ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Data and Methods
Results and Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2019; 15(7):1452-1459. doi:10.7150/ijbs.31957 This issue Cite
Research Paper
SAROTUP: a suite of tools for finding potential target-unrelated peptides from phage display data
Bifang He1,2, Heng Chen1, Ning Li2, Jian Huang2, ![]()
1. School of Medicine, Guizhou University, Guiyang 550025, China
2. Center for Informational Biology, University of Electronic Science and Technology of China, Chengdu 611731, China
Abstract

SAROTUP (Scanner And Reporter Of Target-Unrelated Peptides) 3.1 is a significant upgrade to the widely used SAROTUP web server for the rapid identification of target-unrelated peptides (TUPs) in phage display data. At present, SAROTUP has gathered a suite of tools for finding potential TUPs and other purposes. Besides the TUPScan, the motif-based tool, and three tools based on the BDB database, i.e., MimoScan, MimoSearch, and MimoBlast, three predictors based on support vector machine, i.e., PhD7Faster, SABinder and PSBinder, are integrated into SAROTUP. The current version of SAROTUP contains 27 TUP motifs and 823 TUP sequences. We also developed the standalone SAROTUP application with graphical user interface (GUI) and command line versions for processing deep sequencing phage display data and distributed it as an open source package, which can perform perfectly locally on almost all systems that support C++ with little or no modification. The web interfaces of SAROTUP have also been redesigned to be more self-evident and user-friendly. The latest version of SAROTUP is freely available at
Keywords: target-unrelated peptide, phage display, biopanning, high-throughput sequencing, computational toolkit
Introduction
Phage display is a powerful in vitro selection technique, which enables the identification of high-affinity peptides or antibodies from libraries of phage particles displaying highly diverse peptides [1]. Libraries with a great variety of peptides are subject to one or more rounds of affinity selection, also called biopanning, which contains repetitive rounds of target-binding (selection) and proliferation [2]. The rapid isolation of ligands for a distinct target by biopanning has a wide range of applications extending from epitope determination [2, 3], protein-protein interaction detection [4], to new vaccines, diagnostics and therapeutics development [5-7]. Additionally, the selection from phage-displayed libraries has been increasingly employed in the design of new chemicals [8] and the development of new materials [9].
In recent years, next-generation sequencing (NGS) has substantially contributed to the analysis of phage-displayed screens [10-15]. In sharp contrast with traditional phage display, phage display selections powered by NGS can produce enormous data output and facilitate the finding of specific binders by avoiding iterative selections and restraining the number of false positive hits [11]. However, both classical phage display and next-generation phage display (NGPD) screens are deeply troubled by the emergence of target-unrelated peptides (TUPs). Derda et al. described and confirmed that selection from phage display libraries is driven by two independent pressures [10, 16]: (1) the selection-related pressure which enriches for clones that bind to the desired target or non-target-related components (e.g., protein A/G, bead) of the biopanning system during the selection step [17]; (2) The proliferation-related pressure which enriches clones exhibiting faster proliferation abilities during the amplification step [18-20]. Hence, biopanning output is actually a mixture of true binders and TUPs. Those TUPs binding to other components of the selection system rather than the desired target are categorized as selection-related TUPs (SrTUPs) [17], whereas those TUPs with an amplification advantage are referred as to propagation-related TUPs (PrTUPs) [18]. Obviously, these false positive hits repeatedly arising in biopanning results are not appropriate candidates for the development of new diagnostics, therapeutics, and vaccines and should be excluded from phage display data.
To identify and exclude putative TUPs from phage display data, several experimental approaches have been proposed. Derda and coworkers resorted to deep sequencing and statistical analysis to identify TUP candidates possessing a proliferation advantage. By sequencing the naïve Ph.D.-7 phage display library and the same library after one round of amplification using next generation sequencing platforms, they found a population of fast-propagating clones displaying 770 unique peptides by differential enrichment analysis [10]. Concurrently, Hall and fellows proposed a very efficient and convenient assay based on propagation rates to diagnose PrTUPs, which involves incubating an E. coli culture with the amplified phage of interest and comparing its concentration at 135 min of incubation with that of normal-propagating phage. They demonstrated that at this point the concentration of fast-propagating phage was significantly higher than that of normal-propagating phage [20].
Although these experimental methods are successful in the identification of TUPs, computational methods are playing an cumulatively important part in cleaning TUPs from biopanning results [21] (see Table 1 for more information). The INFO program in the RELIC suite was the first tool to report PrTUPs [22], which is based on information theory. Afterwards, several tools based on database search have been developed. PepBank has a Google-like search function, which can be utilized to find peptides already reported by other research groups [23]. The BDB database is a specialized archive for phage display data and can be used as a comprehensive platform for biologists to clean their panning results [24-27]. However, special tools for precluding target-unrelated peptides are still needed. In 2010, our group developed the first web tool for scanning, reporting and excluding possible TUPs and named it SAROTUP, which is the abbreviation for “Scanner And Reporter Of Target-Unrelated Peptides” [28]. This is a motif-based search tool and can be employed to find those TUPs with previously described motifs. Subsequently, the MimoSearch and MimoBlast tool based on database search were developed and integrated into SAROTUP [26]. To combat PrTUPs, we proposed PhD7Faster for predicting clones propagating faster from the Ph.D.-7 phage display peptide library [29]. We have also developed two support vector machine (SVM) based predictors, SABinder and PSBinder. SABinder allows the detection of streptavidin-binding peptides (SBP) [30], while PSBinder is a predictor for polystyrene surface-binding peptides (PSBP) [31]. However, these programs are unable to analyze large amounts of data derived from NGPD screens.
