SEM (n = 5). *P < 0.05, **P < 0.01.
The effect of MitoQ in CSE-induced NF-κB and NLRP3 inflammasome activation in HUVECs. (A) Expression of protein IκBα, p-IκBα and nuclear p65 was detected by Western blotting. GAPDH and histone as the standards. Quantification analysis of p-IκBα (B) and nuclear p65(C) levels. (D) Immunofluorescence staining of p65 (red) using confocal microscopy (400×). Expression of protein NLRP3 (E), Caspase-1 (F) and IL-1β (G) was detected by Western blotting. Data are expressed as mean ± SEM (n = 5). *P < 0.05, **P < 0.01.
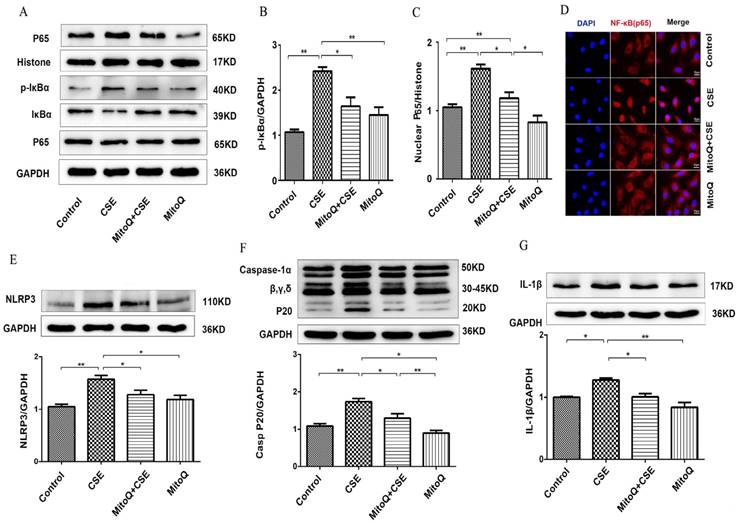
MitoQ inhibited CSE-induced NF-κB and NLRP3 inflammasome activation in human umbilical vein endothelial cells
To identify the effect of MitoQ on inflammation in HUVECs, NF-κB and NLRP3 inflammasome signaling pathways in HUVECs treated with 5% CSE were analyzed. The Western blotting results showed that after MitoQ pretreatment, CSE-induced activation of the cytosolic content of p-IκBα and the expression level of nuclear protein NF-κB p65 significantly decreased (Fig. 5A-C). The immunofluorescent staining results showed that MitoQ significantly decreased CSE-induced NF-κB p65 nuclear translocation (Fig. 5D). Furthermore, we investigated whether NLRP3 inflammasome pathways are involved in the protective role of MitoQ. The expression of the inflammatory cytokines NLRP3, caspase-1 and IL-1β in different groups was also analyzed by Western blotting. The results showed that CSE-induced expression of NLRP3 (Fig. 5E, P < 0.05), cleaved caspase-1 (Fig. 5F, P < 0.05) and IL-1β (Fig. 5G, P < 0.05) markedly decreased after MitoQ treatment. These results indicated that MitoQ inhibited NF-κB pathways and the activation of the NLRP3 inflammasome induced by CSE in HUVECs.