Summary of different TUPs cleaning tools and methods
| Program/method (year published) | Highlights and comments | Limitation(s) | Reference |
|---|---|---|---|
| INFO (2004) | Based on information theory to identify PrTUPs | Unable to access | [22] |
| TUPScan (2010) | Based on known TUP motifs to identify TUPs | Unable to discover TUPs which do not match TUP motifs incorporated in TUPScan | [28] |
| MimoSearch (2012) | Based on database search to find identical peptides to the query peptides | Unable to find peptides which are not stored in BDB | [26] |
| MimoBlast (2012) | Powered by BLASTP to find peptides in the BDB database very similar to the query peptides | Unable to find peptides which are not stored in BDB | [26] |
| PhD7Faster (2014) | SVM-based tool to predict phage clones with proliferation advantages from Ph.D.-7 phage display library | Unable to predict PrTUPs from other types of phage display libraries except Ph.D.-7 phage display library | [29] |
| SABinder (2016) | SVM-based predictor to detect streptavidin-binding peptides | Unable to predict other types of TUPs except SBP | [30] |
| PSBinder (2017) | SVM-based predictor to identify polystyrene surface-binding peptides | Unable to predict other types of TUPs except PSBP | [31] |
Phage display coupled with NGS technology has been used in over twenty reports [11, 13, 15, 32-50]. Many computational methods for converting raw sequencing data to peptide sequences and frequencies [51, 52], target-binding motif analysis [15, 53-55] and finding candidate target-binding ligands [56, 57] have been proposed. While these programs address specificity, selectivity and affinity of peptides, they do not incorporate a procedure to eliminate TUPs. As NGPD data are noisy datasets, TUPs should be excluded by de-noising tools; then target-binding motifs and ligands analyses can be performed. To the best of our knowledge, there is no tool for mining TUPs in large-scale NGPD datasets. Due to the continuing popularity of NGPD, there is a growing demand for TUPs cleaning tools for “big phage display data.”
In this study, we make an important update to the SAROTUP suite. We developed the standalone SAROTUP application with graphical user interface (GUI) and command line version for processing NGS phage display data and distribute it as an open source package, which can perform perfectly locally on almost all systems that support C++ with little or no modification. We also compiled many new TUP motifs and sequences into the motif-based tool and integrated three tools based on SVM into the latest version of SAROTUP. Furthermore, the web interface of SAROTUP has also been redesigned to be more self-evident. SAROTUP has been developed into a suite of tools for TUP detecting and data preprocessing, which is freely available at http://i.uestc.edu.cn/sarotup3.
Data and Methods
New TUP motifs and sequences
SAROTUP, the motif-based tool, was developed in 2010, based on only 23 TUP motifs known till then [28]. In 2011, Vodnik and fellows characterized and revealed already known and new target-unrelated peptides [58]. Whereafter, they confirmed that HWGMWSY was a plastic binder instead of a faster propagating sequence [59]. Recently, Derda and coworkers found 770 parasitic sequences ('parasites') that grew fast during amplification [10]. Furthermore, 29 fast-propagating phage clones were reported which displayed 29 distinct peptides [20, 60]. All TUPs from the above references were incorporated in SAROTUP 3.1. We also analyzed the phage display data in the BDB database released on July 23, 2018 [24]. Those peptides which were selected by four or more completely different targets were suspected TUPs and also included into the TUPScan tool in the SAROTUP suite.
Tools in the SAROTUP suite. SAROTUP contains three categories of tools: motif-based tool(s), machine learning method-based tools and database-based tools. After analyzed by the SAROTUP toolkit, a part of putative TUPs (red pentagrams) in phage display results can be excluded.
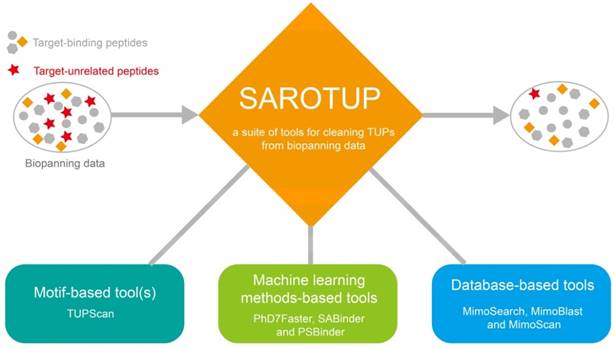
New data analysis tools integrated into SAROTUP
A series of data cleaning tools, which were based on database search and machine learning methods, were integrated into SAROTUP (Figure 1). Among them, MimoSearch is a batched peptide search tool for multiple peptide sequences to search against the BDB database, which is implemented as a CGI program with Perl [26]. The tool empowers biologists to seek out peptides in the database that are identical to their query sequences, as well as to verify if each sequence has been selected with diverse targets. Whereas MimoSearch can only find identical peptides, MimoBlast can find peptides identical to or very similar to the query sequences in BDB, which is powered by BLASTP 2.2.31+ [61] and the BDB database. MimoScan is designed to check if there is any peptide in the BDB database that matches the query patterns. The main algorithm of the tool is to convert query pattern to regular expression, and the latter is used by the MimoScan script to scan all peptides in the BDB database and find matched peptides.
The PhD7Faster tool is a predictor that can be used to predict if phages bearing peptides from the Ph.D.-7 library might grow faster. The positive training data of PhD7Faster 1.0 were peptides with 15 or higher copy numbers in the naïve Ph.D.-7 phage display library after one round of amplification [11]. Ru et al. thought that a fast-growing peptide-displaying phage should have a high copy number after proliferation, but did not consider copy numbers of these clones in the naïve library [29]. Phage clones with displayed peptides found by this way may not have enhanced propagation rates. Therefore, PhD7Faster was redeveloped with parasitic peptides identified by Derda and colleagues (considering peptide abundance in both libraries) [62].
Since streptavidin is frequently used in the biopanning system either as the target or the anchoring molecule, SBP would present in biopanning results in such cases. SABinder was developed based on SVM to predict if peptides might be streptavidin binders [30]. PSBP are also a very common type of TUPs in the screening of phage-displayed libraries. PSBinder [31], a SVM-based predictor, was assembled into SAROTUP to detect and exclude these noisy peptides.
Standalone SAROTUP application
We used open source Qt 5.6 in the creation of the SAROTUP application under the GPL & LGPLv3 licenses, which is a cross-platform application framework widely used for developing application software that can run on various platforms with little or no change in the underlying codebase. A modern GUI for SAROTUP was designed, and all tools in SAROTUP were redeveloped using C++ language. A command line version has also been developed. All versions can be downloaded at http://i.uestc.edu.cn/sarotup3/download.html.
Test dataset construction
The test dataset was collected from [10] (http://www.chem.ualberta.ca/~derda/parasitepaper/rawfiles/NoMuPhD7-GTA-30FuR.txt), which was from the naïve Ph.D.-7 phage display library. Nucleotide sequences and copy numbers in the dataset were trimmed. Those peptides with the same peptide sequences were combined. Finally, a large dataset with 3.05×106 unique peptide sequences was used to test the performance and efficiency of SAROTUP (see .txt file in the Supplementary Information). Each peptide in the dataset contains 7 amino acids.
Results and Discussion
New TUP motifs and sequences compiled into TUPScan
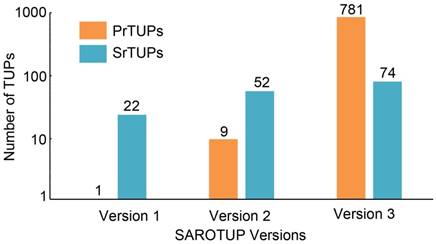
SAROTUP 1.0, released in 2010, contained only twenty-two SrTUPs and one PrTUPs [28]. In 2012, we compiled 52 SrTUPs and 9 PrTUPs into SAROTUP 2.0. At present, 74 SrTUPs and 781 PrTUPs are incorporated into TUPScan of SAROTUP 3.1. Both SrTUPs and PrTUPs have increased substantially, compared with those of previous versions (Figure 2). The algorithm of TUPScan remains largely the same as in the original [28].
Application of the tools in the SAROTUP suite
Each tool in the SAROTUP package can be utilized to process NGPD data locally, while the online version of each tool can only be employed to handle small-scale traditional phage display data. The use of the tools is declared here: (1) Use TUPScan to eliminate peptides matching with any previously known TUP motifs; (2) Use MimoSearch to remove peptides identical to those in the BDB database selected by various kinds of targets; (3) Use MimoBlast to exclude peptides remarkably similar to those in the BDB database with different targets; (4) Use MimoScan to find peptides in the BDB database with known TUP motifs; (5) Use PhD7Faster to predict peptide-displaying clones with enhanced propagation advantages if they are isolated from the popular Ph.D.-7 phage display library (New England Biolabs); (6) Use SABinder to identify and filter peptides that likely bind to streptavidin if the protein just serves as a part of the biopanning system instead of the target molecule; (7) Use PSBinder to detect and report polystyrene surface-binding peptides, thereby removing these false hits from the selected peptides.