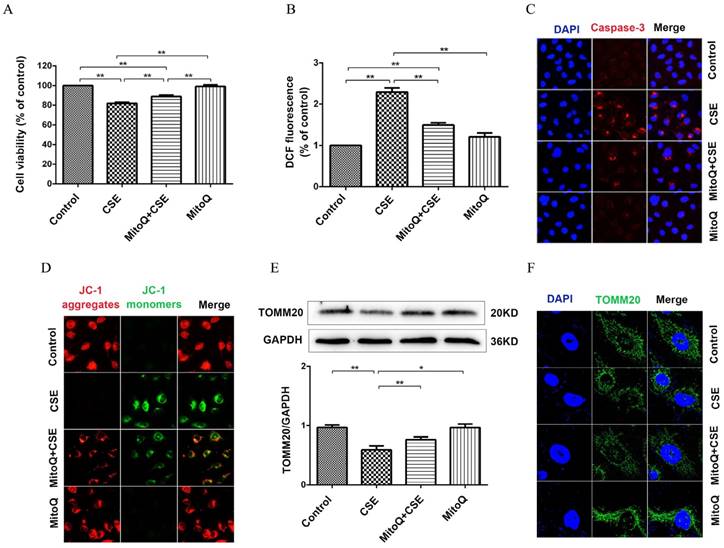
MitoQ inhibited mitochondrial damage induced by CSE exposure
To evaluate the protective mechanism of MitoQ on endothelial barrier function and inflammation, we analyzed mitochondrial function. As shown in Fig. 6A, the CCK-8 assay showed that the CSE-induced decrease in cell viability significantly increased after the MitoQ treatment (Fig. 6A) (P < 0.01). CSE significantly increased the generation of ROS in HUVECs, which was largely reduced after the MitoQ treatment (Fig. 6B) (P < 0.01). Furthermore, the protein expression level of cleaved caspase-3 was analyzed by immunofluorescence. Our results showed that CSE significantly increased the expression level of cleaved caspase-3, which was markedly reversed by the treatment with MitoQ (Fig. 6C). In addition, mitochondrial membrane potential (ΔΨm) was analyzed by JC-1 fluorescence. The MitoQ treatment inhibited the JC-1 fluorescent color change from red to green, which is caused by CSE (Fig. 6D), suggesting that MitoQ inhibited the decrease in ΔΨm and mitochondrial damage. Subsequently, we analyzed the expression of the TOMM20 protein to confirm mitochondrial integrity. Our results showed that MitoQ protected against the decrease in the TOMM20 protein expression level induced by CSE (Fig. 6E). The immunofluorescent staining results are similar to the Western blotting results, indicating that CSE-induced mitochondrial damage was reversed by MitoQ (Fig. 6F). These results indicated that the protection provided by MitoQ against endothelial barrier dysfunction and inflammation is related to the stabilization of mitochondrial function.
MitoQ ameliorated mitochondrial damage induced by CSE in HUVECs. HUVECs were stimulated by 5% CSE and (or) 100nM MitoQ for 24h. (A) Cell viability was measured by CCK8 kit. (B) Reactive oxygen species levels were analyzed by DCFH-DA kit. (C) Immunofluorescence staining (400×) for caspase-3 (red) and nuclei (blue). (D) Mitochondrial membrane potential in HUVECs was analyzed by JC-1 staining (400×). (E) Expression of protein TOMM20 was detected by Western blotting. (F) Expression of TOMM20 was detected by immunofluorescence staining (1000×). Data are expressed as mean ± SEM (n = 5). *P < 0.05, **P < 0.01.
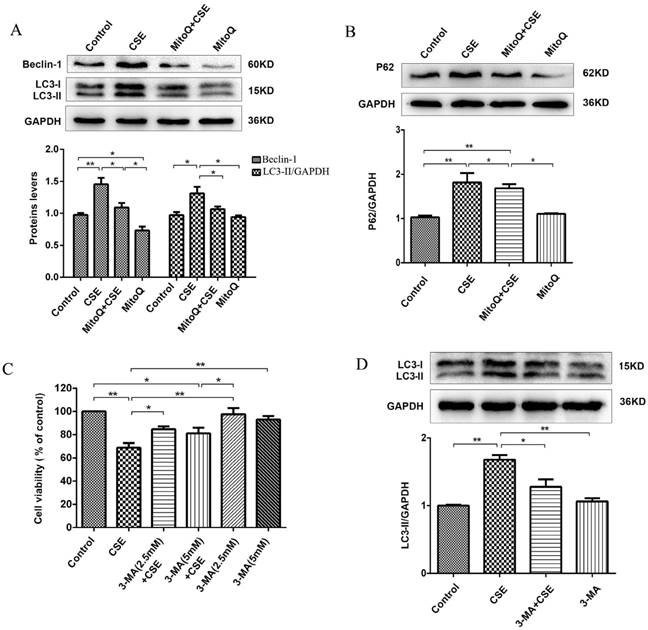
MitoQ inhibited CSE-induced excess autophagy in human umbilical vein endothelial cells
To further identify the protective effect of MitoQ related to autophagy in HUVECs, we analyzed the autophagy level. The Western blotting results showed that 5% CSE significantly upregulated the expression of the proteins Beclin-1 and LC3-II (P < 0.01) and increased the expression level of P62 (P < 0.01) in HUVECs, which was markedly reversed by MitoQ (Fig. 7A and B, P < 0.05). To further examine the specific role of autophagy in the process of CSE-induced injury of HUVECs, 3-MA was added to the cells. The CCK-8 assay demonstrated that treatment with 3-MA at a concentration of 2.5 mmol/L significantly reduced the decrease in cell viability induced by CSE. The 3-MA alone group showed no changes in cell viability (Fig. 7C). Western blotting analysis confirmed the effect of 3-MA on autophagy. The results showed that 3-MA significantly decreased CSE-induced LC3-II protein expression level in HUVECs (Fig. 7D, P < 0.05). Overall, these data indicate that autophagy plays a negative role in CSE-induced HUVEC injury. We infer that MitoQ alleviates mitochondrial damage by inhibiting the production of ROS and the excessive activation of autophagy.
Discussion
Chronic obstructive pulmonary disease is a complex progressive disease characterized by chronic inflammation, irreversible airflow limitation and emphysema. Cigarette smoke is considered to be a major risk factor for the development of COPD [32, 33]. However, the mechanism of pathogenesis is yet to be elucidated. Pulmonary endothelial dysfunction and vascular remodeling have also been reported in the lungs of COPD patients. The pulmonary endothelium plays an important role in the maintenance of vascular barrier function [8-10]. Lung endothelial injury also contributes to the pathogenesis of emphysema [4]. Our results showed that CSE reduced the expression of VE-cadherin and the formation of intercellular gaps; furthermore, F-actin reorganization and increased stress fiber formation induced by CSE in HUVECs further confirmed that CSE significantly induced VE-cadherin disassembly and actin cytoskeleton remodeling, disrupting endothelial barrier integrity in endothelial cells.
MitoQ inhibited CSE-induced autophagy in HUVECs. (A) Expression of LC3 and Beclin-1 were detected by Western blotting and quantification analysis. (B) Expression of protein P62 was detected by Western blotting. (C) Cell viability in HUVECs treated with CSE and (or) 3-MA was measured by CCK8 kit. (D) Expression of LC3 was detected by Western blotting and quantification analysis. Data are expressed as mean ± SEM (n = 5). *P < 0.05, **P < 0.01.
The pathogenesis of COPD is quite complex, and it is generally speculated that inflammatory reactions play an important role in the occurrence and development of COPD [34]. NF-κB is a nuclear transcription factor that is widely present in many cells in the body, and the imbalance of its activation is closely related to the occurrence and development of COPD [35]. Based on the previously mentioned theoretical level, we used different concentrations of CSE to stimulate HUVECs and analyzed the protein expression levels of the NF-κB signaling pathway. It was found that the NF-κB signaling pathway and NF-κB p65 nuclear translocation were clearly activated by 5% CSE. Furthermore, it has also been pointed out that the nucleotide-binding oligomerization domain-like receptor 3 (NLRP3) inflammasome is closely related to the pathological process of COPD [16, 36], and NLRP3 KO significantly inhibits the expression levels of IL-1α and IL-1β in the lung tissue of COPD mice [37]. Similar results were found in our study. CSE induced the activation of the NLRP3 inflammatory corpuscle signaling pathway and stimulated the activation of caspase-1 and the release of IL-1β.