TUPScan: a motif-based data cleaning tool
As TUPScan contains abundant TUP motifs or sequences and its results are fairly straightforward to be understood, we strongly recommend users to employ TUPScan firstly to identify and eliminate peptides matching previously characterized TUP motifs or sequences. However, this tool cannot find TUPs not matching reported TUP motifs. We scanned the testing dataset (from the naïve Ph.D.-7 phage display library) against TUPScan and found that 36197 unique peptides matched TUP motifs (see .xlsx file in the Supplementary Information). Whether TUPs are detected or not by TUPScan, we suggest users to use data cleaning tools based on database search for further filtering hidden TUPs.
MimoSearch, MimoBlast and MimoScan: data analysis tools based on database search
SAROTUP has three data analysis tools based on database search, i.e. MimoSearch, MimoBlast and MimoScan. MimoSearch is capable of finding peptides identical to query sequences in the BDB database. Actually, MimoScan and MimoBlast can also find peptides in the BDB database that are identical to query sequences. However, MimoSearch is the best choice to find peptides isolated by various targets because the target information will be explicitly displayed in its result table. On the contrary, users cannot directly get the target information from the result tables of MimoScan and MimoBlast, but can click the BiopanningDataSet ID linked to the BDB database to find corresponding targets. However, MimoScan and MimoBlast also have their own advantages. For example, MimoScan allows the identification of all sequences in BDB containing the query peptide, and MimoBlast can find all sequences in BDB similar to the query sequence besides the identical ones.
Peptides selected with four or more different targets
| Peptide | Number of Unique Targets | BiopanningDataset numbers | SAROTUP 2.0a |
|---|---|---|---|
| NFMESLPRLGMH | 8 | 8 | - |
| NRPDSAQFWLHH | 8 | 9 | - |
| AETVESC | 7 | 7 | - |
| EPLQLKM | 7 | 9 | - |
| NQDVPLF | 7 | 7 | - |
| GAMHLPWHMGTL | 6 | 6 | - |
| IPTLPSS | 6 | 10 | - |
| IQSPHFF | 6 | 6 | - |
| LTPCDT | 6 | 6 | - |
| TALATSSTYDPH | 6 | 6 | - |
| NHVHRMHATPAY | 5 | 5 | - |
| SGHQLLLNKMPN | 5 | 6 | - |
| SILSTMSPHGAT | 5 | 5 | - |
| YRAPWPP | 5 | 9 | - |
| YSIPKSS | 5 | 5 | - |
| GKPMPPM | 4 | 5 | - |
| SPNFSWLPLGTT | 4 | 4 | - |
| GWSDLHKLPPHT | 4 | 4 | - |
| NSLTPCGRTRDN | 4 | 4 | - |
| SHPWNAQRELSV | 4 | 4 | - |
| NSLTPCGRTRVTSC | 4 | 4 | - |
| NYLHNHPYGTVG | 4 | 4 | - |
| QDVHLTQQSRYT | 4 | 4 | - |
| RETADDLLSLLL | 4 | 4 | - |
| ILANDLTAPGPR | 4 | 4 | - |
| AREYGTRFSLIGGYR | 4 | 4 | - |
| CAREVTLLC | 4 | 6 | - |
| LPPNPTK | 4 | 4 | - |
| CGRTRDN | 4 | 8 | - |
| CGRTRVTSC | 4 | 8 | - |
| CTVRTSADC | 4 | 4 | - |
| LSTHTTESRSMV | 4 | 4 | - |
| SWMPHPRWSPQH | 4 | 4 | - |
| VSRHQSWHPHDL | 4 | 4 | - |
| YQLRPNAESLRF | 4 | 4 | - |
a In this column, '-' means no known TUP motif is found by SAROTUP2.0.
With the number of peptides in the BDB database constantly increasing, these tools have become more powerful. Accordingly, it is practical to mine new TUPs using these tools. MimoSearch, the batched peptide search tool, can be applied to check whether query peptides have been identified in multiple reported biopanning experiments. Due to a phage-displayed library with millions or billions of various peptides, the probability of acquiring an identical peptide with different targets is extremely low. If the same peptide has been isolated from peptide libraries with varied targets, it is more likely to be a TUP than an actual target-binding peptide. The peptide might be obtained as a result of having a propagation advantage or binding to components other than the target in the biopanning system. Such peptides should be excluded in case they would mislead further analysis. We employed MimoSearch to scan all peptides in the BDB databases against itself. As shown in Table 2, 35 new peptides were found to be suspected TUPs as each peptide was identified in the panning against four or more entirely different targets. These peptides were included in TUPScan.
Regardless of the results of MimoSearch, users are encouraged to use MimoBlast to detect any disguised TUP further. As the chance of selecting peptides with high degree of similarity from a large peptide library using different targets remains small, users can utilize MimoBlast to identify possible TUPs. Peptides highly similar to a known TUP sequence may also be TUPs. For example, SVSVGMNPSPRP is probably a TUP because it is almost identical to SVSVGMKPSPRP, which has been isolated by many different targets and is very likely to be a TUP [63]. If the former peptide emerges in the panning results, a BLAST against the BDB database would hint researchers that it may be a TUP.