Oxidative stress in the lungs significantly increases after chronic CS exposure [38, 39]. Mitochondria are important sources of endogenous ROS and play an important role in many physiological and pathological environments [40]. Mitochondrial dysfunction has been shown to be responsible for cellular senescence [41, 42]. Cigarette smoking-induced mitochondrial ROS can accelerate an imbalance in fission and fusion, resulting in mitochondrial fragmentation, which contributes to COPD development[43]; however, increased mitochondrial ROS generation and release further cause inflammatory responses in chronic lung diseases[44]. It has been reported that antioxidant treatments counteract the pro-inflammatory effects of CSE exposure in BBB endothelial cells and protect against endothelial barrier function [45]. Treatment with MitoQ has been shown to decrease ischemia-reperfusion injury in a murine syngeneic heart transplant model and in animal models of myocardial infarction [46, 47]. The antioxidant MitoQ, which is targeted to decrease mitochondrial oxidative damage, has been shown to specifically lead to reduced inflammation and proliferation in airway smooth muscle cells [48]. These results are consistent with the results from our study. This study showed that pretreatment with the mitochondrial-targeting antioxidant MitoQ markedly protected against endothelial barrier dysfunction induced by CSE. Furthermore, the classical NF-κB signaling pathway and the nuclear translocation of NF-κB were reversed by MitoQ.
A large number of studies have shown that activation of the NLRP3 inflammasome contributes to the development of COPD [37, 49]. CSE has been shown to significantly induce activation of the NLRP3 inflammasome in a COPD model [50]. MitoQ has been shown to ameliorate experimental mouse colitis by suppressing NLRP3 inflammasome-mediated inflammatory cytokines [51]. This study demonstrated that MitoQ suppressed CSE-induced NLRP3 inflammasome activation and inhibited caspase-1 activity; furthermore, the maturation and release of IL-1β were also inhibited by MitoQ in HUVECs. These findings suggest that MitoQ protected against endothelial barrier dysfunction and inflammation induced by CSE in our model.
It has been reported that autophagy can remove damaged mitochondria from cells and prolong the lifespan of cells. In contrast, autophagy may also promote apoptosis [52]. Recent studies have indicated that cigarette smoke exposure induces epithelial cell death by autophagy activation in COPD models [53, 54]. In chronic lung disease caused by exposure to silica, autophagy deficiency in macrophages enhances NLRP3 inflammasome activity[55]. In this study, CSE caused mitochondrial dysfunction associated with a decrease in mitochondrial membrane potential and increased mitochondrial ROS production, which were markedly reversed by MitoQ. Interestingly, we also found that MitoQ protected mitochondrial function but decreased the expression levels of the LC3-II and P62 proteins induced by CSE in HUVECs. The treatment of HUVECs with 3-MA significantly decreased the CSE-induced expression level of the LC3-II protein but markedly increased the cell viability of HUVECs. These results suggested that inhibition of autophagy using 3-MA protected against HUVEC injury induced by CSE, indicating that excess autophagy inhibited autophagic flux and aggravated mitochondrial dysfunction in CSE-induced HUVEC injury. Therefore, we speculated that autophagy also plays a negative role in the pathogenesis of CSE-induced HUVEC injury.
In conclusion, these results indicate that CSE impairs endothelial barrier function, as well as NF-κB and NLRP3 inflammasome pathway-dependent inflammation. The mitochondrion-targeting antioxidant MitoQ restores endothelial barrier function, and inflammation is associated with a reduction in mitochondrial damage by inhibiting substantial production of ROS and excess autophagy. This evidence provides new strategies for the treatment of CSE-induced lung injury and suggests that antioxidants and restored autophagy may contribute to the rescue of CSE-induced COPD.
Acknowledgements
This work was supported by grants from the Zhejiang Provincial Natural Science Funding (LY17H010005, LY17H010009, LY17H160193), the National Science Foundation of China (No. 81772450, No.81472165, No. 81600033).