MimoScan can find peptides with query patterns in BDB. It can be used to find other peptides in the BDB database matching with the query TUP motifs, thereby checking how specific the patterns derived from biopanning results are.
Growth of SrTUPs and PrTUPs in SAROTUP. Compared with previous versions of SAROTUP, the current version of SAROTUP (version 3) contains much more TUPs.
PhD7Faster, SABinder and PSBinder: data cleaning tools based on machine learning methods
PhD7Faster, SABinder and PSBinder are predictors developed by our lab for target-unrelated peptides, which are built with machine learning methods. PhD7Faster can predict if phages bearing query peptides from the Ph.D.-7 phage display library might grow faster. SABinder can be used to predict if peptides would bind to streptavidin. The PSBinder tool enables the prediction of polystyrene surface-binding peptides. It is important to keep in mind that users can use PhD7Faster to cull peptides possessing propagation advantages only if they are isolated from the Ph.D.-7 phage display library. Users can employ SABinder to filter SBP when streptavidin acts as a component of the screening system rather than the target. PSBinder can be applied to clean PSBP from phage display data if polystyrene plates exist in the biopanning system but not as the target of interest.
Ph.D.-7 phage display library is one of the most popular combinatorial libraries, and 494 (494/3264 = 15%) sets of phage display data in the BDB database are derived from selections of this library. However, multiple other types of libraries, such as Ph.D.-C7C, Ph.D.-12 and f88-15mer libraries, have been produced and employed for ligand discovery. There are more than 400 types of libraries curated in the BDB databases. The design of PhD7Faster indicates that deep sequencing of other naïve and amplified phage libraries can make it possible to develop computational tools for detecting putative PrTUPs in phage-encoded libraries other than Ph.D.-7 library. Furthermore, bioinformatics tools for predicting peptides binding to other common components (such as biotin, protein A and G and secondary antibody) of the screening system are necessary to be established, as these molecules usually exist for other purposes rather than act as the target of interest.
Performance testing
We employed the testing dataset to evaluate the time requirements of all tools in the SAROTUP suite. Each command line tool was run on a desktop computer with Intel Core i3 Processor and 4GB RAM (Windows system). MimoSearch, PhD7Faster, SABinder and PSBinder were able to complete the analysis of the large dataset within 30 minutes. MimoBlast and MimoScan can accomplish analysis within 1.5 hours. TUPScan can finish the analysis of this dataset within a single hour.
Web interface and standalone SAROTUP
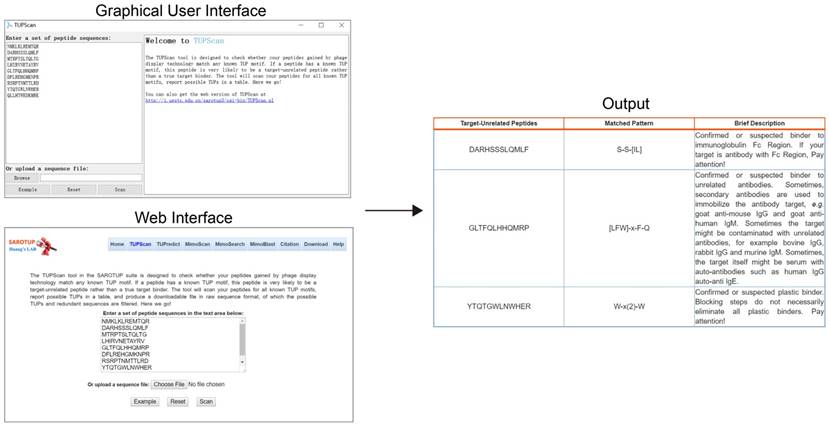
To facilitate the users to use SAROTUP, the web interfaces of SAROTUP have been redesigned to be more self-evident and user-friendly. And a detailed help information has been added to the help page. We also provided a version of SAROTUP with GUI, which was written in C++ and tested on Windows and Ubuntu systems. It is distributed as an open source package and can perform perfectly natively on almost all systems that support C++ with little or no modification. The source code is available at http://i.uestc.edu.cn/sarotup3/versions/Source_code.zip for free. The interface and utilization of the GUI version is similar to that of the web server. Let's take the TUPScan as an example. The GUI, web interface and output of TUPScan are shown in Figure 3. According to feedback from bioinformaticians, a command line version of SAROTUP has also been implemented. We strongly recommend that users use the command line version of these tools if the size of the dataset is very large.
Future development
Contending with TUPs in phage display needs everyone's efforts. Efficient identification of TUPs can be achieved if there is a shared-public database of TUP sequences in which many researchers participate and contribute sequences. We plan to implement such a database in the very near future. SAROTUP will be frequently updated to meet new requirements and demands.
Conclusions
SAROTUP has become a very popular and effective toolkit for TUP identification and prediction over the past few years. More TUP reporting tools have been integrated into the SAROTUP suite. We also developed the standalone version of each tool, which can be used to analyze traditional phage display data as well as NGPD data. This serious upgrade makes SAROTUP as an enhanced and versatile toolkit for scanning and reporting TUPs. The SAROTUP suite will help future reports on the development of new diagnostics, therapeutics, and vaccines. We hope that TUPs analysis will be established as a standard operating procedure in phage display field.
Abbreviations
TUPs: target-unrelated peptides; GUI: graphical user interface; NGS: next-generation sequencing; NGPD: next-generation phage display; SrTUPs: selection-related TUPs; PrTUPs: propagation-related TUPs; SVM: support vector machine; SBP: streptavidin-binding peptides; PSBP: polystyrene surface-binding peptides.
Web interface, GUI and output of TUPScan. The input interface of the standalone TUPScan is quite similar to that of the online one, and their output interfaces are almost the same.
Supplementary Material
Supplementary test peptides.
Supplementary TUPScan results.
Acknowledgements
This work was supported by the National Natural Science Foundation of China [grant number 61571095], the Fundamental Research Funds for the Central Universities of China [ZYGX2005Z006], the Sichuan Science and Technology Program [2018HH0154], and the 2018 Talent Research Program of Guizhou University [702569183301 and 702570183301].
Competing Interests
The authors have declared that no competing interest exists.
References
1. Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315-7
2. Pande J, Szewczyk MM, Grover AK. Phage display: concept, innovations, applications and future. Biotechnology advances. 2010;28:849-58
3. He B, Mao C, Ru B, Han H, Zhou P, Huang J. Epitope mapping of metuximab on CD147 using phage display and molecular docking. Computational and mathematical methods in medicine. 2013;2013:983829
4. Sundell GN, Ivarsson Y. Interaction analysis through proteomic phage display. BioMed research international. 2014;2014:176172
5. Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nature reviews Drug discovery. 2010;9:767-74
6. O'Rourke JP, Daly SM, Triplett KD, Peabody D, Chackerian B, Hall PR. Development of a mimotope vaccine targeting the Staphylococcus aureus quorum sensing pathway. PloS one. 2014;9:e111198
7. Hairul Bahara NH, Tye GJ, Choong YS, Ong EB, Ismail A, Lim TS. Phage display antibodies for diagnostic applications. Biologicals: journal of the International Association of Biological Standardization. 2013;41:209-16
8. Mannocci L, Leimbacher M, Wichert M, Scheuermann J, Neri D. 20 years of DNA-encoded chemical libraries. Chemical communications. 2011;47:12747-53
9. Nguyen TT, Lee HR, Hong SH, Jang JR, Choe WS, Yoo IK. Selective lead adsorption by recombinant Escherichia coli displaying a lead-binding peptide. Applied biochemistry and biotechnology. 2013;169:1188-96
10. Matochko WL, Cory Li S, Tang SK, Derda R. Prospective identification of parasitic sequences in phage display screens. Nucleic acids research. 2014;42:1784-98
11. t Hoen PA, Jirka SM, Ten Broeke BR, Schultes EA, Aguilera B, Pang KH. et al. Phage display screening without repetitious selection rounds. Analytical biochemistry. 2012;421:622-31
12. Christiansen A, Kringelum JV, Hansen CS, Bogh KL, Sullivan E, Patel J. et al. High-throughput sequencing enhanced phage display enables the identification of patient-specific epitope motifs in serum. Scientific reports. 2015;5:12913
13. Ngubane NA, Gresh L, Ioerger TR, Sacchettini JC, Zhang YJ, Rubin EJ. et al. High-throughput sequencing enhanced phage display identifies peptides that bind mycobacteria. PloS one. 2013;8:e77844
14. Jijakli K, Khraiwesh B, Fu W, Luo L, Alzahmi A, Koussa J. et al. The in vitro selection world. Methods. 2016;106:3-13
15. Rentero Rebollo I, Sabisz M, Baeriswyl V, Heinis C. Identification of target-binding peptide motifs by high-throughput sequencing of phage-selected peptides. Nucleic acids research. 2014;42:e169
16. Derda R, Tang SK, Li SC, Ng S, Matochko W, Jafari MR. Diversity of phage-displayed libraries of peptides during panning and amplification. Molecules. 2011;16:1776-803
17. Menendez A, Scott JK. The nature of target-unrelated peptides recovered in the screening of phage-displayed random peptide libraries with antibodies. Analytical biochemistry. 2005;336:145-57