Author Contributions
S.C. and Y.W. Z.L. conducted cell culture experiment. H.Z., R.C. and D.L. performed data analysis. S.C. and H.Z. drafted the manuscript. F.L. and T.J. prepared figures. H.Z. and Z.L. participated in the manuscript preparation. X.K. and L.Y. is the guarantor of this work.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lebowitz MD. Smoking habits and changes in smoking habits as they relate to chronic conditions and respiratory symptoms. American journal of epidemiology. 1977;105:534-43
2. Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. American journal of respiratory and critical care medicine. 2001;163:1256-76
3. Auerbach O, Hammond EC, Garfinkel L, Benante C. Relation of smoking and age to emphysema. Whole-lung section study. The New England journal of medicine. 1972;286:853-7
4. Tarasevicienestewart L, Voelkel NF. Molecular pathogenesis of emphysema. Journal of Clinical Investigation. 2008;118:394-402
5. Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP. et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91:3527-61
6. Harris ES, Nelson WJ. VE-cadherin: at the front, center, and sides of endothelial cell organization and function. Current opinion in cell biology. 2010;22:651-8
7. Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Developmental cell. 2013;26:441-54
8. Ferrer E, Peinado VI, Castaneda J, Prieto-Lloret J, Olea E, Gonzalez-Martin MC. et al. Effects of cigarette smoke and hypoxia on pulmonary circulation in the guinea pig. The European respiratory journal. 2011;38:617-27
9. Peinado VI, Pizarro S, Barberà JA. Pulmonary vascular involvement in COPD. Chest. 2008;134:808-14
10. Wright JL, Churg A. Short-term exposure to cigarette smoke induces endothelial dysfunction in small intrapulmonary arteries: analysis using guinea pig precision cut lung slices. Journal of applied physiology (Bethesda, Md: 1985). 2008;104:1462-9
11. Di Stefano A, Caramori G, Oates T, Capelli A, Lusuardi M, Gnemmi I. et al. Increased expression of nuclear factor-kappaB in bronchial biopsies from smokers and patients with COPD. The European respiratory journal. 2002;20:556-63
12. Deo P, Cheng ZZ, Han WZ, Tong L. The Change and Significance of Nuclear Transcription Factor NF- kB at Different Period in Mice While Gradually Develop COPD. Progress in Modern Biomedicine. 2011
13. Hu W, Zhou PH, Rao T, Zhang XB, Wang W, Zhang LJ. Adrenomedullin attenuates interleukin-1β-induced inflammation and apoptosis in rat Leydig cells via inhibition of NF-κB signaling pathway. Experimental Cell Research. 2015;339:220-30
14. Balwani S, Chaudhuri R, Nandi D, Jaisankar P, Agrawal A, Ghosh B. Regulation of NF-kappaB activation through a novel PI-3K-independent and PKA/Akt-dependent pathway in human umbilical vein endothelial cells. PLoS One. 2012;7:e46528
15. Baldwin AS Jr. Series introduction: the transcription factor NF-kappaB and human disease. The Journal of clinical investigation. 2001;107:3-6
16. dos Santos CC. The role of the inflammasome in ventilator-induced lung injury. American journal of respiratory and critical care medicine. 2012;185:1141-4
17. Leemans JC, Cassel SL, Sutterwala FS. Sensing damage by the NLRP3 inflammasome. Immunological Reviews. 2011;243:152-62
18. Chung KF, Marwick JA. Molecular mechanisms of oxidative stress in airways and lungs with reference to asthma and chronic obstructive pulmonary disease. Annals of the New York Academy of Sciences. 2010;1203:85-91
19. Maes T, Bracke KR, Vermaelen KY, Demedts IK, Joos GF, Pauwels RA. et al. Murine TLR4 is implicated in cigarette smoke-induced pulmonary inflammation. International archives of allergy and immunology. 2006;141:354-68
20. Birrell MA, Eltom S. The role of the NLRP3 inflammasome in the pathogenesis of airway disease. Pharmacology & therapeutics. 2011;130:364-70
21. Lu Q, Sakhatskyy P, Grinnell K, Newton J, Ortiz M, Wang Y. et al. Cigarette smoke causes lung vascular barrier dysfunction via oxidative stress-mediated inhibition of RhoA and focal adhesion kinase. American journal of physiology Lung cellular and molecular physiology. 2011;301:L847-57
22. Lomas-Neira J, Venet F, Chung CS, Thakkar R, Heffernan D, Ayala A. Neutrophil-endothelial interactions mediate angiopoietin-2-associated pulmonary endothelial cell dysfunction in indirect acute lung injury in mice. American journal of respiratory cell and molecular biology. 2014;50:193-200
23. Huang X, Dai Z, Cai L, Sun K, Cho J, Albertine KH. et al. Endothelial p110γPI3K Mediates Endothelial Regeneration and Vascular Repair Following Inflammatory Vascular Injury. Circulation. 2016;133:1093-103
24. Di A, Mehta D, Malik AB. ROS-activated calcium signaling mechanisms regulating endothelial barrier function. Cell calcium. 2016;60:163-71
25. Li X, Fang P, Yang WY, Chan K, Lavallee M, Xu K. et al. Mitochondrial ROS, uncoupled from ATP synthesis, determine endothelial activation for both physiological recruitment of patrolling cells and pathological recruitment of inflammatory cells. Canadian journal of physiology and pharmacology. 2017;95:247-52
26. Dikalov SI, Ungvari Z. Role of mitochondrial oxidative stress in hypertension. American journal of physiology Heart and circulatory physiology. 2013;305:H1417-27
27. Usatyuk PV, Vepa S, Watkins T, He D, Parinandi NL, Natarajan V. Redox regulation of reactive oxygen species-induced p38 MAP kinase activation and barrier dysfunction in lung microvascular endothelial cells. Antioxidants & redox signaling. 2003;5:723-30
28. Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of Endothelial Junctional Permeability. Annals of the New York Academy of Sciences. 2008;1123:134-45
29. Natarajan V. Oxidants and signal transduction in vascular endothelium. The Journal of laboratory and clinical medicine. 1995;125:26-37
30. Coyle CH, Martinez LJ, Coleman MC, Spitz DR, Weintraub NL, Kader KN. Mechanisms of H2O2-induced oxidative stress in endothelial cells. Free Radic Biol Med. 2006;40:2206-13
31. Nanasinkam SP, Lee JD, Sottosantiago S, Stearman RS, Keith RL, Choudhury Q. et al. Prostacyclin prevents pulmonary endothelial cell apoptosis induced by cigarette smoke. American Journal of Respiratory & Critical Care Medicine. 2007;175:676-85
32. Zong D, Li J, Cai S, He S, Liu Q, Jiang J. et al. Notch1 regulates endothelial apoptosis via the ERK pathway in chronic obstructive pulmonary disease. American journal of physiology Cell physiology. 2018;315:C330-C340
33. Zhang M, Tang J, Shan H, Zhang Q, Yang X, Zhang J. et al. p66Shc Mediates Mitochondrial Dysfunction Dependent on PKC Activation in Airway Epithelial Cells Induced by Cigarette Smoke. 2018. 2018:5837123
34. Mitra A, Vishweswaraiah S, Thimraj TA, Maheswarappa M, Krishnarao CS, Sundararaja Lokesh K. et al. Association of Elevated Serum GM-CSF, IFN-gamma, IL-4, and TNF-alpha Concentration with Tobacco Smoke Induced Chronic Obstructive Pulmonary Disease in a South Indian Population. 2018. 2018:2027856
35. Madani A, Alack K, Richter MJ, Kruger K. Immune-regulating effects of exercise on cigarette smoke-induced inflammation. Journal of inflammation research. 2018;11:155-67
36. Bauernfeind F, Ablasser A, Bartok E, Kim S, Schmid-Burgk J, Cavlar T. et al. Inflammasomes: current understanding and open questions. Cellular and molecular life sciences: CMLS. 2011;68:765-83