18. Thomas WD, Golomb M, Smith GP. Corruption of phage display libraries by target-unrelated clones: diagnosis and countermeasures. Analytical biochemistry. 2010;407:237-40
19. Brammer LA, Bolduc B, Kass JL, Felice KM, Noren CJ, Hall MF. A target-unrelated peptide in an M13 phage display library traced to an advantageous mutation in the gene II ribosome-binding site. Analytical biochemistry. 2008;373:88-98
20. Nguyen KT, Adamkiewicz MA, Hebert LE, Zygiel EM, Boyle HR, Martone CM. et al. Identification and characterization of mutant clones with enhanced propagation rates from phage-displayed peptide libraries. Analytical biochemistry. 2014;462:35-43
21. He B, Dzisoo AM, Derda R, Huang J. Development and application of computational methods in phage display technology. Current medicinal chemistry. 2018 [Epub ahead of print]
22. Mandava S, Makowski L, Devarapalli S, Uzubell J, Rodi DJ. RELIC-a bioinformatics server for combinatorial peptide analysis and identification of protein-ligand interaction sites. Proteomics. 2004;4:1439-60
23. Shtatland T, Guettler D, Kossodo M, Pivovarov M, Weissleder R. PepBank-a database of peptides based on sequence text mining and public peptide data sources. BMC bioinformatics. 2007;8:280
24. He B, Chai G, Duan Y, Yan Z, Qiu L, Zhang H. et al. BDB: biopanning data bank. Nucleic acids research. 2016;44:D1127-32
25. Ru B, Huang J, Dai P, Li S, Xia Z, Ding H. et al. MimoDB: a new repository for mimotope data derived from phage display technology. Molecules. 2010;15:8279-88
26. Huang J, Ru B, Zhu P, Nie F, Yang J, Wang X. et al. MimoDB 2.0: a mimotope database and beyond. Nucleic acids research. 2012;40:D271-7
27. He B, Jiang L, Duan Y, Chai G, Fang Y, Kang J. et al. Biopanning data bank 2018: hugging next generation phage display. Database: the journal of biological databases and curation. 2018;2018:1-8
28. Huang J, Ru B, Li S, Lin H, Guo FB. SAROTUP: scanner and reporter of target-unrelated peptides. Journal of biomedicine & biotechnology. 2010;2010:101932
29. Ru B, t Hoen PA, Nie F, Lin H, Guo FB, Huang J. PhD7Faster: predicting clones propagating faster from the Ph.D.-7 phage display peptide library. Journal of bioinformatics and computational biology. 2014;12:1450005
30. He B, Kang J, Ru B, Ding H, Zhou P, Huang J. SABinder: a web service for predicting streptavidin-binding peptides. BioMed research international. 2016;2016:9175143
31. Li N, Kang J, Jiang L, He B, Lin H, Huang J. PSBinder: a web service for predicting polystyrene surface-binding peptides. BioMed research international. 2017;2017:5761517
32. Ernst A, Gfeller D, Kan Z, Seshagiri S, Kim PM, Bader GD. et al. Coevolution of PDZ domain-ligand interactions analyzed by high-throughput phage display and deep sequencing. Molecular bioSystems. 2010;6:1782-90
33. Ravn U, Gueneau F, Baerlocher L, Osteras M, Desmurs M, Malinge P. et al. By-passing in vitro screening-next generation sequencing technologies applied to antibody display and in silico candidate selection. Nucleic acids research. 2010;38:e193
34. Ng S, Lin E, Kitov PI, Tjhung KF, Gerlits OO, Deng L. et al. Genetically encoded fragment-based discovery of glycopeptide ligands for carbohydrate-binding proteins. Journal of the American Chemical Society. 2015;137:5248-51
35. Glanville J, Zhai W, Berka J, Telman D, Huerta G, Mehta GR. et al. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20216-21
36. Ryvkin A, Ashkenazy H, Smelyanski L, Kaplan G, Penn O, Weiss-Ottolenghi Y. et al. Deep Panning: steps towards probing the IgOme. PloS one. 2012;7:e41469
37. Ravn U, Didelot G, Venet S, Ng KT, Gueneau F, Rousseau F. et al. Deep sequencing of phage display libraries to support antibody discovery. Methods. 2013;60:99-110
38. Dias-Neto E, Nunes DN, Giordano RJ, Sun J, Botz GH, Yang K. et al. Next-generation phage display: integrating and comparing available molecular tools to enable cost-effective high-throughput analysis. PloS one. 2009;4:e8338
39. Fowler DM, Araya CL, Fleishman SJ, Kellogg EH, Stephany JJ, Baker D. et al. High-resolution mapping of protein sequence-function relationships. Nature methods. 2010;7:741-6
40. Bawazer LA, Newman AM, Gu Q, Ibish A, Arcila M, Cooper JB. et al. Efficient selection of biomineralizing DNA aptamers using deep sequencing and population clustering. ACS nano. 2014;8:387-95
41. Staquicini FI, Cardo-Vila M, Kolonin MG, Trepel M, Edwards JK, Nunes DN. et al. Vascular ligand-receptor mapping by direct combinatorial selection in cancer patients. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:18637-42
42. Ferrara F, Naranjo LA, D'Angelo S, Kiss C, Bradbury AR. Specific binder for Lightning-Link(R) biotinylated proteins from an antibody phage library. Journal of immunological methods. 2013;395:83-7