37. Pauwels NS, Bracke KR, Dupont LL, Van Pottelberge GR, Provoost S, Vanden Berghe T. et al. Role of IL-1alpha and the Nlrp3/caspase-1/IL-1beta axis in cigarette smoke-induced pulmonary inflammation and COPD. The European respiratory journal. 2011;38:1019-28
38. MacNee W. Pulmonary and systemic oxidant/antioxidant imbalance in chronic obstructive pulmonary disease. Proceedings of the American Thoracic Society. 2005;2:50-60
39. Rahman I, Kilty I. Antioxidant therapeutic targets in COPD. Current Drug Targets. 2006;7:707-20
40. Dan Dunn J, Alvarez LA, Zhang X, Soldati T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox biology. 2015;6:472-85
41. Passos JF, Zglinicki T. Mitochondrial dysfunction and cell senescence-skin deep into mammalian aging. Aging. 2012;4:74-5
42. Seo AY, Joseph AM, Dutta D, Hwang JC, Aris JP, Leeuwenburgh C. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. Journal of cell science. 2010;123:2533-42
43. Boukhenouna S, Wilson MA, Bahmed K, Kosmider B. Reactive Oxygen Species in Chronic Obstructive Pulmonary Disease. 2018. 2018:5730395
44. Yue L, Yao H. Mitochondrial dysfunction in inflammatory responses and cellular senescence: pathogenesis and pharmacological targets for chronic lung diseases. Br J Pharmacol. 2016;173:2305-18
45. Kaisar MA, Prasad S, Cucullo L. Protecting the BBB endothelium against cigarette smoke-induced oxidative stress using popular antioxidants: Are they really beneficial?. Brain research. 2015;1627:90-100
46. Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RAJ, Murphy MP. et al. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. Faseb Journal Official Publication of the Federation of American Societies for Experimental Biology. 2005;19:1088-95
47. Dare AJ, Logan A, Prime TA, Rogatti S, Goddard M, Bolton EM. et al. The mitochondria-targeted anti-oxidant MitoQ decreases ischemia-reperfusion injury in a murine syngeneic heart transplant model. Journal of Heart & Lung Transplantation. 2015;34:1471-80
48. Wiegman CH, Charalambos M, Gulammehdi H, Priyanka N, Clarke CJ, Russell KE. et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. Journal of Allergy & Clinical Immunology. 2015;136:769-80
49. Wang H, Lv C, Wang S, Ying H, Weng Y, Yu W. NLRP3 Inflammasome Involves in the Acute Exacerbation of Patients with Chronic Obstructive Pulmonary Disease. Inflammation. 2018;41:1321-33
50. Sayan M, Mossman BT. The NLRP3 inflammasome in pathogenic particle and fibre-associated lung inflammation and diseases. Particle and fibre toxicology. 2016;13:51
51. Dashdorj A, Jyothi KR, Lim S, Jo A, Nguyen MN, Ha J. et al. Mitochondria-targeted antioxidant MitoQ ameliorates experimental mouse colitis by suppressing NLRP3 inflammasome-mediated inflammatory cytokines. BMC medicine. 2013;11:178
52. Ito S, Araya J, Kurita Y, Kobayashi K, Takasaka N, Yoshida M. et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy. 2015;11:547-59
53. Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T. et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. The Journal of clinical investigation. 2014;124:3987-4003
54. Frank M, Duvezincaubet S, Koob S, Occhipinti A, Jagasia R, Petcherski A. et al. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochimica Et Biophysica Acta. 2012;1823:2297-310
55. Jessop F, Hamilton RF, Rhoderick JF, Shaw PK, Holian A. Autophagy deficiency in macrophages enhances NLRP3 inflammasome activity and chronic lung disease following silica exposure. Toxicol Appl Pharmacol. 2016;309:101-10
Author contact
![]() Corresponding authors: E-mail addresses: kongxxedu.cn (X.Kong), taiyang2630com (L.Yang).
Corresponding authors: E-mail addresses: kongxxedu.cn (X.Kong), taiyang2630com (L.Yang).
Received 2018-9-25
Accepted 2019-1-12
Published 2019-6-2