43. Saggy I, Wine Y, Shefet-Carasso L, Nahary L, Georgiou G, Benhar I. Antibody isolation from immunized animals: comparison of phage display and antibody discovery via V gene repertoire mining. Protein engineering, design & selection: PEDS. 2012;25:539-49
44. Venet S, Kosco-Vilbois M, Fischer N. Comparing CDRH3 diversity captured from secondary lymphoid organs for the generation of recombinant human antibodies. mAbs. 2013;5:690-8
45. Zhang H, Torkamani A, Jones TM, Ruiz DI, Pons J, Lerner RA. Phenotype-information-phenotype cycle for deconvolution of combinatorial antibody libraries selected against complex systems. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:13456-61
46. Rosander A, Guss B, Frykberg L, Bjorkman C, Naslund K, Pringle M. Identification of immunogenic proteins in Treponema phagedenis-like strain V1 from digital dermatitis lesions by phage display. Veterinary microbiology. 2011;153:315-22
47. McLaughlin ME, Sidhu SS. Engineering and analysis of peptide-recognition domain specificities by phage display and deep sequencing. Methods in enzymology. 2013;523:327-49
48. Scott BM, Matochko WL, Gierczak RF, Bhakta V, Derda R, Sheffield WP. Phage display of the serpin alpha-1 proteinase inhibitor randomized at consecutive residues in the reactive centre loop and biopanned with or without thrombin. PloS one. 2014;9:e84491
49. Zimmermann B, Gesell T, Chen D, Lorenz C, Schroeder R. Monitoring genomic sequences during SELEX using high-throughput sequencing: neutral SELEX. PloS one. 2010;5:e9169
50. Thiel WH, Bair T, Wyatt Thiel K, Dassie JP, Rockey WM, Howell CA. et al. Nucleotide bias observed with a short SELEX RNA aptamer library. Nucleic acid therapeutics. 2011;21:253-63
51. Matochko WL, Chu K, Jin B, Lee SW, Whitesides GM, Derda R. Deep sequencing analysis of phage libraries using Illumina platform. Methods. 2012;58:47-55
52. He B, Tjhung K, Bennett N, Chou Y, Rau A, Huang J. et al. Compositional bias in naïve and chemically-modified phage-displayed libraries uncovered by paired-end deep sequencing. Scientific reports. 2018;8:1214
53. Kim T, Tyndel MS, Huang H, Sidhu SS, Bader GD, Gfeller D. et al. MUSI: an integrated system for identifying multiple specificity from very large peptide or nucleic acid data sets. Nucleic acids research. 2012;40:e47
54. Alam KK, Chang JL, Burke DH. FASTAptamer: a bioinformatic toolkit for high-throughput sequence analysis of combinatorial selections. Molecular therapy Nucleic acids. 2015;4:e230
55. Krejci A, Hupp TR, Lexa M, Vojtesek B, Muller P. Hammock: a hidden Markov model-based peptide clustering algorithm to identify protein-interaction consensus motifs in large datasets. Bioinformatics. 2016;32:9-16
56. Fowler DM, Araya CL, Gerard W, Fields S. Enrich: software for analysis of protein function by enrichment and depletion of variants. Bioinformatics. 2011;27:3430-1
57. Brinton LT, Bauknight DK, Dasa SS, Kelly KA. PHASTpep: analysis software for discovery of cell-selective peptides via phage display and next-generation sequencing. PloS one. 2016;11:e0155244
58. Vodnik M, Zager U, Strukelj B, Lunder M. Phage display: selecting straws instead of a needle from a haystack. Molecules. 2011;16:790-817
59. Vodnik M, Strukelj B, Lunder M. HWGMWSY, an unanticipated polystyrene binding peptide from random phage display libraries. Analytical biochemistry. 2012;424:83-6
60. Zygiel EM, Noren KA, Adamkiewicz MA, Aprile RJ, Bowditch HK, Carroll CL. et al. Various mutations compensate for a deleterious lacZalpha insert in the replication enhancer of M13 bacteriophage. PloS one. 2017;12:e0176421
61. Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K. et al. BLAST+: architecture and applications. BMC bioinformatics. 2009;10:421
62. He B, Chen H, Huang J. PhD7Faster 2.0: predicting clones propagating faster from the Ph.D.-7 phage display library by coupling PseAAC and tripeptide composition. PeerJ. 2019 [accepted]
63. Estephan E, Dao J, Saab MB, Panayotov I, Martin M, Larroque C. et al. SVSVGMKPSPRP: a broad range adhesion peptide. Biomedizinische Technik Biomedical engineering. 2012;57:481-9
Author contact
![]() Corresponding author: Jian Huang, University of Electronic Science and Technology of China, Chengdu 611731, China. Tel: +86-28-8320-2351; Fax: +86-28-8320-8238; Email: hjedu.cn
Corresponding author: Jian Huang, University of Electronic Science and Technology of China, Chengdu 611731, China. Tel: +86-28-8320-2351; Fax: +86-28-8320-8238; Email: hjedu.cn
Received 2018-12-3
Accepted 2019-4-9
Published 2019-6-2